
Abstract
Background
Islands have traditionally been the centre of evolutionary biological research, but the dynamics of immigration and differentiation at continental islands have not been well studied. Therefore, we focused on the Japanese archipelago, the continental islands located at the eastern end of the Eurasian continent. While the Japanese archipelago is characterised by high biodiversity and rich freshwater habitats, the origin and formation mechanisms of its freshwater organisms are not clear. In order to clarify the history of the planorbid gastropod fauna, we conducted phylogenetic analysis, divergence time estimation, ancestral state reconstruction, and lineage diversity estimations.
Results
Our analyses revealed the formation process of the planorbid fauna in the Japanese archipelago. Most lineages in the Japanese archipelago have closely related lineages on the continent, and the divergence within the Japanese lineages presumably occurred after the late Pliocene. In addition, each lineage is characterised by different phylogeographical patterns, suggesting that immigration routes from the continent to the Japanese archipelago differ among lineages. Furthermore, a regional lineage diversity plot showed that the present diversity in the Japanese archipelago potentially reflects the differentiation of lineages within the islands after the development of the Japanese archipelago.
Conclusions
Although additional taxon sampling and genetic analysis focused on each lineage are needed, our results suggest that immigration from multiple routes just prior to the development of the Japanese archipelago and subsequent diversification within the islands are major causes of the present-day diversity of the Japanese planorbid fauna.
Similar content being viewed by others

Background
Islands have long been treated as an excellent model system of evolutionary biology, and critical evolutionary mechanisms that generate species diversity such as adaptive radiation have been revealed by studying islands [1,2,3,4]. In considering island biology, immigration is an important event. As in the case of oceanic islands where the biota could not be established without dispersal, dispersal over the ocean or immigration through a land bridge have a large influence on the current biodiversity of continental islands [5,6,7]. On the other hand, speciation and differentiation within an island also contribute greatly to the diversification of the present fauna [8,9,10,11,12,13,14]. The importance of speciation and differentiation within an island have been suggested based on molecular phylogenetic research (e.g. [11, 15,16,17]). However, most of the studies conducted to date have focused on terrestrial organisms, and the origin of the biodiversity of freshwater organisms on islands it is not well known.
We accordingly focused on the freshwater fauna of the continental Japanese archipelago (including the Ryukyu Islands). The Japanese archipelago, which includes continental islands located at the eastern end of the Eurasian continent, is considered to be a global hotspot of biodiversity [18]. It is presumed to have formed mainly from land masses that separated from the Eurasian continent about 15 Ma [19,20,21,22,23]. Then, following the uplift of Fossa Magna after 6.0 Ma [24, 25] and the expansion of the Okinawa Trough (2.0–3.0 Ma) [26,27,28], the cleavage of the southern strait of the Japan Sea occurred 1.7 Ma (Fig. 1) [29,30,31,32]. This event rendered the Japanese archipelago a system of isolated islands. There were frequent connections and disconnections with the continent via land bridges due to sea level changes [22, 32,33,34]. This archipelago and its complex geography are an attractive model system for biogeographical studies, and a lot of research using molecular data has been published in recent years (e.g. [16, 35,36,37,38,39,40,41,42,43]). However, fewer biogeographical studies have been conducted using freshwater organisms in the Japanese archipelago. Most of these studies have focused on freshwater fishes. These studies have revealed the geographical genetic structure and clarified the formation mechanism of strictly freshwater fish within the Japanese archipelago [28, 44,45,46,47,48,49,50,51,52]. However, only a few studies have focused on the entire Eastern Eurasian region (e.g. [53, 54]). There have been several biogeographical studies focused on freshwater insects (e.g. [55,56,57,58]) and crustaceans (e.g. [59,60,61]), but information obtained from these investigations is still limited. In the case of freshwater molluscs, one of the most diverse animal groups in the freshwater system, only a few phylogeographical studies have been conducted (e.g. [62,63,64,65]). In summary, the biogeography of freshwater organisms in the Japanese archipelago, in particular their origin and comprehensive formation mechanisms, is still not well known.

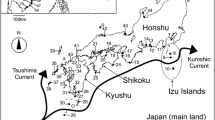
Map of the sample-collection sites. Colours and bars indicate areas where the samples were collected. A paleogeographic map in the lower left box shows geological history of the Japanese archipelago [32]. A sea passage between the continent and archipelago appeared in the southern part of the Japan Sea at 1.7 Ma, and the land bodies of the Japanese archipelago was separated completely from the continent
We accordingly focused on investigating Planorbidae, a group of small freshwater snails. Planorbidae is one of the taxa with the highest species diversity of freshwater molluscs in the Japanese archipelago [66]. Most species in the Japanese archipelago are also found on the Eurasian continent (or closely related species are found) [66,67,68,69]. Because freshwater snails have a low active dispersal ability and high passive dispersal ability [70], their geographical genetic structure may have been strongly influenced by a small number of long-range dispersal and diversification events within the islands. Hence, by conducting biogeographical research over the entire Eastern Eurasia region centred on the Japanese archipelago using planorbid snails, we expect to address the origin and diversification mechanisms of freshwater molluscs with few research cases within the continental Japanese archipelago.
Methods
Species sampling
We sampled 205 individuals from 163 sites from the Japanese archipelago, Russia, Vietnam, Mongolia, China, Hong Kong, Taiwan, South Korea, Philippines, the United States, Thailand, and Poland for analysis (Additional file 1 and Fig. 1). We used three Hygrophila species as outgroups, referring to the prior researches of higher phylogenetic position within Hygrophila [71, 72]. These samples including outgroups were identified by morphological characteristics using catalogues and lists of freshwater molluscs [66,67,68,69, 73]. However, the taxonomy of Planorbidae in East Asia is not fully known, and this investigation is beyond the scope of this research. Therefore, we limited the identification of samples to the genus except for some morphologically clear species. Furthermore, although species that have been hitherto identified as Gyraulus pulcher can be clearly distinguished based on their shell morphology [74, 75], the type specimens have some taxonomical problems [Saito T and Fukuda H, unpublished observations]. Hence, Gyraulus pulcher was provisionally treated as Gyraulus “pulcher”. Other Gyraulus species, except G. albus, G. biwaensis and G. parvus were not indentified here. For another Palearctic (mainly Russian) planorbids we used names of genus-group taxa treated by Starobogatov et al. [67]. However, we treated some subgenus instead of genus, because Starobogatov et al. [67] treated Choanomphalus as a genus including a number of taxa formerly treated as genera, but in contrast, recent Planorbidae molecular phylogenies [76, 77] showed that some subgenera were not closely related to other subgenenera. These are such genera as Vitreoplanorbis and Pseudogyraulus first described as subgenera of the genus Choanomphalus. In any case, all six genera (including at least 9 species) that were recorded in Japan as native were collected from the Japanese archipelago and these genera were also collected from continental Asia except for Camptoceras spp. from South and Southeast Asia. The summarized information of taxon sampling is shown in Table 1. The examined samples were deposited in Tohoku University Museum and Okayama University. Detailed information about the samples is listed in Additional file 1 and in Fig. 1.
Molecular methods
Total DNA was isolated from individual gastropods using Nucleospin tissue (TaKaRa, Shiga Pref., Japan) according to the manufacturer’s instructions. To conduct the phylogenetic analyses of Planorbidae, we sequenced fragments of the mitochondrial cytochrome c oxidase subunit 1 (CO1), the mitochondrial large ribosomal subunit (16S) and the nuclear Histone 3 (H3). The conditions of the polymerase chain reaction (PCR) and primers used are listed in Table 2. The PCR products were purified using Exo-SAP-IT (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). Sequencing was performed using a BigDye™ Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA, USA) and electrophoresed using an ABI 3130xl sequencer (Applied Biosystems, Carlsbad, CA, USA). The obtained CO1, 16S and H3 sequences have been deposited in the DDBJ/EMBL/GenBank database (Additional file 1).
Phylogenetic analyses
There was no gap in the alignment of CO1 and H3 except for a 15-bp insertion (this region was removed) and a 9-bp deletion of a few planorbid species in the CO1 sequences. These sequences were aligned with MUSCLE v3.8 [78]. To eliminate uncertainty of the 16S alignment, trimAl 1.2 [79] was used to select regions of the aligned sequences for analysis (Additional file 2). The phylogenetic trees were obtained using Bayesian inference (BI), maximum likelihood (ML), and neighbour-joining (NJ) methods. Prior to the BI and ML analyses, we used the program Kakusan4–4.0.2011.05.28 [80] to select the appropriate models of sequence evolution (Table 3). Based on these models, ML analysis was performed using RaxML [81] and Phylogears2, v2.2.2012.02.13 [82] software referring to recommended in the manual. For the ML analyses, we assessed nodal support by performing bootstrap analyses with 1000 replications. The BI analysis was performed using MrBayes v3.1.2 [83], with two simultaneous runs. Each run consisted of four simultaneous chains for eight million generations and sampling of trees every 100 generations. We discarded the first 8001 trees as burn-in after examining convergence and effective sample size (ESS) using Tracer v. 1.6 [84]; the remaining samples were used to estimate phylogeny. Then, the topologies of each single-locus tree (Additional files 3, 4, 5) were examined. There were no major inconsistencies among the analysed sequences in supported tree topology of three loci, with the proviso that the H3 tree had low resolution. Accordingly, phylogenies using the combined locus were estimated. The same protocols as in single-locus analysis were used in the combined-locus analysis. The selected model is also listed in Table 3. The ML analysis was performed with 1000 replications of the bootstrap analyses. The two simultaneous runs in the BI analysis consisted of four simultaneous chains for 18,000,000 generations and sampling of trees every 1000 generations. We discarded the first 3101 trees as burn-in after checking by Tracer v. 1.6, and the remaining samples were used to estimate phylogeny.
Divergence time estimation and ancestral state reconstruction
We estimated divergence time and conducted ancestral state reconstruction simultaneously using BEAST2 v. 2.4.4 [85] with same dataset with the following settings: tree prior = Yule process; ngen = 20,000,000; samplefreq = 1000; clock models = uncorrelated lognormal relaxed clock. Substitution models of each partition were set as follows: CO1 = GTR + Γ + I, 16S = GTR + Γ, H3 = GTR + Γ. These models were selected using Kakusan4–4.0.2011.05.28 [80] from available evolutionary models in BEAST2 v. 2.4.4 [85]. In addition, the CO1 model was chosen from models in which the molecular clock rate was considered by Wilke, Schultheiß & Albrecht [86]. This molecular clock rate is an average clock rate among the lineages of Protostomia. This rate is also very close to the reliable clock rate obtained by fossil-based calibration in freshwater molluscus [87]. A molecular clock rate (uniform prior) ranging from 0.0125–0.0206 (substitutions per site and My) was proposed for the COI gene for different Protostomia groups referring to the substitution models GTR + Γ + I (see [86]). The geographical region for the ancestral state reconstruction was determined as follows: Palearctic = I, Oriental = II, Nearctic = III, Japanese Archipelago = IV. In our analysis, six BEAST2 runs were conducted with same settings and combined using LogCombiner v. 2.4.4 (BEAST package). Finally, maximum clade credibility files were annotated in TreeAnnotator v. 2.4.4 (BEAST package; burn-in = 10%) summarizing the entire posterior distribution and ancestral state probability including a total of 108,005 trees after log and tree files were checked with Tracer v. 1.6 [84].
Estimating lineage diversity at internal nodes
To measure lineage diversity at internal nodes in the Japanese archipelago, we conducted three steps of analyses developed by Mahler et al. [88]. In step 1, we estimated the geographical location probabilities of each node in the tree using a Bayesian ancestral state reconstruction that we analysed using BEAST2 v. 2.4.4 [85] (see Section “Divergence time estimation and ancestral state reconstruction”). In step 2, we summed the location probabilities estimated for each region at all earlier nodes, obtaining lineage richness estimates for each region at each time. In step 3, we calculated the product by element of the vector of regional lineage richness at the focal time (from step 2) and the vector of location probabilities from the focal node (from step 1) to obtain the lineage diversity at the focal node. This final sum is the weighted mean of the estimated lineage diversities at each region at each time of our focal node. In addition, to clarify the mechanism of lineage diversification in the Japanese archipelago, we identified branches that occurred within the Japanese archipelago. We did not process nodes with location probabilities less than 0.70.
Results
Phylogenetic relationships
For the molecular phylogenetic analyses, the ESS values visualized in Tracer v. 1.6 were higher than 200. The inferred Bayesian phylogenetic relationships are shown in Fig. 2. All three estimated trees (BI, ML, and NJ) resulted in nearly identical topologies. The planorbid species from the Japanese archipelago included nine major clades, which we refer to as “A” to “I.” The monophyly of the nine clades was almost fully supported by all three methods. Seven of those nine clades consisted of Japanese samples and Eurasian samples, and the remaining two clades (C and I) consisted only of samples from the Japanese archipelago.

The Bayesian phylogenetic tree inferred from a combined dataset of mtDNA and nDNA sequences (CO1, 16S, and H3; 1375 bp). Radix sp., Physella acuta, and Acroloxus sp. are the outgroups chosen for the tree root. Each number and colour at the terminal branch of the tree indicates the sample number, species name and collected region (Fig. 1 and Additional file 1). Numbers at the branch nodes represent BPP, MLBV, and NJ. On the right side, the vertical bars indicate nominal clades
Clades A and B were composed of only Gyraulus spp. and these two clades were sister groups. The samples in clade A were collected from Palearctic, Oriental, and the Japanese archipelago. Although there were some supported monophyletic subclades, which tend to be united with samples in nearby regions, the phylogenetic relationships of inner clade A were not sufficiently resolved because many branches had low support values. The samples in clade B were collected from Palearctic regions and the Japanese archipelago. Clade B was subdivided into two subclades. One subclade was composed of mainly Palearctic samples (with the exception of one sample from the Japanese archipelago), and it was strongly supported. Another subclade consisted of both samples from the Japanese archipelago and the Palearctic region, but it was not sufficiently supported by BI (Bayesian posterior probabilities (BI) = 0.85). Clade C included only G. “pulcher” from the Japanese archipelago. This clade was a sister to the supported clade of European Anisus and Bathyomphalus, but it not sufficiently supported and its sister group was unclear. Clade D consisted of monophyletic G. soritai from the Japanese archipelago and Vitreoplanorbis hyaliniiformis from Palearctic. This clade was a sister to the monophyletic Planorbis planorbis. This monophyly was supported by the BI and ML methods (BI = 1.00, ML bootstrap value (BV) = 75). Clade E was composed of Helicorbis spp. from the Japanese archipelago and Palearctic and Oriental regions, and clade F was composed of Polypylis spp. from the Japanese archipelago and Palearctic and Oriental regions. These clades included some regionally supported subclades. Clade G consisted of Ferrissia spp. from the Japanese archipelago and Palearctic and Oriental regions and F. nipponica from the Japanese archipelago. Continental Ferrissia sp., Japanese F. nipponica, and Ferrissia sp. from Oriental regions and the Japanese archipelago created a well-supported monophyly. In addition, Continental Ferrissia sp. and Japanese F. nipponica were also a monophyly. Clade H consisted of Continental Culmenella sp. and Japanese Cu. prashadi, which were a strongly supported monophyly. Finally, Camptoceras hirasei was a strongly supported monophyly (clade I), and these two lineages exhibited a sister relationship.
Divergence time estimation and ancestral state reconstruction
For the molecular clock analyses, the ESS values visualized in Tracer v. 1.6 were considerably higher than 200. The inferred Bayesian phylogenetic relationships using BEAST2 appear in Fig. 3 and Table 4. The tree topology was nearly consistent with that obtained in the MrBayes and RaxML analyses (Fig. 2). In particular, the major nine clades (clades A through I) were enough supported (BPP > = 0.97) again, and the samples included in these clades were completely consistent with other phylogeny.

Maximum clade credibility tree generated by the BEAST2 analysis from the mtDNA and nDNA sequences (CO1, 16S, and H3; 1375 bp). The outgroups are not shown. On the right side, sample numbers and nominal clades are listed. Colour indicates the region of the collected samples or an estimation of region according to ancestral state reconstruction. The branches with a low location probability (< 0.70) and a high location probability (0.90) are shown using a striped pattern and a grid pattern, respectively. Node bars indicate 95% CI of the divergence time, and pink node bars indicate branches within the Japanese archipelago. The numbers or marks on the left side of each node indicate BPP. The BPP and node bars are only shown for the relatively supported (BPP > 0.90) nodes. The numbers on the right side of the nodes are the nominal clade number. In the lower part of the graph is the geologic time scale
In most cases, divergence first occurred between the lineages of the Asian continent and those of the Japanese Archipelago, and then divergence occurred within the lineages of the Japanese archipelago. The mean divergence times of the lineages of Asian continent and those of the Japanese Archipelago were early to middle Pleistocene (e.g. nodes 1, 9, 14 and 19). In contrast, divergence of the lineages within the Japanese Archipelago occurred after middle Pleistocene (e.g. nodes 8, 13, 18 and 20). These results of the dominant nodes are listed in Table 4. Other all results are listed in Additional file 6.
Lineage diversity
The estimated lineage diversity in the Japanese archipelago seems to have manifested around 3.0 Ma and diversified after 1.7 Ma (Fig. 4).

The lineage diversity plot using the method of Mahler [88]. The vertical axis indicates lineage diversity, and the horizontal axis indicates time. The pie chart shows the ancestral regional state in each node in ancestral state reconstruction analysis (Fig. 3). The colour indicates region. The circles with pink outline showed nodes that have a high probability Japanese ancestral state. The dotted pink line indicates the time that the Japanese archipelago formed as an archipelago by the cleavage of the south strait of the Japan Sea (1.7 Ma) [29,30,31,32]
Discussion
Our phylogeny clearly showed that planorbid snails in the Japanese archipelago do not have a single origin because samples from the Japanese archipelago constitute several monophyletic groups with continental samples at the nominal genus or species level. These clades have different biogeographical patterns, and they appear to be roughly divided into four types: northern clades (Figs. 2 and 3; clades B, D, and H), southern clades (Figs. 2 and 3; clades E and G), widely distributed clades (Figs. 2 and 3; clades A and F), and the endemic Japanese archipelago clades (Figs. 2 and 3; clades C and I). The northern clades consist of samples from the Japanese archipelago and the Palearctic. These samples from the Palearctic were collected in the northern part of the region, and these clades were located mainly in high-latitude areas. In contrast, the southern clade was composed of samples from the Japanese archipelago and Oriental and South Korea. This clade also seems to be distributed in mainly low-latitude areas in eastern Eurasia. On the other hand, widely distributed clades included samples from a wide range of eastern Eurasia. In these clades, regional subclades tend to be formed. Finally, the clades found only in the Japanese archipelago did not have closely related sister group in our analyses. However, Camptoceras spp. from the continent were not collected in this research, although this lineage is clearly distinguished by its shell morphology [89]. As a result, clade I is not discussed in this study. On the contrary, our phylogeny indicates that clade C may be an endemic lineage, although further investigation is needed.
These biogeographical patterns may be related to the origins of the planorbid populations in the Japanese archipelago. Japanese populations included northern clades and southern clades. The former originated on the northern Eurasian continent, and the latter originated on the southern Eurasia continent. The geological history of the Japanese archipelago supports this hypothesis. The Japanese archipelago was often connected with the Eurasian continent via a land or ice bridge [22, 32]. The following three conjunctions are known to be major routes with the continent: the south route via the Ryukyu archipelago, the north route via Sakhalin and the Kuril archipelago, and another route via the Korean Peninsula [22]. The Japanese population in the northern clade may have been established via immigration from the northern route, and likewise the Japanese population in the southern clade may have originated from the southern route. Furthermore, perhaps the Japanese population in widely distributed clades may be derived from the Korean Peninsula route. These biogeographical patterns have been noted from fossil records or comparisons of animal fauna between the Japanese archipelago and the continent (e.g. [90,91,92,93,94]) and, more recently, molecular phylogenetic studies of various taxa have provided certain evidence for this possibility (e.g. [28, 39, 41,42,43, 95]). We estimated the ancestral regional state at each branch that splits into Japanese and continental lineages, but our results were not necessarily clear. The ancestral regional state at the branches of clades B and E was estimated to be region I (Palearctic) and IV (the Japanese archipelago) with a relatively high probability (>.90). In addition, immigration from the Japanese archipelago to the continent was estimated in some branches within some major clades. Although these results appear to fluctuate depending on taxon sampling or regional classification, immigration from the Japanese archipelago to the continent may have occurred. Although immigration from the Japanese archipelago to the continent has often not been taken into consideration in the past, its importance has begun to be pointed out in recent years [28, 96], consistent with our analysis.
We have addressed the question as to when planorbid fauna in the Japanese archipelago were established. Our analysis suggests that the divergence time within major clades except for clade I was around the late Pliocene to the early Pleistocene. In particular, the estimated mean divergence time was concentrated between 2.0 and 2.5 Ma. In these clades, it is not easy to decide which branch emigrated from the continent to the Japanese archipelago. However, our analysis suggests that the foundation of the Japanese population occurred around 2.0–2.5 Ma, and this time scale may result from the development of the Japanese archipelago. The land bodies that formed the Japanese archipelago first separated from the continent 15 Ma [19,20,21,22,23]. During this event, the land bodies of the Japanese archipelago still partially connected with the Asian continent. A sea passage was developed in the southern part of the Japan Sea at 1.7 Ma [29,30,31,32], and the land bodies of the Japanese archipelago was separated completely from the continents (Fig. 1). This dramatic geological event occurred 1.7 Ma [29,30,31,32]. The planorbid fauna present in the Japanese archipelago today appear to be strongly influenced by the immigrations that occurred before Japan became an “island.” However, the estimated divergence time of a lot of the branches between the Japanese archipelago and the continent (e.g., nodes 6, 11, and 17) post-dates this period. These biogeographical patterns are likely to have been formed via immigration through the several connections from the continent after the Japanese archipelago had been established in its present-day location. Actually, since 1.7 Ma the Japanese archipelago has been sometimes connected with the continent [22, 32,33,34]. In addition, incidental long-range dispersal of freshwater snails due to birds, wind, ocean current, and desalination of the ocean should also be considered. In particular, dispersal by birds (i.e., snails attached to birds or eaten by birds) has been shown to be possible experimentally [97,98,99,100]. In fact, our phylogeny also indicated that gene flow of both regions could occur when the Japanese archipelago and the continent were temporally connected.
Despite the influence from the continent noted above, our results suggest that diversification of the Japanese planorbid lineages within the islands may be the main cause of their present-day diversity (Fig. 4). Although the time in this figure only use the mean estimated divergence time in BEAST2, our results suggest that differentiation within the island may have played a key role in lineage diversification despite the high passive dispersal ability of freshwater snails [70] and the frequent connection between the island and the continent. Such diversification within the island is caused by various mechanisms [2,3,4], but it is difficult to identify the mechanism based on our results. Nevertheless, some regional clades in our phylogeny suggest that geographical structure within the Japanese archipelago contributed to the diversification.
The time of the immigration and diversification estimated based our analyses is clearly more recent than that estimated by phylogeography of most freshwater fishes in Japan. Most divergence times within the Japanese archipelago of widely distributed strictly freshwater fish species are estimated to precede 1.7 Ma (e.g. [44, 47, 48, 50, 51, 53, 101]). As a consequence, the colonization from the continent occurred earlier. This difference between freshwater snails and fishes may be derived from differences in their ability and mode of dispersal. As noted above, freshwater snails have low active and high passive dispersal potential [70]. On the other hand, fish have strong active dispersal potential within well-connected river and wetland systems [102, 103], but have limited dispersal potential within vicariant water systems [104,105,106]. Additional taxon sampling and detailed analyses of population genetics are required to clarify the diversification mechanisms and biogeographic history of planorbids. However, our study shows that diversification occurred after the separation of the islands from the continent, an event that was crucial for creating the diversity of freshwater organisms in the Japanese archipelago today.
Conclusions
Our results have shown that most of the planorbid lineages in the Japanese archipelago have closely related groups on the continent. In each lineage, different biogeographical patterns were detected via phylogenetic analysis. In addition, the branches between the Japanese archipelago populations and the continental populations date back to 1.7 Ma before the Japanese archipelago formed as an “archipelago.” On the other hand, our analysis showed that the present diversity of Japanese planorbid lineages is mainly the result of differentiation within the Japanese archipelago. Although additional taxon sampling and genetic analysis focused on each lineage are necessary, our study shows that diversification within the islands is more crucial to creating the present diversity than the diversity that existed when the islands were not separated from the continent.
Abbreviations
- 16S:
-
Large ribosomal subunit
- BI:
-
Bayesian inference
- BPP:
-
Bayesian posterior probabilities
- BV:
-
Bootstrap value
- CO1:
-
Cytochrome c oxidase subunit 1
- ESS:
-
Effective sample size
- H3:
-
Histone 3
- Ma:
-
Mega annum
- ML:
-
Maximum likelihood
- NJ:
-
Neighbour-joining
- PCR:
-
Polymerase chain reaction
References
MacArthur RH, Wilson EO. The theory of island biogeography. Princeton: Princeton University Press; 1967.
Schluter D. The ecology of adaptive radiation. Oxford: Oxford University Press; 2000.
Whittaker RJ, Fernández-Palacios JM. Island biogeography: ecology, evolution, and conservation. Oxford: Oxford University Press; 2007.
Losos JB, Ricklefs RE. Adaptation and diversification on islands. Nature. 2009;457:830–6.
Sanmartín I, Ronquist F. Southern hemisphere biogeography inferred by event-based models: plant versus animal patterns. Syst Biol. 2004;53:216–43.
de Queiroz A. The resurrection of oceanic dispersal in historical biogeography. Trend Ecol Evol. 2005;20:68–73.
Cowie RH, Holland BS. Dispersal is fundamental to biogeography and the evolution of biodiversity on oceanic islands. J Biogeogr. 2006;33:193–8.
Losos JB, Schluter D. Analysis of an evolutionary species-area relationship. Nature. 2000;408:847–50.
Emerson BC, Kolm N. Species diversity can drive speciation. Nature. 2005;434:1015–7.
O’Grady P, DeSalle R. Out of Hawaii: the origin and biogeography of the genus Scaptomyza (Diptera: Drosophilidae). Biol Lett. 2008;4:195–9.
Gifford ME, Larson A. In situ genetic differentiation in a Hispaniolan lizard (Ameiva chrysolaema): a multilocus perspective. Mol Phylogenet Evol. 2008;49:277–91.
Whittaker RJ, Triantis KA, Ladle RJ. A general dynamic theory of oceanic island biogeography. J Biogeogr. 2008;35:977–94.
Chiba S, Cowie RH. Evolution and extinction of land snails on Oceanic Islands. Annu Rev Ecol Evol Syst. 2016;47:123–41.
Borregaard MK, Amorim IR, Borges PAV, Cabral JS, Fernández-Palacios JM, Field R, Heaney LR, Kreft H, Matthewa TJ, Olesen JM, Price J, Rigal F, Steinbauer MJ, Triantis KA, Valente L, Weigelt P, Whittaker RJ. Oceanic island biogeography through the lens of the general dynamic model: assessment and prospect. Biol Rev. 2017;92:830–53.
Parent CE, Caccone A, Petren K. Colonization and diversification of Galápagos terrestrial fauna: a phylogenetic and biogeographical synthesis. Philos Trans R Soc B. 2008;363:3347–61.
Sota T, Nagata N. Diversification in a fluctuating island setting: rapid radiation of Ohomopterus ground beetles in the Japanese Islands. Philos Trans R Soc B. 2008;363:3377–90.
Dietrich M, Wilkinson DA, Soarimalala V, Goodman SM, Dellagi K, Tortosa P. Diversification of an emerging pathogen in a biodiversity hotspot: Leptospira in endemic small mammals of Madagascar. Mol Ecol. 2014;23:2783–96.
Conservation International: Japan. https://www.conservation.org/global/japan/priority_areas/hotspots/asia-pacific/Pages/japanhotspot.aspx (2017). Accessed 10 Dec 2017.
Jolivet L, Tamaki K. Neogene kinematics in the Japan Sea region and volcanic activity of the Northeast Japan arc. Proceedings of the ocean drilling program. Scientific Results. 1992;127(/128):1311–27.
Jolivet L, Tamaki K, Fournier M. Japan Sea, opening history and mechanism: a synthesis. J Geophys Res Solid Earth. 1994;99:22237–59.
Otofuji YI. Large tectonic movement of the Japan arc in late Cenozoic times inferred from paleomagnetism: review and synthesis. Island Arc. 1996;5:229–49.
Yonekura N, Nogami M, Kaizuka S, Chinzei K. The geography of Japan 1 –review–. Tokyo. Tokyo: University Press; 2001.
Martin AK. Double saloon door tectonics in the Japan Sea, Fossa magna, and the Japanese Island arc. Tectonophysics. 2011;498:45–65.
Takeuchi A. Stress field and tectonic process during the Neogene and later period in the northern part of Nagano prefecture, Central Japan. J Geol Soc Jpn. 1977;83:679–91.
Takeuchi A. The post-6Ma diastrophic history of the Japanese Islands. The post-Pliocene sress field and tectonics course in north shin-etu province. Chikyu Monthly, vol. 21; 1999. p. 583–8.
Kamata H, Kodama K. Tectonics of an arc-arc junction: an example from Kyushu Island at the junction of the Southwest Japan arc and the Ryukyu arc. Tectonophysics. 1994;233:69–81.
Nakamura Y, Kameo K, Asahara Y, Ozawa T. Stratigraphy and geologic age of the Neogene Shimajiri Group in Kumejima Island, Ryukyu Islands, southwestern Japan. J Geol Soc Jpn. 1999;105:757–70.
Watanabe K, Takahashi H, Kitamura A, Yokoyama R, Kitagawa T, Takeshima H, Sato S, Yamamoto S, Takehama Y, Mukai T, Ohara K, Iguchi K. Biogeographical history of Japanese freshwater fishes: Phylogeographic approaches and perspectives. Jpn J Ichthyol. 2006;53:1–38.
Kitamura A, Takano O, Takada H, Omote H. Late Pliocene-early Pleistocene paleoceanographic evolution of the sea of Japan. Palaeogeogr Palaeoclimatol Palaeoecol. 2001;172:81–98.
Kitamura A. Molluscan response to past warming in the sea of Japan. Jpn J Benthol. 2002;57:119–30.
Kitamura A, Kimoto K. Reconstruction of the Southern Channel of the Japan Sea at 3.9-1.0 ma. Quat Res. 2004;43:417–34.
Kitamura A, Kimoto K. History of the inflow of the warm Tsushima current into the sea of Japan between 3.5 and 0.8 ma. Palaeogeogr Palaeoclimatol Palaeoecol. 2006;236:355–66.
Ono Y. The northern Landbridge of Japan. Quat Res. 1990;20:183–92.
Furukawa M, Fujitani T. Comparative Study on Pleistocene Paleogeographic Maps of Ryukyu Arc. Bull Faculty Sci Univ Ryukyus. 2014;98:1–8.
Sota T, Hayashi M. Comparative historical biogeography of Plateumaris leaf beetles Coleoptera: Chrysomelidae in Japan: interplay between fossil and molecular data. J Biogeogr. 2007;34:977–93.
Schoville SD, Uchifune T, Machida R. Colliding fragment islands transport independent lineages of endemic rock-crawlers (Grylloblattodea: Grylloblattidae) in the Japanese archipelago. Mol Phylogenet Evol. 2013;66:915–27.
Nakamura K, Denda T, Kokubugata G, Suwa R, Yang TYA, Peng CI, Yokota M. Phylogeography of Ophiorrhiza japonica (Rubiaceae) in continental islands, the Ryukyu archipelago, Japan. J Biogeogr. 2010;37:1907–18.
Kameda Y, Kato M. Terrestrial invasion of pomatiopsid gastropods in the heavy-snow region of the Japanese archipelago. BMC Evol Biol. 2011;11:118.
McKay BD. A new timeframe for the diversification of Japan’s mammals. J Biogeogr. 2012;39:1134–43.
Dufresnes C, Litvinchunk SN, Borzée A, Jang Y, Li JT, Miura I, Perrin N, Stöck M. Phylogeography reveals an ancient cryptic radiation in east-Asian tree frogs (Hyla japonica group) and complex relationships between continental and island lineages. BMC Evol Biol. 2016;16:253.
Sato JJ. A review of the processes of mammalian faunal assembly in Japan: insights from molecular Phylogenetics. In: Motokawa M, Kajihara H, editors. Species diversity of animals in Japan. Tokyo: Springer; 2017. p. 49–116.
Okamoto T. Historical biogeography of the terrestrial reptiles of Japan: a comparative analysis of geographic ranges and molecular phylogenies. In: Motokawa M, Kajihara H, editors. Species diversity of animals in Japan. Tokyo: Springer; 2017. p. 135–64.
Tojo K, Sekiné K, Suzuki T, Saito R, Takenaka M. The species and genetic diversities of insects in Japan, with special reference to the aquatic insects. In: Motokawa M, Kajihara H, editors. Species diversity of animals in Japan. Tokyo: Springer; 2017a. p. 229–48.
Setiamarga DHE, Miya M, Yamanoue Y, Azuma Y, Inoue JG, Ishiguro NB, Mabuchi K, Nishida M. Divergence time of the two regional medaka population in Japan as a new time scale for comparative genomics of vertebrates. Biol Lett. 2009;5:812–6.
Saitoh K, Chen WJ, Mayden RL. Extensive hybridization and tetrapolyploidy in spined loach fish. Mol Phylogenet Evol. 2010;56:1001–10.
Miyake T, Nakajima J, Onikura N, Ikemoto S, Iguchi K, Komaru A, Kawamura K. The genetic status of two subspecies of Rhodeus atremius, an endangered bitterling in Japan. Conserv Genet. 2011;12:383–400.
Kakioka R, Kokita T, Tabata R, Mori S, Watanabe K. The origins of limnetic forms and cryptic divergence in Gnathopogon fishes (Cyprinidae) in Japan. Environ Biol Fish. 2013;96:631–44.
Watanabe K, Mori S, Tanaka T, Kanagawa N, Itai T, Kitamura J, Suzuki N, Tominaga K, Kakioka R, Tabata R, Abe T, Tashiro Y, Hashimoto Y, Nakajima J, Onikura N. Genetic population structure of Hemigrammocypris rasborella (Cyprinidae) inferred from mtDNA sequences. Ichthyol Res. 2014;61:352–60.
Komiya T, Fujita-Yanagibayashi S, Watanabe K. Multiple colonization of Lake Biwa by Sarcocheilichthys fishes and their population history. Environ Biol Fish. 2014;97:741–55.
Kitanishi S, Hayakawa A, Takamura K, Nakajima J, Kawaguchi Y, Onikura N, Mukai T. Phylogeography of Opsariichthys platypus in Japan based on mitochondrial DNA sequences. Ichthyol Res. 2016;63:506–18.
Nakagawa H, Seki S, Ishikawa T, Watanabe K. Genetic population structure of the Japanese torrent catfish Liobagrus reinii (Amblycipitidae) inferred from mitochondrial cytochrome b variations. Ichthyol Res. 2016;63:333–46.
Tabata R, Kakioka R, Tominaga K, Komiya T, Watanabe K. Phylogeny and historical demography of endemic fishes in Lake Biwa: the ancient lake as a promoter of evolution and diversification of freshwater fishes in western Japan. Ecol Evol. 2016;6:2601–23.
Takada M, Tachihara K, Kon T, Yamamoto G, Iguchi K, Miya M, Nishida M. Biogeography and evolution of the Carassius auratus-complex in East Asia. BMC Evol Biol. 2010;10:7.
Gao Y, Wang SY, Luo J, Murphy RW, Du R, Wu SF, Zhu CL, Li Y, Poyarkov AD, Nguyen SN, Luan PT, Zhang YP. Quaternary palaeoenvironmental oscillations drove the evolution of the Eurasian Carassius auratus complex (Cypriniformes, Cyprinidae). J Biogeogr. 2012;39:2264–78.
Kato Y, Morii Y, Tojo K. Molecular phylogeographic analysis of east Asian cryptoperlan stoneflies (Insecta: Plecoptera, Peltoperlidae). Limnology. 2013;14:179–94.
Suzuki T, Kitano T, Tojo K. Contrasting genetic structure of closely related giant water bugs: phylogeography of Appasus japonicus and Appasus major (Insecta: Heteroptera, Belostomatidae). Mol Phylogenet Evol. 2014;72:7–16.
Saito R, Jo J, Sekiné K, Bae YJ, Tojo K. Phylogenetic analyses of the isonychiid mayflies (Ephemeroptera: Isonychiidae) in the northeast palearctic region. Entomol Res. 2016;46:246–59.
Saito R, Tojo K. Complex geographic- and habitat-based niche partitioning of an east Asian habitat generalist mayfly Isonychia japonica (Ephemeropter:Isonychiidae) with reference to differences in genetic structure. Freshw Sci. 2016;35:712–23.
Makino W, Tanabe AS. Extreme population genetic differentiation and secondary contact in the freshwater copepod Acanthodiaptomus pacificus in the Japanese archipelago. Mol Ecol. 2009;18:3699–713.
Koizumi I, Usio N, Kawai T, Azuma N, Masuda R. Loss of genetic diversity means loss of geological information: the endangered Japanese crayfish exhibits Remarkble historical footprints. PLoS One. 2012;7:e33986.
Tomikawa K, Nakano T, Sato A, Onodera Y, Ohtaka A. A molecular phylogeny of Pseudocrangonyx from Japan, including a new subterranean species (Crustacea, Amphipoda, Pseudocrangonyctidae). Zoosystematics Evol. 2016;92:187–202.
Miura O, Köhler F, Lee T, Li J, Ó Foighil D. Rare, divergent Korean Semisulcospira spp. mitochondrial haplotypes have Japanese sister lineages. J Molluscan Stud. 2013;79:86–9.
Köhler F. Rampant taxonomic incongruence in a mitochondrial phylogeny of Semisulcospira freshwater snails from Japan (Cerithioidea: Semisulcospiridae). J Molluscan Stud. 2016;82:268–81.
Hirano T, Saito T, Chiba S. Phylogeny of freshwater viviparid snails in Japan. J Molluscan Stud. 2015;81:435–41.
Sano I, Shirai A, Kondo T, Miyazaki J. Phylogenetic relationships of Japanese Unionoida (Mollusca: Bivalvia) based on mitochondrial 16S rDNA sequences. J Water Resour Prot. 2017;9:493–509.
Masuda O, Uchiyama R. Freshwater mollusks of Japan 2: freshwater mollusks of Japan. Including brackish water species. Tokyo: Pisces; 2004. p. 2004.
Starobogatov YI, Prozorova LA, Bogatov VV, Molluscs SEM. In: Bogatov VV, Tsalolikhin SJ, editors. Key to Freshwater Invertebrates of Russia and adjacent lands, Vol. 6: Molluscus, Polychaetes, Nemerteans. St. Petersburg: Nauka; 2004. p. 10–491.
Meier-Brook C. Taxonomic studies on Gyraulus (Gastropoda: Planorbidae). Malacologia. 1983;24:1–113.
Madsen H, Hung NM. An overview of freshwater snails in Asia with main focus on Vietnam. Acta Trop. 2014;140:105–17.
Kappes H, Haase P. Slow, but steady: dispersal of freshwater molluscs. Aquat Sci. 2012;74:1–14.
Jörger KM, Stöger I, Kano Y, Fukuda H, Knebelsberger T, Schrödl M. On the origin of Acochlidia and other enigmatic euthyneuran gastropods, with implications for the systematics of Heterobranchia. BMC Evol Biol. 2010;10:323.
Dayrat B, Conrad M, Balayan S, White TR, Albrecht C, Golding R, Gomes SR, Harasewych MG, de Frias Martins AM. Phylogenetic relationships and evolution of pulmonate gastropods (Mollusca): new insights from increased taxon sampling. Mol Phylogenet Evol. 2011;59:425–37.
Piechocki A, Wydrowska BW. Guide to Freshwater and Marine Mollusca of Poland. Poznań: Bogucki Wydawnictwo Naukowe; 2016.
Fukuda H. Gyraulus pulcher. In: Okayama Prefecture editor. Red data book in Okayama 2009. Okayama: Okayama Prefecture; 2010. 328.
Saito T, Hirano T, Uchida S, Yamazaki D. New records of Gyraulus sp. (Gastropoda: Planorbidae) in Ishigaki and Iriomote islands. Molluscan Divers. 2017;5:79–82.
Jørgensen A, Kristensen TK, Stothard JR. An investigation of the “Ancyloplanorbidae” (Gastropoda, Pulmonata, Hyglophila): preliminary evidence from DNA sequence data. Mol Phylogenet Evol. 2004;32:778–87.
Albrecht C, Kuhn K, Streit B. A molecular phylogeny of Planorboidea (Gastropoda, Pulmonata): insight from enhanced taxon sampling. Zool Scr. 2007;36:27–39.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Tanabe AS. Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Mol Ecol Resour. 2011;11:914–21.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90.
Tanabe AS. Phylogears version 2.2.2012.02.13. 2012. http://www.fifthdimension.jp/ Accessed 4 Oct 2017.
Ronquist F, Huelsenbeck JP. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Rambaut A, Drummond AJ, Suchard M. Tracer v1.6. 2013. http://tree.bio.ed.ac.uk/software/tracer/. Accessed 4 Oct 2017.
Bouckaert R, Heled J, Kühnert D, Vaughan TG, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ. BEAST2: A software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:e1003537.
Wilke T, Schultheiß R, Albrecht C. As time Goes by: a simple Fool's guide to molecular clock approaches in invertebrates. Am Malacol Bull. 2009;27:25–45.
Schultheiß R, Bocxlaer VB, Riedel F, von Rintelen T, Albrecht C. Disjunct distributions of freshwater snails testify to a central role of the Congo system in shaping biogeographical patterns in Africa. BMC Evol Biol. 2014;14:42.
Mahler DL, Revell LJ, Glor RE, Losos JB. Ecological opportunity and the rate of morphological evolution in the diversification of greater Antilllean anoles. Evolution. 2010;64:2731–45.
Hubendick B. On the genus Camptoceras. Arkiv for Zoologi. 1967;20:165–73.
Dobson M. Patterns of distribution in Japanese land mammals. Mammal Rev. 1994;24:94–111.
Dobson M, Kawamura Y. Origin of the Japanese land mammal fauna: allocation of extant species to historically-based categories. Quat Res Tokyo. 1998;37:385–95.
Ota H. Geographic patterns of endemism and speciation in amphibians and reptiles of the Ryukyu archipelago, Japan, with special reference to their paleogeographical implications. Res Popul Ecol. 1998;40:189–204.
Ota H. The current geographic faunal pattern of reptiles and amphibians of the Ryukyu archipelago and adjacent regions. Tropics. 2000;10:51–62.
Konishi S, Yoshikawa S. Immigration times of the two proboscidean species, Stegodon orientalis and Palaeoloxodon naumanni, into the Japanese Islands and the formation of land bridge. Earth Sci. 1999;53:125–34.
Tojo K, Sekiné K, Takenaka M, Isaka Y, Komaki S, Suzuki T, Schoville SD. Species diversity of insects in Japan: their origins and diversification processes. Entomol Sci. 2017b;20:357–81.
Hikida T. Biogeography of reptiles in islands in East Asia—geographic distributions from views of molecular and morphological studies. Seibutu Kagaku. 2003;54:205–20.
Boag DA. Dispersal in pond snails: potential role of waterfowl. Can J Zool. 1986;64:904–9.
Sonohara T. An assumption of possible dispersal of Corbicula fluminea (Bivalvia: Corbiculidae) by waterfowl immigration. Chiribotan. 2005;36:31–2.
van Leeuwen CHA, van der Velde G, van Lith B, Klaassen M. Experimental quantification of long distance dispersal potential of aquatic snails in the gut of migratory birds. PLoS One. 2012;7:e32292.
van Leeuwen CHA, van der Velde G. Prerequisites for flying snails: external transport potential of aquatic snails by waterbirds. Freshw Sci. 2012;31:963–72.
Tominaga K, Nakajima J, Watanabe K. Cryptic divergence and phylogeography of the pike gudgeon Pseudogobio esocinus (Teleostei: Crprinidae): a comprehensive case of feshwater phylogeography in Japan. Ichthyol Res. 2016;63:79–93.
Fagan WF. Connectivity, fragmentation, and extinction risk in dendritic metapopulations. Ecology. 2002;83:3243–9.
Lowe W, Likens G. Linking scales in stream ecology. BioScience. 2006;56:591–7.
Beisner BE, Peres PR, Lindstrom ES, Barnett A, Longhi ML. The role of enivironmental and spatial processes in structuring lake communities from bacteria to fish. Ecology. 2006;87:2985–91.
Shurin JB, Cottenie K, Hillebrand H. Spatial autocorrelation and dispersal limitation in freshwater organisms. Oecologia. 2009;159:151–9.
De Bie T, De Meester L, Brendonck L, Martens K, Goddeeris B, Ercken D, Hampel H, Denys L, Vanhecke L, Van der Gucht K, Van Wichelen J, Vyverman W, Declerck SAJ. Body size and dispersal mode as key traits determining metacommunity structure of aquatic organisms. Ecol Lett. 2012;15:740–7.
Habe T. The list of Japanese freshwater mollusks:2. Hitachiobi. 1990;55:3–9 (in Japanese).
Saito T, Do VT, Prozorova L, Hirano T, Fukuda H, Chiba S. Endangered freshwater limpets in Japan are actually alien invasive species. Conserv Genet. 2018;19:947–58.
Mori S. Classification of the Japanese Planorbidae. Mem Coll Sci, Kyoto Imp Univ. 1938;14:279–300.
Folmer O, Black M, Hoeh W, Lutz RA, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994;3:294–9.
Palumbi SR, Martin A, Romano S, Mcmillan WO, Stice L, Grabowski G. The simple Fool’s guide to PCR. Honolulu: University of Hawaii Press; 1991.
Uit de Weerd DR, Gittenberger E. Phylogeny of the land snail family Clausiliidae (Gastropoda: Pulmonata). Mol Phylogenet Evol. 2013;67:201–16.
Acknowledgements
We are grateful to Mukai Y for supporting samples collecting, Uchida S, Aota T, and Wada S for supporting laboratory works and analysis. We also thank to Makino W, Sato T, Nagao K, Yamakami R, Ye B, and Otani JU for providing samples or sample’s information, Urabe J, Bogatov VV, Suzuki T, Yamaki K, Tanangonan JB, Seo T, Akiyama K, Saito N, Shiraishi K, Okayama Prefectural Nature Conservation Center, and three drivers for help with field work. Finally, we thank two anonymous reviewers for insightful comments on a previous version of the manuscript.
Funding
This study was funded in part by JSPS Research Fellow Grant Number 16 J04692 and JPSP KAKENHI Grant Number 17H04611.
Availability of data and materials
DNA sequences are available in GenBank under accession numbers LC428844 - LC429227, LC429392 - LC429587. The other data sets supporting the results of this article are included within the article and its Additional files.
Author information
Authors and Affiliations
Contributions
TakS and SC conceived and designed the study with contribution from YC and HF. TakS and TH coordinated the taxon sampling with major contribution from LP, YM, and VD. TakS, YM, HF, LP, AD, VD, YK, TatS, PS, DY and TH performed the field survey. TakS conducted molecular experiment with major contributions from YK and TH. TakS took the lead in analyzing the dataset and writing manuscript with major contributions from TH, DY and SC. All authors read, reviewed, edited and commented on the manuscript, and then approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Sample information of Planorbidae in this study. See also Fig. 1. TUMC samples were deposited in the Tohoku University Museum Collection, Tohoku University; OKCABM samples were deposited in the Laboratory of Conservation of Aquatic Biodiversity, Faculity of Agriculture, Okayama University. (XLSX 26 kb)
Additional file 2:
Aligned sequences of 16S after selecting by trimAl. (TXT 88 kb)
Additional file 3:
The Bayesian phylogenetic tree inferred from mtocondorial CO1. Each number and colour at the terminal branch of the tree indicates the sample number, species name and collected region (Fig. 1 and Additional file 1). Numbers at the branch nodes represent BPP, MLBV, and NJ. On the right side, the vertical bars indicate nominal clades. (PDF 267 kb)
Additional file 4:
The Bayesian phylogenetic tree inferred from 16S. Each number and colour at the terminal branch of the tree indicates the sample number, species name and collected region (Fig. 1 and Additional file 1). Numbers at the branch nodes represent BPP, MLBV, and NJ. On the right side, the vertical bars indicate nominal clades. (PDF 267 kb)
Additional file 5:
The Bayesian phylogenetic tree inferred from H3. Each number and colour at the terminal branch of the tree indicates the sample number, species name and collected region (Fig. 1 and Additional file 1). Numbers at the branch nodes represent BPP, MLBV, and NJ. On the right side, the vertical bars indicate nominal clades. (PDF 263 kb)
Additional file 6:
Detailed information of divergence time estimation, ancestral state reconstruction, and lineage diversity estimation. See also Table 4. (XLSX 56 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Saito, T., Hirano, T., Prozorova, L. et al. Phylogeography of freshwater planorbid snails reveals diversification patterns in Eurasian continental islands. BMC Evol Biol 18, 164 (2018). https://doi.org/10.1186/s12862-018-1273-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-018-1273-3