
Abstract
Background
Allelopathy in rice (Oryza sativa) is a chemically induced response that is elevated by the exogenous application of chemical compounds and barnyard grass root exudates. An in-depth understanding of the response mechanisms of rice to chemical induction is necessary for the identification of target genes for increasing the allelopathic potential of rice. However, no previous studies have evaluated the transcriptomic changes associated with allelopathy in rice in response to barnyard grass exudates treatment. Thus, the aim of the present study was to reveal differentially expressed genes (DEGs) in allelopathic and non-allelopathic rice seedlings treated with barnyard grass exudates to identify target allelopathy genes.
Results
The inhibitory effect of the culture solutions on the allelopathic rice accession PI312777 (PI) and the non-allelopathic rice accession Lemont (LE) significantly increased (P < 0.05) after treatment with barnyard grass root exudates. The RNA sequencing results revealed that 14,891 genes in PI(+B) vs. LE(+B), 12,505 genes in PI(+B) vs. PI(−B), and 5857 genes in LE(+B) vs. LE(−B) were differentially expressed following root exudates treatment. These DEGs were classified into three categories and 32 functional groups, i.e., 12 groups in the biological process category, 12 groups in the cellular component category, and eight groups in the molecular function category. There were 5857 and 2846 upregulated genes and 135 and 50 upregulated Gene Ontology terms (P < 0.05) in the biological process category in PI(+B) vs. PI(−B) and LE(+B) vs. LE(−B), respectively. These results indicated that the allelopathic accession PI is more sensitive than the non-allelopathic accession LE to exogenous root exudates treatment. Genes related to rice allelochemical-related biosynthesis pathways, particularly the shikimic acid and acetic acid pathways, were significantly differentially expressed in both rice accessions. These findings suggested that phenolic acids, fatty acids, and flavonoids, which constitute the downstream metabolites of the shikimic acid and acetic acid pathways, are significantly expressed in response to root exudates of barnyard grass.
Conclusions
The allelopathic potential of both rice accessions could be significantly enhanced by barnyard grass root exudates application. Furthermore, genes related to the biosynthesis pathways of reported rice allelochemicals were significantly differentially expressed in both accessions. Phenylalanine ammonia lyase was determined to be a potential target for the regulation of chemical induction.
Similar content being viewed by others


Background
Since Dilday and his colleagues (Dilday et al. 1989) observed apparent allelopathic activity in certain rice (Oryza sativa L.) varieties in response to the aquatic weed ducksalad [Heteranthera limosa (Sw.) Wild] in field tests, there has been great interest in the application of rice allelopathy for controlling paddy weeds, thus reducing herbicide usage in rice production. However, three decades after Dilday’s study, rice allelopathy has yet to be widely applied in practice. The main reason is that rice allelopathy is a quantitative trait that is heavily influenced by environmental conditions (Dilday et al. 2000; Shin et al. 2000; Fang et al. 2010; Kato-Noguchi 2011; Wang et al. 2010; He et al. 2012). Rice allelopathic activity is polygenically controlled (Ebana et al. 2001; Jensen et al. 2001, 2008; He et al. 2004; Junaedi et al. 2007). To date, no candidate gene has been used for the genetic breeding of allelopathic rice cultivars. Although only 3–5% of screened rice accessions have been found to certain degree of allelopathic effect on one or more weed species (Dilday et al. 2000), Olofsdotter (1998) suggested that allelopathic genes are also present in modern varieties of rice. However, the high allelopathic activity of wild types may be reduced or lost in crops during hybridization and selection for other characteristics (Putnam and Duke 1974; Dilday et al. 2000). Studies have shown that abiotic stress, such as nitrogen (N), phosphorous (P), and potassium (K) deficiency (He et al. 2008; Wang et al. 2010; Fang et al. 2010) and UV-B irradiation (Shin et al. 2000), and biotic stress, such as the co-cultivation of rice and barnyard grass (Kato-Noguchi 2011; He et al. 2012), could enhance the allelopathic potential of allelopathic rice accessions. Recent studies show that allelopathy can be induced by exogenous application of chemicals, such as exogenous phenolic acids, methyl jasmonate, and methyl salicylate (Kong et al. 2004a; Bi et al. 2007; Fang et al. 2009; Xu et al. 2010; Kato-Noguchi 2011). Our previous study confirmed that rice allelopathy in both an allelopathic accession PI312777 (PI) and a non-allelopathic accession Lemont (LE) can be effectively elevated by treatment with barnyard grass root exudates in a hydroponic system (Zhang et al. 2018). Therefore, an in-depth understanding of chemical-induced mechanism of rice allelopathy is necessary for identification of target genes to increase rice allelopathic potential.
RNA sequencing (RNA-Seq) is an approach used to deduce and quantify transcriptomes (Wang et al. 2009) and quantitatively determine the RNA expression levels of transcripts under different conditions. RNA-Seq has recently been used to track gene expression changes in rice in response to abiotic stress, including zinc, P, and N deficiency, anoxia, and drought (Nguyen et al. 2013; Yang et al. 2015; Nanda et al. 2017; Deng et al. 2018), high temperature and cold stress (Shen et al. 2014; Dametto et al. 2015; Mangrauthia et al. 2018), drought and salinity stress (Shankar et al. 2016; Zhou et al. 2016; Zhang et al. 2016), cadmium and arsenic stress (Chakrabarty et al. 2009; He et al. 2015), as well as the characteristics of rice root heterosis and crown tissue under different conditions (Zhai et al. 2013; Nanda et al. 2017). However, no studies have evaluated the transcriptomic changes in allelopathy in response to chemical induction in rice. Previous studies showed that rice accessions expressed allelopathic effect on barnyard grass (Echinochloa crus-galli) at 3–7 leaf stages. (Hassan et al. 1998; Li et al. 2015). Vergara (1992) reported that rice yield was not significantly affected when paddy weeds were removed within 30 days after transplantation. Increasing rice allelopathic potential as early as possible can enhance the ability of rice seedlings to resist paddy weeds. In our previous work, we successfully elevated the inhibition of culture solutions and leaf extracts of accessions PI and LE by induction of barnyard grass root exudates (Zhang et al. 2018). LE, as a non-allelopathic accession, has been confirmed to possess increased allelopathic potential under induction treatment. Therefore, the aim of this paper is to reveal the differential expression of various genes in the PI and LE rice accessions after treatment with barnyard grass exudates to identify target genes for future research.
Methods
Preparation of the barnyard grass root exudates solution
Root exudates of barnyard grass [Echinochloa crus-galli (L.) Beauv] was obtained as described in our previous paper (Zhang et al. 2018). In brief, 100 uniform seedlings at the one-leaf stage were cultured in 10 L Hoagland’s solution (Hoagland and Arnon 1950). At the five-leaf stage, the culture solutions were collected and filtered using a Buchner funnel apparatus with suction filtration equipment. The filtrate was concentrated to less than 100 mL by rotary evaporation at 40 °C ± 1 °C and then stored at 4 °C in a refrigerator for 24 h. In this process, the inorganic salts and lower soluble compounds were precipitated and removed by filtration. The filtrate was filtered through a 0.22-μm membrane and diluted to a volume of 100 mL (1 mL concentrate is equivalent to one seedling). This solution was then used as the inducing solution for subsequent experiments.
Allelopathic induction experiments
The induction experiment of the allelopathic rice accession PI and non-allelopathic rice accession LE was conducted as described in our previous paper (Zhang et al. 2018). Briefly, 50 uniform rice seedlings of each accession at the 1-leaf stage were cultured in 10 L Hoagland’s solution. At the 3-leaf stage, the culture solution was replaced with 5 L Hoagland’s solution (pH 6.0) containing 75 mL of barnyard grass root exudates; these treatments are denoted as PI(+B) and LE(+B). The control treatment consisted of 5 L Hoagland’s solution (pH 6.0) lacking barnyard grass root exudates; these treatments are denoted as PI(−B) and LE(−B). Each treatment had three replicates. After two days of treatment, the roots were harvested and immediately placed into liquid nitrogen and stored at − 80 °C until total RNA extraction. The culture solutions were also collected for inhibitory bioassays and the determination of phenolic acid contents.
Inhibitory bioassay of rice culture solutions
The culture solutions collected above were filtered using a Buchner funnel apparatus with suction filtration equipment and then filtered through a 0.22-μm membrane. The change in rice allelopathy was estimated based on the inhibitory rate of the rice culture solutions on lettuce (Lactuca sativa L.) root length in a laboratory bioassay, as described previously (He et al. 2009; Zhang et al. 2018). Briefly, five germinated lettuce seeds were placed in a 250 mL beaker lined with filter paper, and 5 mL of the test solutions was added. The control consisted of 5 mL of Hoagland’s solution. The beakers were covered with perforated Parafilm and placed into an incubator at 25 °C ± 2 °C with 12 h (8:00–20:00) of 70 μmol m− 2 s− 1 light intensity. The experiments were replicated four times. After 3 days, the root lengths were measured. The relative inhibition rate (IR) was used to assess the effect of the test solutions compared to the control and was calculated as follows: IR (%) = [(control – treatment)/control] × 100%.
Determination of phenolic acid concentrations in the rice culture solutions
The contents of seven phenolic allelochemicals in the rice culture solutions, namely, protocatechuic acid, 4-hydroxybenzoic acid, syringic acid, vanillic acid, salicylic acid, ferulic acid, and cinnamic acid, were determined by solid-phase extraction and high-performance liquid chromatography (SPE-HPLC), as described previously (Li et al. 2017). The instrument was an Agilent 1206 HPLC (Agilent Technologies, USA) equipped with a C18 reversed-phase column (ZORBAX SB-C18, 150 mm × 4.6 mm, 5 μm). The mobile phase was a mixture of methanol (A) and 1% phosphoric acid (B) with the following gradient elution program: A:B = 27:73 (9 min), A:B = 30:70 (2 min), and A:B = 50:50 (4 min). The flow rate of the mobile phase was 1.6 mL min− 1 and was detected at 280 nm. The injection volume was 5 μL.
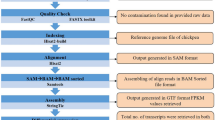
RNA extraction and sequencing
Total RNA was extracted from the roots using TRIzol reagent (Tiangen Bio Co., Ltd., Beijing, China). RNA degradation and contamination were monitored on 1% agarose gels. RNA purity was evaluated using a NanoPhotometer spectrophotometer (IMPLEN, CA, USA). The RNA integrity was assessed using the RNA Nano 6000 Assay Kit for the Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). mRNA library preparation and sequencing were out-sourced (Allwegene Tech., Beijing, China). The 12 libraries were sequenced on an Illumina HiSeq 2500 platform, and paired-end 150 bp reads were generated. Reads containing more than 10% poly-N and more than 50% low-quality reads (Q ≤ 20) were removed from the raw data using Trimmomatic v0.33 (Bolger et al. 2014). Concurrently, the Q20 and Q30 values, GC content, and sequence duplication level of the clean data were calculated. All downstream analyses were based on clean, high-quality data.
Alignment and assembly of the RNA sequencing reads
Since non-allelopathic accession LE is a Japonica Group variety and could have enhanced allelopathic potential under induction treatment (Gealy et al. 2003; Zhang et al. 2018), we chose the Japonica Group as a reference genome annotation so that the results would be more suitable for conventional rice cultivars. RNA-Seq reads were aligned to the rice reference genome (Oryza sativa Japonica Group, Ensembl_IRGSP_1.0.34) using TopHat 2 (v2.1.0) (Kim et al. 2013). Each read was mapped with Cufflinks (v2.1.1), which assembled the alignments in Sequence Alignment/Map format into transfrags (Trapnell et al. 2010). The assembly files were then merged with reference transcriptome annotations for further analysis (Trapnell et al. 2012).
Differential expression analysis
The differential expression of each gene was calculated by quantifying the Illumina reads based on Fragments Per Kilobase of transcript per million fragments mapped (FPKM) (Mortazavi et al. 2008).
Pairwise differential expression analyses were performed using the DEGseq package (Anders and Huber 2012) in the R statistical environment (Team 2014). Genes with an FDR-adjusted P-value < 0.05 as determined by DEGseq were assigned as differentially expressed. Heatmaps for the differentially expressed genes (DEGs) were constructed using the Pheatmap v. 1.0.8 package (Kolde 2015).
Functional annotation and classification
Gene Ontology (GO, http://www.geneontology.org/) enrichment analysis of the DEGs was implemented using GOseq (v. 1.22) (Young et al. 2010) using Wallenius’ noncentral hypergeometric distribution, which can adjust for gene length bias in DEGs. Affected pathways were determined using Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.kegg.jp) (Kanehisa et al. 2008). We used KOBAS (Mao et al. 2005) software to annotate and identify the enriched KEGG pathways of the DEGs.
Real-time quantitative PCR (RT-qPCR)
The expression patterns of 10 genes associated with secondary metabolite biosynthesis pathways related to rice allelochemicals, including phenolic acids, fatty acids, terpenoids, steroids, and flavones (Mattice et al. 1998; Kim et al. 2000; Kato-Noguchi et al. 2002; Kong et al. 2004b; Macias et al. 2006; Seal et al. 2004; He et al. 2012), were analyzed using RT-qPCR (gene-specific primers are provided in Additional file 1: Table S1). Total RNA was isolated from the roots using TRIzol (Tiangen Bio Co., Ltd., Beijing, China), and cDNA was synthesized using the PrimeScript™ RT reagent Kit with a gDNA Eraser. RT-qPCR was conducted using SYBR® Premix Ex Taq™ II according to the manufacturer’s instructions (Takara, Tokyo, Japan). The actin gene (5-TGTAAGCAACTGGGATGA-3 and 5-CCTTCGTAGATTGGGACT -3) was used as an internal standard. The PCR program was as follows: 30 s of predenaturation at 95 °C, followed by 40 cycles of 5 s at 95 °C and 40 s at 60 °C and steps for dissociation curve generation (10 s at 95 °C, 60 s at 60 °C, and 15 s at 95 °C). The relative gene expression of each target gene was determined using the 2 −ΔΔCt method (Livak and Schmittgen 2001).
Statistical analysis
All data were analyzed using DPS (v. 7.05) statistical software (Tang and Feng 2007). Statistical significance was determined using one-way analysis of variance (ANOVA) using least significant difference (LSD) tests at the 5% level of probability.
Results
Inhibitory effect of rice culture solutions and phenolic acid contents
Treatment with barnyard grass root exudates significantly increased the inhibitory effect of the two rice culture solutions on lettuce root length (Fig. 1). The inhibitory rates were increased by 14.67% in PI (increased from 23.17% to 37.84%) and 14.75% in LE (increased from 7.34% to 22.09%) compared to the control. The contents of five phenolic allelochemicals were significantly elevated in the PI culture solution. In the LE culture solution, the contents of three phenolic allelochemicals were significantly elevated, while the content of one phenolic allelochemical was significantly decreased (Table 1). The total contents of the seven selected phenolic allelochemicals were significantly higher in both the PI and LE culture solutions than in the control.

Inhibitory rate of the rice culture solutions on lettuce root length. The bars represent the standard errors of the mean (n = 4). Different letters indicate significant differences at P < 0.05
Transcriptome sequencing
The transcriptomic responses of the two rice cultivars to barnyard grass root exudates treatment were assayed by RNA sequencing. In total, more than 311 million raw reads from 12 libraries with an average of 25 million reads per condition were obtained (Additional file 2: Table S2). More than 94% high-quality reads with an average Phred quality score of > 30 at each base position were obtained and used for downstream analyses. TopHat was used to map the reference genome. A total of 80.33–79.92% (average 80.11%) of the reads from PI(−B), 80.54–80.16% (average 80.36%) of the reads from PI(+B), 73.82–73.02% (average 73.03%) of the reads from LE(−B), and 81.24–80.10% of the reads (average 80.76%) from LE(+B) were mapped to the reference genome. The percentage of multiple mapped reads was < 2%, and approximately 79% of the reads were uniquely located in the genome (Table 2). In PI(−B), 95.27% of the reads were mapped to exonic regions, 1.36% of the reads were mapped to intronic regions, and 3.37% of the reads were mapped to intergenic regions. In PI(+B), 95.64% of the reads were mapped to exonic regions, 1.05% of the reads were mapped to intronic regions, and 3.31% of the reads were mapped to intergenic regions. In LE(−B), 95.44% of the reads were mapped to exonic regions, 1.31% of the reads were mapped to intronic regions, and 3.26% of the reads were mapped to intergenic regions. In LE(+B), 95.19% of the reads were mapped to exonic regions, 1.31% of the reads were mapped to intronic regions, and 3.51% of the reads were mapped to intergenic regions (Additional file 3: Table S3).
DEGs specific to exudates treatment
Gene expression levels were calculated using FPKM. Only the genes with FPKM values > 1 were analyzed. Correlations between the treatments and controls were investigated using cluster analysis in Pheatmap. The gene expression levels of the different rice cultivars varied largely between the treatments and controls (Fig. 2), as indicated by clustering analysis of the various transcripts.

Hierarchical cluster analysis of the DEGs. The color key represents FPKM-normalized log2-transformed counts. PI(+B) and LE(+B) represent the PI and LE accessions treated with barnyard grass root exudates. PI(−B) and LE(−B) represent the corresponding no-induction controls
Comparisons of transcript abundances between PI and LE revealed that 17,103 genes were differentially expressed in PI(−B) vs. LE(−B). Of these, 8656 genes were upregulated, and 8447 genes were downregulated. In addition, 14,891 genes were differentially expressed following root exudates treatment in PI(+B) vs. LE(+B). Of these, 7364 genes were upregulated, and 7527 genes were downregulated (Fig. 3). A total of 4819 upregulated genes and 4644 downregulated genes were shared between PI and LE.

Common and specific DEGs in the two rice cultivars after induction treatment by barnyard grass root exudates. Note: upregulated DEGs are in red, and downregulated genes are in black. PI(+B) and LE(+B) represent the PI and LE accessions treated with barnyard grass root exudates. PI(−B) and LE(−B) represent the corresponding no-induction controls
Comparisons of transcript abundance between exudate-treated PI or LE and the controls revealed that 12,505 genes were differentially expressed in PI(+B) vs. PI(−B). Of these, 5857 genes were upregulated and 6648 genes were downregulated. In LE(+B) vs. LE(−B), 6374 genes were differentially expressed following root exudates treatment. Of these, 2846 genes were upregulated, and 3528 genes were downregulated (Fig. 3). A total of 578 upregulated genes and 1134 downregulated genes were shared between PI and LE.
Functional classification by GO
Based on the GO analysis, the 12,505 DEGs of PI(+B) vs. PI(−B) and 6374 DEGs of LE(+B) vs. LE(−B) were classified into three categories with 32 functional groups, specifically 12 groups in the biological process category (Fig. 4a), 12 groups in the cellular component category (Fig. 4b), and eight groups in the molecular function category (Fig. 4c).

GO annotation clusters of DEGs. GO functional enrichment analysis of DEGs in the roots of different allelopathic rice accessions. Based on sequence homology, 18,879 genes were distributed among the three main categories: a, biological process; b, cellular component; and c, molecular function
GO terms in the biological processes category
We further identified the GO terms in the biological processes category. Fifty and 135 GO terms in this category were significantly upregulated (P < 0.05) in LE and PI, respectively (Additional file 4: Table S4 and Additional file 5: Table S5), compared to the corresponding controls. The common GO terms in the two rice accessions included regulation of metabolic process, regulation of primary metabolic process, regulation of cellular biosynthetic process, and regulation of biosynthetic process. Some notable differences in GO terms were also identified between the two rice accessions. For instance, the glucose catabolic process, ATP synthesis coupled electron transport, electron transport chain, amino acid activation, and biosynthetic process terms were highly enriched in PI(+B) compared to PI(−B).
KEGG pathway mapping and DEGs related to the biosynthesis of secondary metabolites and rice allelochemicals
To identify the metabolic pathways enriched by the DEGs in the two rice accessions, pathway-based analysis was performed using the KEGG pathway database. In PI, 2159 protein sequences were classified into 123 biological pathways, including ketone synthesis and degradation, ribosomes, propionic acid, pantothenic acid metabolism, phenylalanine metabolism and coenzyme A (Co-A) biosynthesis, metabolism of amino acid biosynthesis, and oleic acid. In LE, 1872 gene and protein sequences were classified into 122 significant biological pathways, mainly the citric acid (TCA) cycle, phenylalanine metabolism, terpenoid metabolism, starch and sucrose metabolism, ribosomes, arginine biosynthesis, and metabolism of amino sugars and sugar nucleotides pathways.
In PI(−B) vs. LE(−B) and PI(+B) vs. LE(+B), there were 569 and 529 DEGs, respectively, related to the biosynthesis of secondary metabolites. Specifically, in PI(−B) vs. LE(−B) and PI(+B) vs. LE(+B), 169 and 153 DEGs, respectively, were related to the shikimic acid pathway for phenolic acid synthesis; 177 and 162 DEGs, respectively, were related to the acetic acid pathway for fatty acid production; 94 and 76 DEGs, respectively, were related to the mevalonic acid (MVA) pathway for terpenoid and steroid formation; and 13 and 14 DEGs, respectively, were related to flavonoid synthesis, which involves a combination of the acetic acid and shikimic acid biosynthesis pathways (Table 3). The number of DEGs related to rice allelochemicals between the two treatments were in the order of: acetic acid pathway > shikimic acid pathway > MVA pathway for both PI(−B) vs. LE(−B) and in PI(+B) vs. LE(+B).
In PI(+B) vs. PI(−B) and LE(+B) vs. LE(−B), there were 484 and 317 DEGs, respectively, related to the biosynthesis of secondary metabolites (Table 3). Specifically, in PI(+B) vs. PI(−B) and LE(+B) vs. LE(−B), 154 and 115 DEGs, respectively, were related to the shikimic acid pathway; 158 and 90 DEGs, respectively, were related to the acetic acid pathway; 65 and 51 DEGs, respectively, were related to the MVA pathway for terpenoid and steroid formation; and 13 and 9 DEGs, respectively, were related to flavonoid synthesis (Table 3). The DEGs between the two treatments were in the order of acetic acid pathway > shikimic acid pathway > MVA pathway in PI(+B) vs. PI(−B) and shikimic acid pathway > acetic acid pathway > MVA pathway in LE(+B) vs. LE(−B).
Confirmation by RT-qPCR
RT-qPCR was used to confirm the RNA gene expression patterns of 10 genes related to rice allelochemical biosynthesis (both in PI and LE) and to validate our RNA sequencing results. A good linear correlation was observed between the gene expression fold-changes obtained using RT-qPCR and RNA sequencing (R2 = 0.8656) (Fig. 5).

Comparison of the log2-value of fold-change (log2FC) of 10 selected transcripts determined using RNA-Seq and RT-qPCR
Discussion
Increase in rice allelopathy following treatment with barnyard grass root exudates
In this study, treatment with barnyard grass root exudates significantly increased the inhibitory rates of the rice culture solutions on lettuce root growth in both the allelopathic accession PI and non-allelopathic accession LE (Fig. 1). We also discovered that the total contents of phenolic acids previously reported as significant rice allelochemicals were significantly increased in the culture solutions of both PI and LE compared to the controls (Table 1). These results are consistent with our previous findings (Zhang et al. 2018) and once again confirm that induction with barnyard grass root exudates increases rice allelopathy. Although the genetic allelopathy was significantly higher in PI than in LE, the induced allelopathy was roughly equal between the two accessions. This finding further supports that induced allelopathy in rice is an important strategy for enhancing rice allelopathic potential.
RNA-Seq results
A comparison of the transcript abundances revealed that treatment with root exudates resulted in 17,103 genes that were differentially expressed in PI(−B) vs. LE(−B) and 14,891 genes that were differentially expressed in PI(+B) vs. LE(+B). The total DEGs, number of upregulated DEGs and number of downregulated DEGs in PI(+B) vs. PI(−B) were nearly the same or more than two times greater than those in LE(+B) vs. LE(−B) (Fig. 3). Additionally, 135 and 50 GO terms in the biological processes category were significantly upregulated (P < 0.05) in PI(+B) vs. PI(−B) and in LE(+B) vs. LE(−B), respectively (Additional file 4: Table S4, Additional file 5: Table S5). These results indicated that the allelopathic rice accession PI responds more actively than the non-allelopathic rice accession LE to root exudates treatment. Previously reported morphological, physiological, and biochemical results indicated that PI is more sensitive than LE under various biotic and abiotic stresses (Shin et al. 2000; He et al. 2008; Fang et al. 2010; Wang et al. 2010; Kato-Noguchi 2011; He et al. 2012). We suggest that this result derives from the genetic traits of the allelopathic rice accessions.
DEGs related to the biosynthesis of secondary metabolites and rice allelochemicals
KEGG pathway mapping (Fig. 6) indicated that, under induction treatment, genes related to transcription, such as basal transcription factors and RNA polymerase II, genes related to translation, such as translation initiation factors and ribosome export factors, and genes related to protein modification processes, such as correct folding and ER-associated degradation, were upregulated in both rice accessions. Fukuda et al. (2007) reported that transketolase was upregulated in rice roots exposed to low P, high Al and low pH. The enolase gene of plant roots is usually upregulated under stress conditions such as high salinity, drought, cold and hypoxia (Lal et al. 1998; Forsthoefel et al. 1995). The citrate synthase activity of rye (Secale cereale L.) increased by 30% under Al stress (Li et al. 2000). Low temperature caused putative aconitate hydratase downregulation and ATP synthase upregulation in rice leaves and malate dehydrogenase upregulation in rice roots (Yan et al. 2006; Cui et al. 2005; Lee et al. 2009). In our results, transketolase (OS04G0266900), enolase (OS06G0136600), citrate synthase (OS02G0232400), and malate dehydrogenase (OS04G0551200) genes were upregulated, and a putative aconitate hydratase (OS03G0136900) gene was downregulated in both rice accessions under induction treatment. This behavior is consistent with previous reports of rice under stress conditions.

Schematic molecular pathway depicting the changes in the rice root transcriptome under induction treatment by barnyard grass root exudates. The red color represents upregulated genes, and the green color represents downregulated genes. AUX/IAA, auxin-responsive protein IAA; ARF, auxin response factor; AHP, histidine-containing phosphotransfer protein; NPR1, regulatory protein NPR1; TGA, transcription factor TGA; I: hydroxymethylglutaryl-CoA reductase; II: 1-deoxy-D-xylulose-5-phosphate synthase; III: mevalonate kinase; IV: isopentenyl-diphosphate delta-isomerase; V: diphosphomevalonate decarboxylase; 4CL, 4-coumarate-CoA ligase; CCR, cinnamoyl-CoA reductase
In this study, we are concerned with changes in secondary metabolism related to rice allelochemicals. The rice allelochemicals reported in the current literature are primarily secondary metabolites, such as phenolic acids, fatty acids, terpenoids, steroids, flavones, and hydroxamic acids (Mattice et al. 1998; Kim et al. 2000; Kato-Noguchi et al. 2002; Kong et al. 2004b; Macias et al. 2006; Seal et al. 2004; He et al. 2012; Zhang et al. 2018). Phenolic compounds are synthesized from the shikimic acid pathway; fatty acids are synthesized from the acetic acid pathway; terpenoids and steroids are synthesized from the MVA pathway, and flavonoids are synthesized via a combination of the shikimic acid pathway to supply the C6-C3 moiety and the acetic acid pathway to supply the C6 moiety (Dixon and Paiva 1995; Buchanan et al. 2000). In the present study, the genes related to these three biosynthetic pathways, particularly genes in the shikimic acid and acetic acid pathways, were significantly differentially expressed in both the rice accessions (Table 3). Three genes in the phenylpropanoid biosynthesis pathway were upregulated in both rice accessions under induction treatment; however, five genes in the terpenoid biosynthesis pathway were upregulated in PI and downregulated in LE. Moreover, some genes in the plant hormone signal transduction pathway, such as auxin-responsive protein IAA (AUX/IAA) and histidine-containing phosphotransfer protein (AHP), which are related to cell enlargement and cell division, and regulatory protein NPR1 (NPR1) and transcription factor TGA (TGA), which are related to salicylic acid, were also upregulated after induction treatment (Fig. 6). These findings suggest that the response of rice to barnyard grass root exudates treatment involves physiological and biochemical processes, as well as biosynthesis pathways of secondary metabolites. Phenolic acids, fatty acids, flavonoids, and terpenoids, which are the downstream metabolites of the shikimic acid, acetic acid and MVA pathways, were significantly expressed in rice under barnyard grass root exudates treatment and further resulted in changes in rice allelopathy. Recently, publications have reported that potential signaling chemicals of barnyard grass root exudates are benzoxazinoids, (−)-loliolide and jasmonic acid (Guo et al. 2017; Kong et al. 2018). Increasing the allelopathic potential of conventional cultivars by the induction method may be a promising and practicable approach in the application of rice allelopathy.
Phenolic acids and the associated biosynthesis pathways
Phenolic acids constitute one type of rice allelochemical. In contrast to the findings of Olofsdotter et al. (2002), who miscalculated the importance of phenolic acids in allelopathy, our previous report found that phenolic acids constitute significant allelochemicals in rice (Li et al. 2017). Phenolic acids are synthesized from the conversion of phenylalanine and tyrosine to phenylpropanoid (C6-C3) intermediates (Herbert 1989; Dixon and Paiva 1995; Buchanan et al. 2000; Crozier et al. 2006). Shikimic acid is a key intermediate in the biosynthesis of phenylalanine and tyrosine, and the enzyme phenylalanine ammonia lyase (PAL) is an initial key enzyme in the biosynthesis of phenolic compounds and catalyzes the conversion of phenylalanine to cinnamic acid and other subsequent phenolic acids (Dixon and Paiva 1995; Buchanan et al. 2000). Another downstream function of phenylalanine and cinnamic acid is supplying the C6-C3 moiety of flavonoids (Herbert 1989; Dixon and Paiva 1995; Buchanan et al. 2000; Crozier et al. 2006). Genes related to the shikimic acid pathway, such as genes associated with phenylpropanoid biosynthesis, phenylalanine metabolism, and phenylalanine and tyrosine biosynthesis, were significantly differentially expressed in both PI and LE compared to the control (Table 3). PAL, 4-coumarate-CoA ligase (4CL), and cinnamoyl-CoA reductase (CCR) were upregulated in both rice accessions under induction treatment (Fig. 6). PAL is always upregulated in PI and LE upon exposure to stress conditions, such as N, P, K, UV-B, and water stress (Shin et al. 2000; He et al. 2008; Fang et al. 2010; Wang et al. 2010). In a previous study, the roots of PI and LE rice co-cultured with barnyard grass at a ratio of 1:1 exhibited 5.06-fold and 2.24-fold up-regulation of PAL, respectively, compared to mono-cultured controls; furthermore, the contents of phenolic allelochemicals were 3.97 times and 1.5 times higher in the PI and LE co-cultured with barnyard grass, respectively, than in the mono-cultured controls (He et al. 2012). Our previous results demonstrated that PAL in PI and LE was upregulated in a hydroponic system in response to barnyard grass root exudates treatment, and this upregulation was accompanied by increases in the phenolic acids in the rice leaves and roots and the culture solution (Zhang et al. 2018). Fang et al. (2013, 2015) provided additional evidence that barnyard grass growth inhibition was significantly higher in a PAL-overexpression line of PI and significantly lower in a PAL-RNA interference line of PI compared to a PI control under identical conditions. It is thus evident that PAL and phenolic acids contribute significantly to the observed increase in rice allelopathy under barnyard grass root exudates treatment.
Conclusions
The allelopathic potential of both allelopathic accession PI and non-allelopathic accession LE could be significantly enhanced by application of barnyard grass root exudates in a hydroponic system. Genes related to the biosynthesis pathways of reported rice allelochemicals were significantly differentially expressed in both rice accessions under barnyard grass root exudates treatment. PAL demonstrates great potential for applications in the regulation of chemical induction.
Abbreviations
- DEGs:
-
Differentially expressed genes
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KO:
-
KEGG Orthology
- LE:
-
Non-allelopathic rice accession Lemont
- LE(+B):
-
The non-allelopathic accession LE treated with barnyard grass root exudates
- LE(−B):
-
The non-allelopathic accession LE no induction controls
- MVA:
-
Mevalonic acid
- PAL:
-
Phenylalanine ammonia lyase
- PI:
-
Allelopathic rice accession PI312777
- PI(+B):
-
The allelopathic accession PI treated with barnyard grass root exudates
- PI(−B):
-
The allelopathic accession PI no induction controls
- RNA-Seq:
-
RNA sequencing
- RT-qPCR:
-
Real-time quantitative polymerase chain reaction
- SPE-HPLC:
-
Solid-phase extraction and high-performance liquid chromatography
References
Anders S, Huber W (2012) Differential expression of RNA-Seq data at the gene level–the DESeq package. European Molecular Biology Laboratory (EMBL), Heidelberg
Bi HH, Zeng RS, Su LM, An M, Luo SM (2007) Rice allelopathy induced by methyl jasmonate and methyl salicylate. J Chem Ecol 33(5):1089–1103
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120
Buchanan BB, Gruissem W, Jones RL (2000) Biochemistry & molecular biology of plants. American Society of Plant Physiologists, Maryland
Chakrabarty D, Trivedi PK, Misra P, Tiwari M, Shri M, Shukla D, Kumar S, Rai A, Pandey A, Nigam D, Tripathi RD, Tuli R (2009) Comparative transcriptome analysis of arsenate and arsenite stresses in rice seedlings. Chemosphere 74(5):688–702
Crozier A, Clifford MN, Ashihara H (2006) Plant secondary metabolites: occurrence, structure and role in the human diet. Blackwell, London
Cui S, Huang F, Wang J, Ma X, Cheng Y, Liu J (2005) A proteomic analysis of cold stress responses in rice seedlings. Proteomics 5(12):3162–3172
Dametto A, Sperotto RA, Adamski JM, Blasi ÉAR, Cargnelutti D, Oliveira LFV, Ricachenevsky FK, Fregonezi JN, Mariath JEA, Cruz RP, Margis R, Fett JP (2015) Cold tolerance in rice germinating seeds revealed by deep RNAseq analysis of contrasting indica genotypes. Plant Sci 138:1–12
Deng QW, Luo XD, Chen YL, Zhou Y, Zhang FT, Hu BL, Xie JK (2018) Transcriptome analysis of phosphorus stress responsiveness in the seedlings of Dongxiang wild rice (Oryza rufipogon Griff.). Biol Res doi.org/10.1186/s40659-018-0155-x
Dilday RH, Mattice JD, Moldenhauer KAK (2000) An overview of rice allelopathy in the USA. In: Kim KU, Shin DH (eds) Rice Allelopathy. Proceedings of the Workshop in Rice Allelopathy. International Rice Research Institute, Manila, pp 15–26
Dilday RH, Nastasi P, Smith RJ Jr (1989) Allelopathic observations in rice (Oryza sativa L.) to ducksalad (Heteranthera limosa). J Arkansas Acad Sci 43(1):21–22
Dixon RA, Paiva NL (1995) Stress-induced phenylpropanoid metabolism. Plant Cell 7:1085–1097
Ebana K, Yan WG, Dilday RH, Namai H, Okuno K (2001) Analysis of QTL associated with the allelopathic effect of rice using water soluble extracts. Breed Sci 51:47–51
Fang C, Li Y, Li C, Li B, Ren Y, Zheng H, Zeng X, Shen L, Lin W (2015) Identification and comparative analysis of micrornas in barnyardgrass (Echinochloa crus-galli) in response to rice allelopathy. Plant Cell Environ 38(7):1368–1381
Fang C, Zhuang Y, Xu T, Li Y, Li Y, Lin W (2013) Changes in rice allelopathy and rhizosphere microflora by inhibiting rice phenylalanine ammonia-lyase gene expression. J Chem Ecol 39(2):204–212
Fang CX, He HB, Wang QS, Qiu L, Wang HB, Zhuang YE, Xiong J, Lin WX (2010) Genomic analysis of allelopathic response to low nitrogen and barnyardgrass competition in rice (Oryza sativa L.). Plant Growth Regul 61(3):277–286
Fang CX, Xiong J, Qiu L, Wang HB, Song BQ, He HB, Lin RY, Lin WX (2009) Analysis of gene expressions associated with increased allelopathy in rice (Oryza sativa L.) induced by exogenous salicylic acid. Plant Growth Regul 57(2):163–172
Forsthoefel NR, Cushman MAF, Cushman JC (1995) Posttranscriptional and posttranslational control of enolase expression in the facultative Crassulacean acid metabolism plant Mesembryanthemum crystallinum L. Plant Physiol 108(3):1185–1195
Fukuda T, Saito A, Wasaki J, Shinano T, Osaki M (2007) Metabolic alterations proposed by proteome in rice roots grown under low P and high Al concentration under low pH. Plant Sci 172:1157–1165
Gealy DR, Wailes EJ, Estorninos LE Jr, Chavez RSC (2003) Rice cultivar differences in suppression of barnyardgrass (Echinochloa crus-galli) and economics of reduced propanil rates. Weed Sci 51(4):601–609
Guo L, Qiu J, Ye C, Jin G, Mao L, Zhang H et al (2017) Echinochloa crus-galli genome analysis provides insight into its adaptation and invasiveness as a weed. Nat Commun 8:1031
Hassan SM, Aidy IR, Bastawisi AO, Draz AE (1998) Weed management using allelopathic rice varieties in Egypt. In: Olofsdotter M (ed) Allelopathy in rice. Proceedings of the Workshop in Rice Allelopathy. International Rice Research Institute, Manila, pp 27–37
He F, Liu Q, Zheng L, Cui Y, Shen Z, Zheng L (2015) RNA-Seq analysis of rice roots reveals the involvement of post-transcriptional regulation in response to cadmium stress. Front Plant Sci 6:1136
He HB, Wang HB, Fang CX, Lin YY, Zeng CM, Wu LZ, Guo WC, Lin WX (2009) Herbicidal effect of a combination of oxygenic terpenoids on Echinochloa crus-galli. Weed Res 49:183–192
He HB, Wang HB, Fang CX, Wu H, Guo XK, Lin ZH, Lin WX (2012) Barnyard grass stress up regulates the biosynthesis of phenolic compounds in allelopathic rice. J Plant Physiol 169(17):1747–1753
He HB, Wang HB, Zeng CM, Cai ZX, Xiong J, Qiu L, Fang CX, Lin WX (2008) Physio-biochemical and molecular response of rice (Oryza sativa L.) cultivars with different allelopathic potentials to potassium deficiency. Chin J Eco-Agri 16(4):878–882
He HQ, Shen LH, Xiong J, Jia XL, Lin WX, Wu H (2004) Conditional genetic effect of allelopathy in rice (Oryza sativa L.) under different environmental conditions. Plant Growth Regul 44(3):211–218
Herbert RB (1989) The biosynthesis of secondary metabolites, 2nd edn. Chapman and Hall, New York
Hoagland DR, Arnon DI (1950) The water culture method of growing plants without soil. California Agricultural Experiment Station, Circular–347. College of Agriculture, University of California, Berkeley
Jensen LB, Courtois B, Olofsdotter M (2008) Quantitative trait loci analysis of allelopathy in rice. Crop Sci 48(4):1459–1469
Jensen LB, Courtois B, Shen LS, Li ZK, Olofsdotter M, Mauleon RP (2001) Locating genes controlling allelopathic effects against barnyardgrass in upland rice. Agron J 93:21–26
Junaedi A, Jung WS, Chung IM, Kim KH (2007) Differentially expressed genes of potentially allelopathic rice in response against barnyardgrass. J Crop Sci Biotech 10(4):231–236
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36:D480–D484
Kato-Noguchi H (2011) Barnyard grass-induced rice allelopathy and momilactone B. J Plant Physiol 168:1016–1020
Kato-Noguchi H, Ino T, Sata N, Yamamura S (2002) Isolation and identification of a potent allelopathic substance in rice root exudates. Physiol Plantarum 115:401–405
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36
Kim KU, Shin DH, Lee IJ, Kim HY (2000) Rice allelopathy in Korea. In: Kim KU, Shin DH (eds) Rice allelopathy. Proceedings of the Workshop in Rice Allelopathy. Kyungpook National University, Taegu, pp 57–82
Kolde R (2015) pheatmap: Pretty Heatmaps. R package version 1.0.8
Kong C, Hu F, Zhang C, Xu X (2004a) Inducible effects of methyl jasmonate on allelochemicals from rice. Acta Ecol Sinica 24(2):177–180
Kong C, Xu X, Liang W, Zhou Y, Hu F (2004b) Non-phenolic allelochemicals in root exudates of an allelopathic rice variety and their identification and weed-suppressive activity. Acta Ecol Sinica 24(7):1317–1322
Kong CH, Zhang SZ, Li YH, Xia ZC, Yang XF, Meiners SJ, Wang P (2018) Plant neighbor detection and allelochemical response are driven by root-secreted signaling chemicals. Nat Commun 9:3867
Lal SK, Lee C, Sachs MM (1998) Differential regulation of enolase during anaerobiosis in maize. Plant Physiol 118(4):1285–1293
Lee DG, Ahsan N, Lee SH, Lee JJ, Bahk JD, Kang KY, Lee BH (2009) Chilling stress-induced proteomic changes in rice roots. J Plant Physiol 166(1):1–11
Li JY, Guo XK, Zhang Q, Liu CH, Lin ZH, Yu ZM, Wu H, He HB (2015) A novel screening method for rice allelopathic potential: the inhibitory-circle method. Weed Res 55:441–448
Li JY, Zhang Q, Yang XY, Hu WW, Lin RL, He HB (2017) A reappraisal of the content and the differences of phenolic acids between allelopathic and non-allelopathic rice accessions. Allelopathy J 40(1):35–46
Li XF, Ma JF, Matsumoto H (2000) Pattern of aluminum-induced secretion of organic acids differs between rye and wheat. Plant Physiol 123(4):1537–1544
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2 –∆∆CT method. Methods 25:402–408
Macias FA, Chinchilla N, Varela RM, Molinillo JM (2006) Bioactive steroids from Oryza sativa L. Steroids 71(7):603–608
Mangrauthia SK, Sailaja B, Pusuluri M, Jena B, Prasantha VV, Agarwala S, Senguttuvela P, Sarlaa N, Babu VR, Subrahmanyama D, Voleti SR (2018) Deep sequencing of small RNAs reveals ribosomal origin of microRNAs in Oryza sativa and their regulatory role in high temperature. Gene Rep 11:270–278
Mao X, Cai T, Olyarchuk JG, Wei L (2005) Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21:3787–3793
Mattice J, Lavy T, Skulman B, Dilday RH (1998) Searching for allelochemicals in rice that control ducksalad. In: Olofsdotter M (ed) Allelopathy in rice, Proceedings of the Workshop in Rice Allelopathy. International Rice Research Institute, Manila, pp 81–98
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Nanda AK, Pujol V, Wissuwa M (2017) Patterns of stress response and tolerance based on transcriptome profiling of rice crown tissue under zinc deficiency. J Exp Bot 68:1715–1729
Nguyen MX, Moon S, Jung KH (2013) Genome-wide expression analysis of rice aquaporin genes and development of a functional gene network mediated by aquaporin expression in roots. Planta 238(4):669–681
Olofsdotter M (1998) Allelopathy in rice. In: Olofsdotter M (ed) Allelopathy in rice, Proceedings of the Workshop in Rice Allelopathy. International Rice Research Institute, Manila, pp 1–5
Olofsdotter M, Rebulanan M, Madrid A, Dali W, Navarez D, Olk DC (2002) Why phenolic acids are unlikely primary allelochemicals in rice. J Chem Ecol 28:229–242
Putnam AR, Duke WO (1974) Biological suppression of weeds: evidence for allelopathy in accessions of cucumber. Sci 185:370–372
Seal AN, Pratley JE, Haig T, An M (2004) Identification and quantitation of compounds in a series of allelopathic and non-allelopathic rice root exudates. J Chem Ecol 30(8):1647–1662
Shankar R, Bhattacharjee A, Jain M (2016) Transcriptome analysis in different rice cultivars provides novel insights into desiccation and salinity stress responses. Sci Rep 6:23719
Shen C, Li D, He R, Fang Z, Xia Y, Gao J, Shen H, Cao M (2014) Comparative transcriptome analysis of RNA-seq data for cold-tolerant and cold-sensitive rice genotypes under cold stress. J Plant Biol 57:337–348
Shin DH, Kim KU, Sohn DS, Kang SG, Kim HY, Lee IJ, Kim MU (2000) Regulation of gene expression related to allelopathy. In: Kim KU, Shin DH (eds) Rice allelopathy, Proceedings of the workshop in Rice allelopathy. Kyungpook National University, Taegu, pp 109–124
Tang QY, Feng MG (2007) DPS data processing system: experimental design, statistical analysis, and data mining. Science Press, Beijing
Team RC (2014) R: a language and environment for statistical computing. R Foundation for Statistical computing, Vienna URL http://www.R-project.org/
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc 7:562–578
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515
Vergara BS (1992) A farmer's primer on growing rice. International Rice Research Institute, Manila, Philippines
Wang HB, He HB, Ye CY, Lu JC, Chen RS, Liu CH, Guo CK, Lin WX (2010) Molecular physiological mechanism of increased weed suppression ability of allelopathic rice mediated by low phosphorus stress. Allelopathy J 25(1):239–248
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10(1):57–63
Xu GF, Zhang FD, Li TL, Wu D, Zhang YH (2010) Induced effects of exogenous phenolic acids on allelopathy of a wild rice accession (Oryza longistaminata, S37). Rice Sci 17(2):135–140
Yan SP, Zhang QY, Tang ZC, Su WA, Sun WN (2006) Comparative proteomic analysis provides new insights into chilling stress responses in rice. Mol Cell Proteomics 5(3):484–496
Yang S, Hao D, Song Z, Yang GZ, Wang L, Su YH (2015) RNA-Seq analysis of differentially expressed genes in rice under varied nitrogen supplies. Gene 555(2):305–317
Young MD, Wakefield MJ, Smyth GK, Oshlack A (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11:R14
Zhai R, Feng Y, Wang H, Zhan X, Shen X, Wu W, Zhang Y, Chen D, Dai G, Yang Z, Cao L, Cheng S (2013) Transcriptome analysis of rice root heterosis by RNA-Seq. BMC Genomics 14:19
Zhang F, Luo X, Zhou Y, Xie J (2016) Genome-wide identification of conserved microRNA and their response to drought stress in Dongxiang wild rice (Oryza rufipogon Griff.). Biotechnol Lett 38(4):711–721
Zhang Q, Li L, Li J, Wang H, Fang C, Yang X, He H (2018) Increasing rice allelopathy by induction of barnyard grass (Echinochloa crus-galli) root exudates. J Plant Growth Regul 37(3):745–754
Zhou Y, Yang P, Cui F, Zhang F, Luo X, Xie J (2016) Transcriptome analysis of salt stress responsiveness in the seedlings of Dongxiang wild rice (Oryza rufipogon Griff.). PLoS One 11:e0146242
Acknowledgments
This work was supported by National Natural Science Foundation (31370380, 31701802, 31871556), China, and Fujian-Taiwan Joint Innovative Center for Germplasm Resources and Cultivation of Crop (FJ 2011 Program, no. 2015-75), the Outstanding Youth Scientific Fund of Fujian Agriculture and Forestry University (xjq201805). We wish to thank Prof. R. S. Zeng, the president of the World Congress of Allelopathy (2014-2017) for his careful corrections and valuable suggestions on the revision.
Funding
This work was supported by National Natural Science Foundation (31370380, 31701802, 31871556), China, and Fujian-Taiwan Joint Innovative Center for Germplasm Resources and Cultivation of Crop (FJ 2011 Program, no. 2015–75), the Outstanding Youth Scientific Fund of Fujian Agriculture and Forestry University (xjq201805). The funding body had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its supplementary information files.
Author information
Authors and Affiliations
Contributions
QZ, CXF, and HBH conceptualized and designed the experiment, managed the project and wrote the manuscript. XYZ and CZG conducted the metabolite analyses and analysed the data. SXL, LL, and JYL carried out the experiments and prepared the experimental materials. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Primer (XLSX 10 kb)
Additional file 2:
Table S2. Sequencing data (XLSX 9 kb)
Additional file 3:
Table S3. Number of mapped reads (XLSX 9 kb)
Additional file 4:
Table S4. Up-regulated genes in LE(+B) compared with LE(−B). GO terms for biological processes enriched with a P < 0.05 are provided. (XLSX 11 kb)
Additional file 5:
Table S5. Up-regulated genes in PI(+B) vs. PI(−B). GO terms for biological processes enriched with P < 0.05 are provided. (XLSX 16 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, Q., Zheng, XY., Lin, SX. et al. Transcriptome analysis reveals that barnyard grass exudates increase the allelopathic potential of allelopathic and non-allelopathic rice (Oryza sativa) accessions. Rice 12, 30 (2019). https://doi.org/10.1186/s12284-019-0290-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-019-0290-1