
Abstract
Stress-induced hyperglycemia has been considered an adaptive mechanism to stress up to the first intensive insulin therapy trial, which showed a 34% reduction in relative risk of in-hospital mortality when normalizing blood glucose levels. Further trials had conflicting results and, at present, stress-induced hyperglycemia management remains non-consensual. These findings could be explained by discrepancies in trials, notably regarding the approach to treat hyperglycemia: high versus restrictive caloric intake. Stress-induced hyperglycemia is a frequent complication during intensive care unit stay and is associated with a higher mortality. It results from an imbalance between insulin and counter-regulatory hormones, increased neoglucogenesis, and the cytokine-induced insulin-resistant state of tissues. In this review, we summarize detrimental effects of hyperglycemia on organs in the critically ill (peripheric and central nervous, liver, immune system, kidney, and cardiovascular system). Finally, we show clinical and experimental evidence of potential benefits from glucose and insulin administration, notably on metabolism, immunity, and the cardiovascular system.
Similar content being viewed by others


Introduction
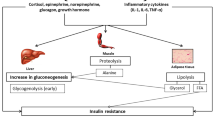
In an ICU, stress induces insulin resistance and overproduction of glucose, resulting in a syndrome called stress-induced hyperglycemia (SIH) [1]. SIH is common during critical illness and is associated with high mortality [1–3]. Its incidence is approximately 50% in septic shock [1] and 13% in surgical patients [4]. Up to the beginning of the 21st century, hyperglycemia was considered an adaptive mechanism to stress. In 2001, the landmark Leuven study by Van den Berghe and colleagues [4] reported a 34% relative risk reduction of in-hospital mortality when blood glucose was maintained at between 80 and 110 mg/dL. Since then, glucose metabolism during critical illness has been the focus of an increasing number of experimental and clinical studies. A decade later, after seven additional major randomized control trials, physicians remain confused about how to manage SIH. The heterogeneity of studies includes differences in population, ICU setting, staff experience, feeding strategy, blood glucose monitoring, variability, definition of hypoglycemia, insulin protocol, infusion site, its continuation after ICU discharge, and finally differences in the choice of the relevant major outcome. In the two ‘positive’ trials from Leuven, mean non-protein daily caloric intake was approximately 20 kcal/kg per day, essentially via glucose administration initially given intravenously: up to 200 to 300 g/day in the 2001 trial, with a median total daily insulin administration of 71 units (confidence interval of 48 to 100). By contrast, in NICE-SUGAR (Normoglycemia in Intensive Care Evaluation and Surviving Using Glucose Algorithm Regulation), which suggested increased mortality with intensive insulin therapy, caloric intake was 11.04 ± 6.08 kcal/kg per day, with 19.5% given intravenously, and cumulative mean daily dose of insulin was 50.2 ± 38.1 units per day [5]. Thus, there were two markedly different therapeutic approaches - that is, intensive gluco- and insulin therapy (liberal glucose intake, or the Leuven approach) and intensive insulin therapy (IIT) (restrictive glucose intake, or the NICE-SUGAR approach) [6]. The aim of this review is to discuss experimental evidence of organ injury and insulin sensitivity during SIH and expose differences in strategies for its control that include a liberal or a rather restrictive glucose intake.
The risk associated with hyperglycemia
After several large clinical trials, there is still no consensus on what blood glucose level (BGL) is ‘too much’. Whereas the Leuven trial demonstrated deleterious effects from uncontrolled glucose levels, subsequent trials comparing strategies to control BGL reported conflicting results [4, 5, 7–11].
SIH is undoubtedly associated with mortality in stroke [12], brain injury [2], and myocardial infarction [13] patients; trauma, cardiothoracic surgery, thermally injured, and mixed ICU patients [3]; and non-ICU hospitalized patients. The aim of this section is to summarize current knowledge about the mechanisms of hyperglycemia toxicity.
Pathogenesis of stress-induced hyperglycemia
Critical illness is characterized by an imbalance between insulin and endogenous or exogenous counter-regulatory hormones (glucagon and glucocorticoids). As a result, glucose production is increased and its storage is decreased secondary to downregulated glycogen synthesis and enhanced glycogenolysis [1]. In animal models, adrenaline infusion induces hyperglycemia via stimulation of hepatic and renal neoglucogenesis [14]. This pathway represents the major source of endogenous glucose during critical illness [1].
SIH is also the consequence of insulin resistance [1]. Experimental data on the mechanisms of sepsis-induced acute insulin resistance are scarce. Most of the knowledge about the mechanisms of acute insulin resistance comes from trauma/hemorrhage experimental models and studies in type 2 diabetes [15]. Under healthy conditions, insulin binding to its receptor results in the phosphorylation of insulin receptor substrates, which transmit insulin metabolic and growth signals. One major effector of the metabolic pathway is glucose transporter family (GLUT) 4, which facilitates glucose transport across cell membranes in muscle and adipose tissue. During injury, inhibitor of kappa B kinase and Jun B pathways are activated, leading to expression of inflammatory markers such as TNF. First, Jun B negatively regulates insulin receptor substrate and then TNF downregulates GLUT 4 gene transcription [16]. These mechanisms could account for insulin resistance during sepsis [17].
Whether this mechanism of response to aggression is deleterious during critical illness is still a matter of debate [18]. In fact, insulin resistance is variable from one tissue to another and appears to be moderate in the heart and diaphragm but is major in skeletal muscles and adipocytes [19]. In fact, despite this insulin resistance, glucose utilization is enhanced in sepsis secondary to different GLUT overexpression [20].
Glucose transport across cell membranes is the rate-limiting step of cellular glucose metabolism. Each GLUT is characterized mostly by organ specificity, insulin sensitivity, and the Michaelis constant (Km). Km is defined as the transporter’s specific value of glycemia for which 50% of transport capacities are reached. These characteristics (Table 1) could explain differences in organ sensitivity to insulin and modulation of glucose uptake with glycemia. This heterogeneity may be seen as a protective effect of vital organs while placing other organs in a ‘hibernating state’.
Hyperglycemia is responsible for reactive oxygen species (ROS) overproduction in diabetes via four major pathways: advanced glycated endproduct release, de novo indirect activation of C kinase protein, increased polyol, and hexosamine pathway flux redox homeostasis [21]. On one hand, some ROS production can be essential for immune cell ‘respiratory burst’ to kill pathogens or for endothelial cell functions. On the other hand, during hyperglycemia, excess ROS may worsen organ failure [22].
Hyperglycemia and the central nervous system
Brain cells are dependent on glucose to maintain their membrane ionic gradient. However, the detrimental cerebral effects of hyperglycemia have been observed in critical illness [2, 12, 23].
Brain glucose metabolism has some particularities:
-
Glucose crosses blood–brain barrier and cellular membranes via a high-affinity and insulin-insensitive process involving GLUT 1 and GLUT 3 [24].
-
Neurons and astrocytes cooperate to metabolize carbohydrates, as suggested by lactate shuttles between these cells.
-
During hypoxemic or hypoperfusion stress, GLUT 1 and 3 are upregulated (up to 300% in trauma) with a subsequent increase in glucose uptake [25].
Hyperglycemia has been shown to enhance the breakdown of the blood–brain barrier via induction of matrix metalloproteinase [26] and to induce apoptosis [23], mostly via enhanced superoxide production. Indeed, in epidemiologic studies on stroke, hyperglycemia is associated with edema, infarct size, mortality in non-diabetic patients, and poor functional status at 1 year [12]. Nevertheless, trials aiming at controlling BGL in stroke, subarachnoid hemorrhage [27], brain injury [28], and neuro-intensive care [29] patients did not report improved outcome with tight glucose control.
Hyperglycemia and the peripheral nervous system
Neuromyopathy is a frequent complication of critical illness, such as septic shock and acute respiratory distress syndrome, with an incidence of up to 50% of patients with these conditions. In the first Leuven study, the risk of developing a critical illness neuromyopathy (CINM) was lowered from 49% to 25% (P < 0.0001) in the interventional group, facilitating weaning from mechanical ventilation. The mechanisms of hyperglycemic neuromyopathy are poorly understood and may involve activation of apoptotic and inflammation pathways in response to acute hyperglycemia in muscles [30] or ROS overproduction as suggested in type 2 diabetic neuropathy or CINM pathogenesis [21].
Hyperglycemia and other organs
Liver
In resting conditions, GLUT 2 is the predominant transporter for glucose in hepatic parenchymal cells [24, 31]. This low-affinity transporter modulates glucose transport proportionally to BGL. After lipopolysaccharide (LPS) stimulation, GLUT 2 decreases, whereas GLUT 1 increases [31], resulting in an enhanced insulin- and glycemia-independent uptake of up to 2.4-fold [31, 32]. Analyses of liver cells from the control group of the Leuven study revealed dramatic lesions to the mitochondria. These mitochondria abnormalities may result from excessive glucose uptake, with subsequent overproduction of ROS [33], which could have been diminished with BGL normalization.
Immune system
In the Leuven study, patients with normoglycemia had almost 50% fewer bloodstream infections (7.8% versus 4.2%, P = 0.003) [4]. Indeed, each step of the immune response to stress is altered with hyperglycemia. First, in diabetes, chronic high BGL induces overexpression of surface and circulating cell adhesion molecules [34–36], whereas LPS challenge is less effective in upregulating cell adhesion molecules [34]. This enhanced overall immune cell adhesion paradoxically results in a less effective chemotactism and transmigration capacity of immune cells [37]. Second, worse polymorphonuclear killing capacities against pathogens, as assessed by concentrations of lysosomal enzyme or burst respiratory intensity, are observed when BGL is poorly controlled with a dose-effect relationship in diabetes [38]. Finally, the production of chemokines and other pro-inflammatory factors is decreased under hyperglycemic conditions [39].
Kidney
In septic shock, GLUT 2 and 3 expressions are decreased in the tubular epithelial cells of the kidney, whereas GLUT 1 expression is increased. This may account for enhanced glycosuria and acute renal failure during septic shock [40]. In the Leuven study, renal replacement therapy was twice less frequent in patients with normoglycemia, whereas insulin per se was associated with worse renal outcome [41]. This kidney protection may result directly from lower BGL, since high BGL directly inhibits transcription of an anti-apoptotic gene in renal tubules [42] or from improvement in lipid profile, ROS production, and endothelial protection [43].
Heart and endothelium
SIH has been shown to be an important prognostic factor in acute coronary syndromes [13]. The heart has a remarkable ability to switch from free fatty acid oxidation to carbohydrate oxidation under hypoxemic conditions [44]. During acute myocardial infarction, SIH activates T cells in the atherosclerotic plaque and increases tissue levels of inflammatory markers and nitric oxide and ROS production, resulting in endothelial dysfunction [45]. Consequently, coronary blood flow and reserve during myocardial infarction are impaired [46]. Furthermore, acute hyperglycemia increases infarct size and suppresses cardioprotective signal transduction via mitochondrial potassium ATP channel inhibition [47].
In shock, although BGLs are high, glucose represents only 12% of substrate oxidation by cardiomyocytes [48]. Therefore, one could argue that hyperglycemia without insulin infusion does not confer a metabolic benefit and presents rather deleterious consequences on an ischemic heart.
Evidence for gluco-insulinotherapy
Clinical trial calendar
See Table 2.
Analysis of critical differences between trials
In the Leuven studies, patients were given intravenous glucose at 8 to 12 g/hour, with mean intravenous glucose feeding of 120 g during the first 15 hours, to a goal of 200 to 260 g/day afterward. This study shows higher intravenous glucose and insulin administration than in any other study (Table 3), whereas epidemiologic studies have shown that both are correlated with an increased mortality [3, 41, 52].
Other trials of glucose control in the ICU used lower glucose intake and insulin doses. In none of these studies did the experimental intervention achieve maintenance of normal BGL like in the Leuven study. NICE-SUGAR is the only study showing increased mortality with tight BGL control. In that study, total caloric intake was much lower than in the Leuven study. In the Specialized Relative Insulin and Nutrition Tables (SPRINT) study, a 35% lowering of hospital mortality for patients with a long stay in the ICU (P = 0.02) was observed after implementation of tight glucose control when glucose was administered enterally to allow a caloric intake of 25 kcal/kg per day. Likewise, the cumulative insulin dose per day was close to that observed in the experimental group of the Leuven study (67.2 units in SPRINT versus 71 units). These findings are in line with the latest IIT meta-analysis by Marik and Preiser [6], who suggested that intravenous calorie administration plays a pivotal role for improvement of outcome during IIT. In contrast, the last Leuven trial, EPaNIC (Early versus late Parenteral Nutrition in Intensive Care), showed that parenteral nutrition administration to achieve a caloric intake of 20 to 25 kcal/kg per day might be detrimental. This raises the question of the effect of an exclusive and important glucose infusion during IIT in critical illness.
It was then suggested that, in the Leuven trial, difference in observed mortality was secondary to a higher mortality in the control group due to an excessive glucose load. Nevertheless, control mortality in the Leuven study matched the mortality expected from estimation of the EuroSCORE (European System for Cardiac Operative Risk Evaluation). Secondly, a recent meta-analysis suggested that intravenous glucose intake was an independent predictive factor for good outcome in the Leuven studies [6]. But whether blood glucose control or insulin administration mediated positive effects in this study was not studied.
In 2003, Van den Berghe and colleagues [41] performed a post-hoc analysis of their first study. The authors showed that both total amount of infused insulin and glycemic control were associated with lower mortality (independently of age, delayed ICU admission, Acute Physiology and Chronic Health Evaluation II score, reason for ICU admission, history of malignancy or diabetes, and at-admission hyperglycemia). The strength of association between the mortality rate and the mean BGL seemed to be stronger than with the total daily infused insulin [41]. Nevertheless, no statistical comparison was made between these factors in this study. Furthermore, the respective effects of these two entwined factors could be analyzed only in an interventional study comparing gluco-insulinotherapy versus tight glycemic control. Then a recent study by Arabi and colleagues [53] with a 2 × 2 factorial design compared IIT and permissive underfeeding (60% to 70% of daily recommended caloric intake versus 90% to 100%). Their study showed no mortality differences between groups but was underpowered and non-blinded, and the therapeutic goals were not achieved [53].
Finally, in 2011, the Leuven group performed the EPaNIC study that evaluated the timing of parenteral nutrition introduction. In that study, a strategy of early parenteral nutrition initiation was performed with administration of 400 kcal (100 g) at day 1 and 800 kcal at day 2 exclusively via intravenous glucose administration, and then a relay with mixed parenteral and enteral nutrition was performed to achieve calculated daily physiological caloric intake [54]. The control group received minimal glucose administration and enteral nutrition was started at day 2 if oral intake was insufficient. Results showed an increased rate of complications in the parenteral nutrition group (infection and cholestasis), whereas the late initiation of parenteral nutrition resulted in a shorter duration of renal replacement therapy, mechanical ventilation, and stay in the ICU. In that study, the amount of administered glucose was three times lower than in the 2001 study, and insulin doses were also lower in both groups: 31 insulin units (interquartile range (IQR) 19 to 48) in the control versus 58 insulin units (IQR 40 to 85) in the experimental group. Furthermore, parenteral nutrition contains lipid at recommended doses that could present detrimental effects as fat oxidation is a high oxygen-consuming metabolic pathway. A post-hoc analysis of EPaNIC concerning the first 2 days in the ICU in that study before introduction of parenteral nutrition might be of interest to clinicians and help them determine whether high glucose administration during IIT is beneficial for patients. We will present clinical and experimental evidence that may support the use of a glucose-insulin administration strategy.
Is gluco-insulinotherapy associated with a decreased incidence of hypoglycemia?
The clinical signs of hypoglycemia are commonly masked in sedated patients. Thus, in clinical trials, hypoglycemia was defined empirically by a BGL value of less than 40 mg/dL. Its incidence varied from 5.1% to 18.7% in patients with IIT and from 0.5% to 4.1% in control groups. Seizures and comas have occasionally been observed following severe hypoglycemic episodes without establishing a clear causal relationship [55]. Neuronal death during or following hypoglycemia has also been found in both animal and human models, but hypoglycemia does not seem to affect neurocognitive development in children [56] but may contribute to long-term cognitive impairment following critical illness in adults [57]. The existence of a direct causal link between hypoglycemia and mortality remains controversial, and hypoglycemia could reflect only a more severe illness. Some epidemiologic studies have found that only early or spontaneous hypoglycemia was independently associated with death in critically ill patients [58, 59]. Preventive interventions are thus warranted in such a situation. Whether an increased daily amount of carbohydrate administered would decrease the risk of hypoglycermia during tight BGL is unknown.
In a retrospective study by Arabi and colleagues [60], glucose intake was not a risk or protective factor of hypoglycemia whereas insulin daily dosage was an evident risk factor (73.5 ± 36.7 in the group presenting hypoglycemia versus 47.5 ± 51.8; P < 0.0001) [60]. Actually, caloric intake lowering (gastroparesis, intravenous glucose, or enteral nutrition lowering) without insulin adjustment may be one of the most frequent risk factors for hypoglycemia [54, 55, 60, 61], and no study evaluated the effect of gluco-insulinotherapy on hypoglycemia rate. The recent EPaNIC study showed a decreased rate of hypoglycemia during IIT when early parenteral nutrition was initiated (1.9% versus 3.5%, P = 0.001), suggesting a possible protective role of gluco-insulinotherapy to be explored in an interventional study [54].
Effects of high insulin and glucose intake on organs
Gluco-insulinotherapy consists of a high amount of glucose infused and higher insulinemia. Effects of insulin on glycemia lowering are mediated mostly by an increase in cellular uptake of glucose through GLUT 4 translocation to the membrane. GLUT 4 is located mostly on adipocytes and skeletal muscle cells [1]. Thus, mostly GLUT 4-expressing cells consume glucose administered intravenously during IIT.
During early sepsis, metabolic stress resulted in glycogenolysis and depleted energetic reserves as shown in skeletal muscle biopsies [33, 62]. This ATP depletion was correlated with poor outcome, and in addition recovery from sepsis was preceded by normalization of the phosphocreatine/ATP ratio [62]. Indeed, energy depletion during sepsis could be a risk factor for CINM, an ICU complication associated with higher mortality [63]. In fact, skeletal muscle protein levels were higher in the IIT group consistently with anabolic effects of insulin [33]. Insulin augments the number of GLUT 4 receptors in adipose and skeletal muscle cells and the glucose uptake by myocytes [64]. This higher amount of energetic substrate may be a protective factor against energy depletion and sarcopenia and may lower the risk to develop CINM. These findings are in line with the observed lower rate of CINM and the late improvement in survival in the Leuven trials [4, 11].
Experiments from the Leuven cohort showed that maintenance of normal BGL protected the liver and skeletal muscles. In the liver, glucose uptake through GLUT 2 is independent of BGLs. Indeed, untreated hyperglycemia was associated with severely damaged mitochondria with altered complex I and IV activities [33]. Furthermore, insulin administration during injury partly suppresses gluconeogenesis [65]. Gluconeogenesis is an active process occurring mostly in the liver that requires four molecules of ATP and two molecules of GTP. This energy requirement may enhance hypoxic injury in liver during stress and could be counteracted with insulin administration [64–66].
Whether gluco-insulinotherapy rather than BGL control protects the liver from hypoxic injury has been studied in few human and experimental studies. One study conducted by a different group showed beneficial effects of insulin independently of BGLs on hepatocyte apoptosis, cytolysis, and expression of inflammatory markers [65]. Further studies in the Leuven group did not reproduce these results and showed a lower blood level of transaminases in burn-injured rabbits with BGL control rather than insulin administration [67]. Liver injury may be mediated by a mitochondriopathy in reaction to cellular hyperglycemia and enhanced glycolysis and is likely to mediate organ damage [68]. As insulin sensitivity is not overcome during IIT in the liver, glucose uptake by hepatocytes is likely to be dependent on glycemia rather than insulinemia [66].
In sepsis models, insulin has been shown to improve immune cell function independently of glycemia. It inhibits the apoptosis of activated macrophages, may modulate antigen presentation, and improves chemotaxis and phagocytic properties. Finally, insulin may modulate the balance between lymphocyte T helper type and lymphocyte T helper type 2 cells, favoring anti-inflammation and repair function [69]. Such effects of insulin on the immune system may account for the reduced rate of bloodstream infection during IIT [4].
During sepsis, the heart shows little or no insulin resistance [70] and lowers its glucose consumption [48, 70]. In porcine models, glucose and insulin infusion favored glucose and lactate utilization. This results in improvement in inotropic function without higher oxygen consumption observed in different studies [67, 71]. In fact, during acute coronary syndrome, glucose insulin potassium therapy was associated with substantial survival and may prevent arrhythmias [72]. However, the benefit of glucose insulin potassium infusion in patients with myocardial infarction remains controversial. In the ICU, IIT was not associated with reduced time or doses of inotropic support [4] and was associated with a higher incidence of cardiovascular death in NICE-SUGAR [5], but the Leuven study introduced IIT with an important amount of intravenous glucose administered to cardiovascular post-surgical patients. Such an inotropic effect may have improved organ perfusion and contributed to the lower renal failure rate and the better outcome in these patients.
In summary, gluco-insulinotherapy may present protective effects on muscle or improve immune or cardiac function. Contrary to Marik and Bellomo [18] in their recent comment, we hypothesize that gluco-insulinotherapy may be a more beneficial rather than a restrictive strategy. This issue needs to be further studied.
Conclusions
The era of glucose control in the ICU started in 2001. Untreated SIH no doubt favors morbidity and mortality. Critical analyses of randomized controlled trials have suggested that glucose control is more likely to be associated with survival benefit when strict normal glucose levels are achieved and early high glucose intake is provided. An interventional study evaluating liberal and restrictive glucose intake during IIT is warranted to provide reliable evidence.
Abbreviations
- BGL:
-
blood glucose level
- CINM:
-
critical illness neuromyopathy
- EPaNIC:
-
Early versus late Parenteral Nutrition in Intensive Care
- GLUT:
-
glucose transporter
- IIT:
-
intensive insulin therapy
- LPS:
-
lipopolysaccharide
- NICE-SUGAR:
-
Normoglycemia in Intensive Care Evaluation and Surviving Using Glucose Algorithm Regulation
- ROS:
-
reactive oxygen species
- SIH:
-
stress-induced hyperglycemia
- SPRINT:
-
Specialized Relative Insulin and Nutrition Tables
- TNF:
-
tumor necrosis factor.
References
McCowen KC, Malhotra A, Bistrian BR: Stress-induced hyperglycemia. Crit Care Clin. 2001, 17: 107-124. 10.1016/S0749-0704(05)70154-8.
Salim A, Hadjizacharia P, Dubose J, Brown C, Inaba K, Chan LS, Margulies D: Persistent hyperglycemia in severe traumatic brain injury: an independent predictor of outcome. Am Surg. 2009, 75: 25-29.
Finney SJ, Zekveld C, Elia A, Evans TW: Glucose control and mortality in critically ill patients. JAMA. 2003, 290: 2041-2047. 10.1001/jama.290.15.2041.
van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R: Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001, 345: 1359-1367. 10.1056/NEJMoa011300.
Finfer S, Chittock DR, Su SY, Blair D, Foster D, Dhingra V, Bellomo R, Cook D, Dodek P, Henderson WR, Hébert PC, Heritier S, Heyland DK, McArthur C, McDonald E, Mitchell I, Myburgh JA, Norton R, Potter J, Robinson BG, Ronco JJ, NICE-SUGAR Study Investigators: Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009, 360: 1283-1297.
Marik PE, Preiser JC: Toward understanding tight glycemic control in the ICU: a systematic review and metaanalysis. Chest. 2010, 137: 544-551. 10.1378/chest.09-1737.
Annane D, Cariou A, Maxime V, Azoulay E, D’honneur G, Timsit JF, Cohen Y, Wolf M, Fartoukh M, Adrie C, Santré C, Bollaert PE, Mathonet A, Amathieu R, Tabah A, Clec’h C, Mayaux J, Lejeune J, Chevret S, COIITSS Study Investigators: Corticosteroid treatment and intensive insulin therapy for septic shock in adults: a randomized controlled trial. JAMA. 2010, 303: 341-348.
Arabi YM, Dabbagh OC, Tamim HM, Al-Shimemeri AA, Memish ZA, Haddad SH, Syed SJ, Giridhar HR, Rishu AH, Al-Daker MO, Kahoul SH, Britts RJ, Sakkijha MH: Intensive versus conventional insulin therapy: a randomized controlled trial in medical and surgical critically ill patients. Crit Care Med. 2008, 36: 3190-3197. 10.1097/CCM.0b013e31818f21aa.
De La Rosa GC, Donado JH, Restrepo AH, Quintero AM, González LG, Saldarriaga NE, Bedoya M, Toro JM, Velásquez JB, Valencia JC, Arango CM, Aleman PH, Vasquez EM, Chavarriaga JC, Yepes A, Pulido W, Cadavid CA, Grupo de Investigacion en Cuidado intensivo: GICI-HPTU: Strict glycaemic control in patients hospitalised in a mixed medical and surgical intensive care unit: a randomised clinical trial. Crit Care. 2008, 12: R120-10.1186/cc7017.
Preiser JC, Devos P, Ruiz-Santana S, Mélot C, Annane D, Groeneveld J, Iapichino G, Leverve X, Nitenberg G, Singer P, Wernerman J, Joannidis M, Stecher A, Chioléro R: A prospective randomised multi-centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: the Glucontrol study. Intensive Care Med. 2009, 35: 1738-1748. 10.1007/s00134-009-1585-2.
Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, Van Wijngaerden E, Bobbaers H, Bouillon R: Intensive insulin therapy in the medical ICU. N Engl J Med. 2006, 354: 449-461. 10.1056/NEJMoa052521.
Baird TA, Parsons MW, Phanh T, Butcher KS, Desmond PM, Tress BM, Colman PG, Chambers BR, Davis SM: Persistent poststroke hyperglycemia is independently associated with infarct expansion and worse clinical outcome. Stroke. 2003, 34: 2208-2214. 10.1161/01.STR.0000085087.41330.FF.
Capes SE, Hunt D, Malmberg K, Gerstein HC: Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000, 355: 773-778. 10.1016/S0140-6736(99)08415-9.
McGuinness OP, Snowden RT, Moran C, Neal DW, Fujiwara T, Cherrington AD: Impact of acute epinephrine removal on hepatic glucose metabolism during stress hormone infusion. Metabolism. 1999, 48: 910-914. 10.1016/S0026-0495(99)90228-7.
Li L, Messina JL: Acute insulin resistance following injury. Trends Endocrinol Metab. 2009, 20: 429-435. 10.1016/j.tem.2009.06.004.
Qi C, Pekala PH: Tumor necrosis factor-alpha-induced insulin resistance in adipocytes. Proc Soc Exp Biol Med. 2000, 223: 128-135. 10.1046/j.1525-1373.2000.22318.x.
Stephens JM, Bagby GJ, Pekala PH, Shepherd RE, Spitzer JJ, Lang CH: Differential regulation of glucose transporter gene expression in adipose tissue or septic rats. Biochem Biophys Res Commun. 1992, 183: 417-422. 10.1016/0006-291X(92)90497-9.
Marik PE, Bellomo R: Stress hyperglycemia: an essential survival response!. Crit Care. 2013, 17: 305-10.1186/cc12514.
Thompson LH, Kim HT, Ma Y, Kokorina NA, Messina JL: Acute, muscle-type specific insulin resistance following injury. Mol Med. 2008, 14: 715-723.
Meszaros K, Lang CH, Bagby GJ, Spitzer JJ: Contribution of different organs to increased glucose consumption after endotoxin administration. J Biol Chem. 1987, 262: 10965-10970.
Brownlee M: Biochemistry and molecular cell biology of diabetic complications. Nature. 2001, 414: 813-820. 10.1038/414813a.
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M: Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000, 404: 787-790. 10.1038/35008121.
Polito A, Brouland JP, Porcher R, Sonneville R, Siami S, Stevens RD, Guidoux C, Maxime V, de la Grandmaison GL, Chrétien FC, Gray F, Annane D, Sharshar T: Hyperglycaemia and apoptosis of microglial cells in human septic shock. Crit Care. 2011, 15: R131-10.1186/cc10244.
Thorens B, Mueckler M: Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab. 2010, 298: E141-E145. 10.1152/ajpendo.00712.2009.
Zhang WW, Zhang L, Hou WK, Xu YX, Xu H, Lou FC, Zhang Y, Wang Q: Dynamic expression of glucose transporters 1 and 3 in the brain of diabetic rats with cerebral ischemia reperfusion. Chin Med J (Engl). 2009, 122: 1996-2001.
Kamada H, Yu F, Nito C, Chan PH: Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: relation to blood–brain barrier dysfunction. Stroke. 2007, 38: 1044-1049. 10.1161/01.STR.0000258041.75739.cb.
Bilotta F, Spinelli A, Giovannini F, Doronzio A, Delfini R, Rosa G: The effect of intensive insulin therapy on infection rate, vasospasm, neurologic outcome, and mortality in neurointensive care unit after intracranial aneurysm clipping in patients with acute subarachnoid hemorrhage: a randomized prospective pilot trial. J Neurosurg Anesthesiol. 2007, 19: 156-160. 10.1097/ANA.0b013e3180338e69.
Bilotta F, Caramia R, Cernak I, Paoloni FP, Doronzio A, Cuzzone V, Santoro A, Rosa G: Intensive insulin therapy after severe traumatic brain injury: a randomized clinical trial. Neurocrit Care. 2008, 9: 159-166. 10.1007/s12028-008-9084-9.
Azevedo JR, Lima ER, Cossetti RJ, Azevedo RP: Intensive insulin therapy versus conventional glycemic control in patients with acute neurological injury: a prospective controlled trial. Arq Neuropsiquiatr. 2007, 65: 733-738. 10.1590/S0004-282X2007000500001.
Russell ST, Rajani S, Dhadda RS, Tisdale MJ: Mechanism of induction of muscle protein loss by hyperglycaemia. Exp Cell Res. 2009, 315: 16-25. 10.1016/j.yexcr.2008.10.002.
Spolarics Z, Pekala PH, Bagby GJ, Spitzer JJ: Brief endotoxemia markedly increases expression of GLUT1 glucose transporter in Kupffer, hepatic endothelial and parenchymal cells. Biochem Biophys Res Commun. 1993, 193: 1211-1215. 10.1006/bbrc.1993.1754.
Battelino T, Goto M, Krzisnik C, Zeller WP: Tissue glucose transport and glucose transporters in suckling rats with endotoxic shock. Shock. 1996, 6: 259-262.
Vanhorebeek I, De Vos R, Mesotten D, Wouters PJ, De Wolf-Peeters C, Van den Berghe G: Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet. 2005, 365: 53-59. 10.1016/S0140-6736(04)17665-4.
Andreasen AS, Pedersen-Skovsgaard T, Berg RM, Svendsen KD, Feldt-Rasmussen B, Pedersen BK, Moller K: Type 2 diabetes mellitus is associated with impaired cytokine response and adhesion molecule expression in human endotoxemia. Intensive Care Med. 2010, 36: 1548-1555. 10.1007/s00134-010-1845-1.
Quagliaro L, Piconi L, Assaloni R, Da Ros R, Maier A, Zuodar G, Ceriello A: Intermittent high glucose enhances ICAM-1, VCAM-1 and E-selectin expression in human umbilical vein endothelial cells in culture: the distinct role of protein kinase C and mitochondrial superoxide production. Atherosclerosis. 2005, 183: 259-267. 10.1016/j.atherosclerosis.2005.03.015.
Mochizuki K, Shimada M, Tanaka Y, Fukaya N, Goda T: Reduced expression of beta2 integrin genes in rat peripheral leukocytes by inhibiting postprandial hyperglycemia. Biosci Biotechnol Biochem. 2010, 74: 2470-2474. 10.1271/bbb.100554.
Turina M, Fry DE, Polk HC: Acute hyperglycemia and the innate immune system: clinical, cellular, and molecular aspects. Crit Care Med. 2005, 33: 1624-1633. 10.1097/01.CCM.0000170106.61978.D8.
McManus LM, Bloodworth RC, Prihoda TJ, Blodgett JL, Pinckard RN: Agonist-dependent failure of neutrophil function in diabetes correlates with extent of hyperglycemia. J Leukoc Biol. 2001, 70: 395-404.
Stegenga ME, van der Crabben SN, Dessing MC, Pater JM, van den Pangaart PS, de Vos AF, Tanck MW, Roos D, Sauerwein HP, van der Poll T: Effect of acute hyperglycaemia and/or hyperinsulinaemia on proinflammatory gene expression, cytokine production and neutrophil function in humans. Diabet Med. 2008, 25: 157-164. 10.1111/j.1464-5491.2007.02348.x.
Schmidt C, Hocherl K, Bucher M: Regulation of renal glucose transporters during severe inflammation. Am J Physiol Renal Physiol. 2007, 292: F804-F811.
Van den Berghe G, Wouters PJ, Bouillon R, Weekers F, Verwaest C, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P: Outcome benefit of intensive insulin therapy in the critically ill: insulin dose versus glycemic control. Crit Care Med. 2003, 31: 359-366. 10.1097/01.CCM.0000045568.12881.10.
Ortiz A, Ziyadeh FN, Neilson EG: Expression of apoptosis-regulatory genes in renal proximal tubular epithelial cells exposed to high ambient glucose and in diabetic kidneys. J Investig Med. 1997, 45: 50-56.
Schetz M, Vanhorebeek I, Wouters PJ, Wilmer A, Van den Berghe G: Tight blood glucose control is renoprotective in critically ill patients. J Am Soc Nephrol. 2008, 19: 571-578. 10.1681/ASN.2006101091.
Beadle RM, Frenneaux M: Modification of myocardial substrate utilisation: a new therapeutic paradigm in cardiovascular disease. Heart. 2010, 96: 824-830. 10.1136/hrt.2009.190256.
Langouche L, Vanhorebeek I, Vlasselaers D, Vander Perre S, Wouters PJ, Skogstrand K, Hansen TK, Van den Berghe G: Intensive insulin therapy protects the endothelium of critically ill patients. J Clin Invest. 2005, 115: 2277-2286. 10.1172/JCI25385.
Jessani SS, Lane DA, Shantsila E, Watson T, Millane TA, Lip GY: Impaired glucose tolerance and endothelial damage, as assessed by levels of von Willebrand factor and circulating endothelial cells, following acute myocardial infarction. Ann Med. 2009, 41: 608-618. 10.1080/07853890903159256.
LaDisa JF, Krolikowski JG, Pagel PS, Warltier DC, Kersten JR: Cardioprotection by glucose-insulin-potassium: dependence on KATP channel opening and blood glucose concentration before ischemia. Am J Physiol Heart Circ Physiol. 2004, 287: H601-H607. 10.1152/ajpheart.00122.2004.
Dhainaut JF, Huyghebaert MF, Monsallier JF, Lefevre G, Dall’Ava-Santucci J, Brunet F, Villemant D, Carli A, Raichvarg D: Coronary hemodynamics and myocardial metabolism of lactate, free fatty acids, glucose, and ketones in patients with septic shock. Circulation. 1987, 75: 533-541. 10.1161/01.CIR.75.3.533.
Krinsley JS: Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients. Mayo Clin Proc. 2003, 78: 1471-1478. 10.4065/78.12.1471.
Brunkhorst FM, Engel C, Bloos F, Meier-Hellmann A, Ragaller M, Weiler N, Moerer O, Gruendling M, Oppert M, Grond S, Olthoff D, Jaschinski U, John S, Rossaint R, Welte T, Schaefer M, Kern P, Kuhnt E, Kiehntopf M, Hartog C, Natanson C, Loeffler M, Reinhart K, German Competence Network Sepsis (SepNet): Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008, 358: 125-139. 10.1056/NEJMoa070716.
Chase JG, Pretty CG, Pfeifer L, Shaw GM, Preiser JC, Le Compte AJ, Lin J, Hewett D, Moorhead KT, Desaive T: Organ failure and tight glycemic control in the SPRINT study. Crit Care. 2010, 14: R154-10.1186/cc9224.
der Voort PH, Feenstra RA, Bakker AJ, Heide L, Boerma EC, van der Horst IC: Intravenous glucose intake independently related to intensive care unit and hospital mortality: an argument for glucose toxicity in critically ill patients. Clin Endocrinol (Oxf). 2006, 64: 141-145. 10.1111/j.1365-2265.2006.02437.x.
Arabi YM, Tamim HM, Dhar GS, Al-Dawood A, Al-Sultan M, Sakkijha MH, Kahoul SH, Brits R: Permissive underfeeding and intensive insulin therapy in critically ill patients: a randomized controlled trial. Am J Clin Nutr. 2011, 93: 569-577. 10.3945/ajcn.110.005074.
Casaer MP, Mesotten D, Hermans G, Wouters PJ, Schetz M, Meyfroidt G, Van Cromphaut S, Ingels C, Meersseman P, Muller J, Vlasselaers D, Debaveye Y, Desmet L, Dubois J, Van Assche A, Vanderheyden S, Wilmer A, Van den Berghe G: Early versus late parenteral nutrition in critically ill adults. N Engl J Med. 2011, 365: 506-517. 10.1056/NEJMoa1102662.
Vriesendorp TM, DeVries JH, van Santen S, Moeniralam HS, de Jonge E, Roos YB, Schultz MJ, Rosendaal FR, Hoekstra JB: Evaluation of short-term consequences of hypoglycemia in an intensive care unit. Crit Care Med. 2006, 34: 2714-2718. 10.1097/01.CCM.0000241155.36689.91.
Mesotten D, Gielen M, Sterken C, Claessens K, Hermans G, Vlasselaers D, Lemiere J, Lagae L, Gewillig M, Eyskens B, Vanhorebeek I, Wouters PJ, Van den Berghe G: Neurocognitive development of children 4 years after critical illness and treatment with tight glucose control: a randomized controlled trial. JAMA. 2012, 308: 1641-1650. 10.1001/jama.2012.12424.
Duning T, van den Heuvel I, Dickmann A, Volkert T, Wempe C, Reinholz J, Lohmann H, Freise H, Ellger B: Hypoglycemia aggravates critical illness-induced neurocognitive dysfunction. Diabetes Care. 2010, 33: 639-644. 10.2337/dc09-1740.
Bagshaw SM, Bellomo R, Jacka MJ, Egi M, Hart GK, George C: The impact of early hypoglycemia and blood glucose variability on outcome in critical illness. Crit Care. 2009, 13: R91-10.1186/cc7921.
Kosiborod M, Inzucchi SE, Goyal A, Krumholz HM, Masoudi FA, Xiao L, Spertus JA: Relationship between spontaneous and iatrogenic hypoglycemia and mortality in patients hospitalized with acute myocardial infarction. JAMA. 2009, 301: 1556-1564. 10.1001/jama.2009.496.
Arabi YM, Tamim HM, Rishu AH: Hypoglycemia with intensive insulin therapy in critically ill patients: predisposing factors and association with mortality. Crit Care Med. 2009, 37: 2536-2544. 10.1097/CCM.0b013e3181a381ad.
Krinsley JS, Grover A: Severe hypoglycemia in critically ill patients: risk factors and outcomes. Crit Care Med. 2007, 35: 2262-2267. 10.1097/01.CCM.0000282073.98414.4B.
Carré JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P, Stotz M, Singer M: Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med. 2010, 182: 745-751. 10.1164/rccm.201003-0326OC.
Sharshar T, Bastuji-Garin S, Stevens RD, Durand MC, Malissin I, Rodriguez P, Cerf C, Outin H, De Jonghe B: Presence and severity of intensive care unit-acquired paresis at time of awakening are associated with increased intensive care unit and hospital mortality. Crit Care Med. 2009, 37: 3047-3053. 10.1097/CCM.0b013e3181b027e9.
Langouche L, Vander Perre S, Wouters PJ, D’Hoore A, Hansen TK, Van den Berghe G: Effect of intensive insulin therapy on insulin sensitivity in the critically ill. J Clin Endocrinol Metab. 2007, 92: 3890-3897. 10.1210/jc.2007-0813.
Jeschke MG, Rensing H, Klein D, Schubert T, Mautes AE, Bolder U, Croner RS: Insulin prevents liver damage and preserves liver function in lipopolysaccharide-induced endotoxemic rats. J Hepatol. 2005, 42: 870-879. 10.1016/j.jhep.2004.12.036.
Mesotten D, Delhanty PJ, Vanderhoydonc F, Hardman KV, Weekers F, Baxter RC, Van Den Berghe G: Regulation of insulin-like growth factor binding protein-1 during protracted critical illness. J Clin Endocrinol Metab. 2002, 87: 5516-5523. 10.1210/jc.2002-020664.
Ellger B, Debaveye Y, Vanhorebeek I, Langouche L, Giulietti A, Van Etten E, Herijgers P, Mathieu C, Van den Berghe G: Survival benefits of intensive insulin therapy in critical illness: impact of maintaining normoglycemia versus glycemia-independent actions of insulin. Diabetes. 2006, 55: 1096-1105. 10.2337/diabetes.55.04.06.db05-1434.
Vanhorebeek I, Ellger B, De Vos R, Boussemaere M, Debaveye Y, Perre SV, Rabbani N, Thornalley PJ, Van den Berghe G: Tissue-specific glucose toxicity induces mitochondrial damage in a burn injury model of critical illness. Crit Care Med. 2009, 37: 1355-1364. 10.1097/CCM.0b013e31819cec17.
Deng HP, Chai JK: The effects and mechanisms of insulin on systemic inflammatory response and immune cells in severe trauma, burn injury, and sepsis. Int Immunopharmacol. 2009, 9: 1251-1259. 10.1016/j.intimp.2009.07.009.
Lang CH, Dobrescu C, Meszaros K: Insulin-mediated glucose uptake by individual tissues during sepsis. Metabolism. 1990, 39: 1096-1107. 10.1016/0026-0495(90)90172-9.
Holger JS, Dries DJ, Barringer KW, Peake BJ, Flottemesch TJ, Marini JJ: Cardiovascular and metabolic effects of high-dose insulin in a porcine septic shock model. Acad Emerg Med. 2010, 17: 429-435. 10.1111/j.1553-2712.2010.00695.x.
Malmberg K, Norhammar A, Ryden L: Insulin treatment post myocardial infarction: the DIGAMI study. Adv Exp Med Biol. 2001, 498: 279-284. 10.1007/978-1-4615-1321-6_35.
Acknowledgments
We thank Fouad Fadel for helpful discussion and Mathilde Vinet for language editing.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Aurélien Mazeraud, Andrea Polito and Djillali Annane contributed equally to this work.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Mazeraud, A., Polito, A. & Annane, D. Experimental and clinical evidences for glucose control in intensive care: is infused glucose the key point for study interpretation?. Crit Care 18, 232 (2014). https://doi.org/10.1186/cc13998
Published:
DOI: https://doi.org/10.1186/cc13998