
Abstract
The relationship between osteoblast-specific insulin signaling, osteocalcin activation and gluco-metabolic homeostasis has proven to be complex and potentially inconsistent across animal-model systems and in humans. Moreover, the impact of postnatally acquired, osteoblast-specific insulin deficiency on the pancreas-to-skeleton-to-pancreas circuit has not been studied. To explore this relationship, we created a model of postnatal elimination of insulin signaling in osteoprogenitors. Osteoprogenitor-selective ablation of the insulin receptor was induced after ~10 weeks of age in IRl°x/lox/Osx-Cre+/− genotypic male and female mice (designated postnatal-OIRKO). At ~21 weeks of age, mice were then phenotypically and metabolically characterized. Postnatal-OIRKO mice demonstrated a significant reduction in circulating concentrations of undercarboxylated osteocalcin (ucOC), in both males and females compared with control littermates. However, no differences were observed between postnatal-OIRKO and control mice in: body composition (lean or fat mass); fasting serum insulin; HbA1c; glucose dynamics during glucose tolerance testing; or in pancreatic islet area or islet morphology, demonstrating that while ucOC is impacted by insulin signaling in osteoprogenitors, there appears to be little to no relationship between osteocalcin, or its derivative (ucOC), and glucose homeostasis in this model.
Similar content being viewed by others
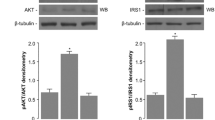


Introduction
Over the last decade, since osteocalcin was first proposed to be a regulator of glucose metabolism1,2,3, a number of controversies have arisen in the field to suggest that mouse models may not satisfactorily represent human osteocalcin physiology - and not all rodent models may fully support the overall bone-energy relationship4,5,6. Initial studies proposed that in mice, insulin signaling in osteoblasts, if disrupted prenatally, was linked to a feed-forward mechanism to enhance insulin secretion by activating osteocalcin to undercarboxylated osteocalcin (ucOC) through enhanced osteoclast activity, and in turn, ucOC regulated glucose homeostasis by directly enhancing pancreatic production of insulin via the GPRC6A receptor2,3,7. Yet, in contrast to the initial studies, when the insulin receptor (IR) is ablated prenatally at an earlier stage in ontogenesis (i.e., in osteoprogenitors), no significant abnormalities in fasting blood glucose or insulin levels, or in glucose or insulin dynamics after a glucose challenge, are observed, in either male or female mice8.
Therefore, to examine further this potential link between insulin signaling in osteoblastic cells and the generation of ucOC as a regulator of glucose homeostasis, we developed a mouse model designed to mitigate any potential prenatal effects that may impact this metabolic circuit through selectively eliminating the insulin receptor in osteoprogenitors only in mature mice. This model of a postnatally-acquired deficiency in insulin signaling also better mimics when insulin deficiency, insulin resistance and/or glucose dysregulation would arise in mice and humans, leading to glucose intolerance and ultimately frank diabetes. Herein, we hypothesized that disruption of insulin signaling in osteoprogenitors in mature mice would impact production of ucOC and potentially impact body composition and glucose homeostasis.
Results
In mice, insulin signaling in osteoblasts has been linked by others to a feed-forward mechanism to “activate” osteocalcin into undercarboxylated osteocalcin, which, in turn, regulates glucose homeostasis by signaling through the GPRC6A on osteogenic cells2,3,9. To interrogate this proposed pathway in mature mice, the metabolic phenotype of mice possessing a postnatal osteoprogenitor-specific IR knock-out (postnatal-OIRKO) was characterized.
Weight and body composition of the postnatal-OIRKO mouse
At study end, body weight (BW) was significantly greater for males, compared with females, both for control mice and for OIRKO mice (Control: M vs. F; 28.7 ± 0.9 vs. 22.2 ± 1.4 gm, p ≤ 0.001. OIRKO: M vs. F; 28.4 ± 1.4 vs. 23.5 ± 1.0 gm, p ≤ 0.001). However, within each sex, there were no differences in BW between control and postnatal-OIRKO genotypes. Similarly, as shown in Fig. 1, body composition by DXA also demonstrated significant differences between male and female mice, both for the control and the OIRKO cohorts, for lean mass and total mass (Fig. 1A,C). Specifically, male mice consistently exhibited higher lean and total mass, compared with females. However, again, no differences were identified between control genotypes and the postnatal-OIRKO mice for either sex. Additionally, there were no significant differences between fat mass or percent (%) fat mass (Fig. 1B,D), either between males and females, or between control and OIRKO genotypes. Bone mineral density (BMD) and bone mineral content (BMC) were also assessed and no significant differences were noted between control mice and postnatal-OIRKO mice for either sex (data not shown).

Body Composition Total body lean mass (A), fat mass (B), total mass (C), and percent fat mass (D), as analyzed by DXA, are shown for control male (M:•), OIRKO male (M:⚬), control female (F:▴), and OIRKO female (F:▵) mice. For between group comparisons, p values are designated as follows: (*), p ≤ 0.05; (**), p ≤ 0.01; (***), p ≤ 0.001.
Skeletal biomarkers in the postnatal-OIRKO mouse
As shown in Fig. 2, when comparing serum levels for both undercarboxylated (Fig. 3A, ucOC) and carboxylated osteocalcin (Fig. 2B, cOC) in males vs. females, ucOC and cOC values were significantly lower in males compared with females, whether control or OIRKO genotype (Fig. 2A,B). Moreover, for both males and females, ucOC levels were lower in OIRKO animals, compared with controls (Fig. 2A; males: p ≤ 0.05; females: p ≤ 0.01), whereas cOC did not differ between OIRKO and control mice. A strong correlation between individual ucOC and cOC values was also evident (Fig. 2C). However, serum concentrations of P1NP, a systemic biomarker of bone formation, OPG, an inhibitor of osteoclastogenesis, and RatLaps, a marker of bone resorption, did not differ across the four groups (see Fig. 2D-F).

Bone Biomarkers Measurement of ucOC (A), cOC (B), RatLaps (D), P1NP (E), and OPG (F) are shown for control male (M:•), OIRKO male (M:⚬), control female (F:▴), and OIRKO female (F:▵) mice. For between group comparisons, p values are designated as follows: (*), p ≤ 0.05; (**), p ≤ 0.01; (***), p ≤ 0.001. A strong correlation between ucOC and cOC was present, in 2 C.

Metabolic Biomarkers Fasting serum insulin (A) and HbA1c (B) are presented, for control male (M:•), OIRKO male (M:⚬), control female (F:▴), and OIRKO female (F:▵) mice. Results for ipGTT are shown in 4 C and 4D. For between group comparisons, p values are designated as follows: (*), p ≤ 0.05; (**), p ≤ 0.01; (***), p ≤ 0.001.
Metabolic phenotyping of the postnatal-OIRKO mouse
As shown in Fig. 3, no significant differences were identified in male or female postnatal-OIRKO mice compared to sex-matched control mice for: fasting serum insulin (Fig. 3A); fed insulin levels (data not shown); HbA1c (Fig. 3B); or glucose dynamics during glucose tolerance testing (Fig. 3C,D). Among control mice alone, ipGTT AUC was lower for females, compared with males (Fig. 3D).
Assessment of pancreatic islets of postnatal-OIRKO mice
As shown in Fig. 4A, islets from both control and postnatal-OIRKO islets stained positive for markers of β-cells (insulin) and α-cells (glucagon), suggesting no qualitative alterations in the cellular composition of islets in the postnatal-OIRKO mouse. Moreover, morphometric analysis of pancreatic islet area and islet circularity, an indirect method of estimating the overall spherical shape of islets which is correlated with islet functionality, did not demonstrate any statistically significant differences between postnatal-OIRKO and control mice (Fig. 4B,C)10.

Pancreatic islet assessment. (A) Representative immunofluorescent microphotographs of IR+/+ (Wildtype) and IRf/f (postnatal-OIRKO) male islets stained for Insulin (Ins, green), Glucagon (GCG, red) and Dapi (blue). Scale bar = 100 μm. (B) Morphometric analysis of pancreatic islets: mean islet area (left panel) and islet circularity (right panel) assessed with Zen2.3 software, in n = 60–85 islets, in n = 3 mice/group (M, male; F, female). A circularity value of 1.0 indicates a perfect circle. As the value approaches 0.0, it indicates an increasingly elongated polygon. Data are expressed as mean ± SEM.
Discussion
In the postnatal-OIRKO mouse, ucOC levels were reduced in both male and female mice compared to control mice, consistent with previous studies2,3. Originally, it was proposed that OPG expression was diminished in mice in which the IR was downregulated in osteoblasts, thereby promoting osteoclastic activity which then in turn caused decarboxylation of cOC to ucOC3. In the current study, ucOC serum concentrations were highly correlated to cOC concentrations (Fig. 2C), but did not correlate to peripheral markers of osteoclast activity (i.e., RatLAPs) or to circulating concentrations of an inhibitor of osteoclasts, OPG (data not shown). Furthermore, RatLaps and OPG were not significantly different amongst sexes or genotypes (Fig. 2D,F). Thus the etiology of decreased ucOC in postnatal-OIRKO male and female mice is not fully understood.
The consistent finding that insulin-signaling in osteoblastic cells is involved in ucOC physiology, is also supported by findings from human studies. We and others have demonstrated that in humans with type 1 diabetes, a condition of insulin deficiency, serum concentrations of ucOC are positively associated with insulin exposure, either exogenously administered or endogenously produced, as assessed by c-peptide levels11,12. Because this association holds true from shortly after the diagnosis of type 1 diabetes to many years after the diagnosis, some have proposed the ucOC might be a useful surrogate marker for residual β-cell function in type 1 diabetes12. Post-hoc analyses examining correlations between fasting insulin levels and ucOC (R2 = −0.259; p = 0.02) or cOC (R2 = −0.363; p = 0.005) in control and postnatal OIRKO mice demonstrates an inverse correlation between fasting insulin levels and ucOC and cOC, suggesting that insulin signaling in osteoprogenitors regulates the production of ucOC and cOC.
Body composition, as determined by DXA, did not differ between control mice and postnatal-OIRKO mice. This finding differs from prior reports wherein prenatal reduction in IR expression in osteoblasts and osteoprogenitors, respectively, produced both overweight and underweight mice, respectively2,8. It is not clear how these variations in body weight, fat mass, and size might contribute, at least in part, to the variable metabolic phenotypes observed in each mouse model. In contrast to previous studies showing lower or normal levels of insulin in prenatal models of osteoblast or osteoprogenitor deletion of the IR, postnatal deletion showed fasting insulin levels trending higher in both male and female postnatal OIRKO mice compared to controls, consistent with other mouse models in which the IR was deleted in specific tissues, such as liver and β-cells, resulting in elevated insulin levels13,14. No significant differences were found in glucose dynamics in the postnatal-OIRKO mouse, similar to what was observed in the prenatal-OIRKO mouse8, but inconsistent with the two originally published studies which described glucose dysregulation when the IR was deleted in osteoblasts prenatally2,3. Furthermore, examination of pancreatic islets from control and postnatal-OIRKO mice revealed similar pancreatic islet size, architecture and morphology. This is in contrast to a prior study which revealed that when the IR was deleted prenatally in osteoblasts, islet size and morphology, as well as insulin content was diminished3.
Interestingly, discrepancies in glucose homeostasis have also been reported in mouse models wherein mediators of insulin signaling (i.e., FoxO transcription factors) have been eliminated in osteoblasts or osteoprogenitors. For instance, deletion of FoxO1 (which would mimic active insulin signaling) in osteoblasts, improves glucose tolerance compared to control mice and also is associated with increased insulin levels15. In contrast, deletion of FoxO1,3,4 in osteoprogenitors results in no alterations in insulin secretion and no dysregulation of glucose homeostasis16. Furthermore, variable metabolic phenotypes have been observed in models in which the purported receptor for ucOC, GPRC6A, has been disrupted. Studies in which exon II of GPRC6A was deleted demonstrated a metabolic phenotype17. However, later studies using different deletion constructs of GPRC6A (i.e., exon VI and full locus) have failed to demonstrate glucose dysregulation, alterations in insulin levels, or changes in body composition18,19,20,21.
Currently, the reasons for the variable metabolic phenotypes in the various mouse models used to explore the feed-forward mechanism of energy homeostasis and ucOC is unclear. Possible confounders may include the well-recognized variability of glucose and insulin dynamics that exists amongst inbred mouse models, between sexes, and the changes that occur over the lifespan22,23. Furthermore, the same genetic deletion in various inbred backgrounds have produced different metabolic phenotypes24,25. For instance, heterozygous knock-down of the IR and IRS1 in three different genetic backgrounds (C57BL/6, 129/Sv and DBA/2) resulted in extremely different metabolic phenotypes from frank hyperglycemia and hyperinsulinemia (C57BL/6) to essentially no metabolic phenotype (129 Sv)25. In addition, differences in glucose dynamics in various models and strains may only be further unmasked if the mouse model is challenged, such as may occur in diet-induced obesity26. Many of these potential confounders may not have been adequately controlled for in prior experimental designs. Other considerations may relate to when in development insulin signaling disruption in osteoblasts occurs (prenatally vs. postnatally); at what stage of osteoblastogenesis the disruption manifests (progenitor vs mature osteoblast, etc.); as well as possible issues with certain Cre-recombinase mouse models and the lack-of specificity for targeting only bone cells27,28.
Conclusions
While there has been wide-spread acceptance of the feed-forward mechanism by which insulin signaling in osteoblasts promotes insulin production and alters glucose metabolism via ucOC through the GPRC6A receptor in β-cells, we and others have now generated several additional mouse models that bring this mechanism into question as a generalizable phenomenon. Future studies will be needed to clarify if the feed-forward mechanism is only contextual in nature and if insulin signaling throughout osteogenesis influences metabolic pathways and energy only within certain windows of osteoblastic development and on certain genetic backgrounds.
Research Design and Methods
Animals and experimental design
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kentucky (#2015–1348). All methods were carried out in accordance with relevant guidelines and regulations. As we have previously described8, osteoprogenitor-selective ablation of the insulin receptor was accomplished by crossing mice homozygous for a floxed insulin receptor (IR) allele in which loxP sites flank exon 4 of the IR gene (designated IRlox/lox) with heterozygous Osterix (Osx)-Cre transgenic mice (B6.Cg-Tg(Sp7-tTA,tetO-EGFP/cre)1Amc/J; The Jackson Laboratory, Bar Harbor, ME). F1 progeny were crossed to produce IRlox/lox/Cre+/− and IRlox/lox/Cre−/− mice which were then bred to produce experimental animals (designated Osteoprogenitor Insulin Receptor Knock-out, OIRKO; with genotype IRlox/lox/Cre+/−). IRlox/lox/Cre−/− mice (IR flox) and IR/Cre+/+ mice (Cre+) littermates were designated as control genotypes.
Cre transgene expression is repressed by doxycycline (Tet-OFF)29. Therefore, for these postnatal studies, breeding animals and their progeny were maintained on chow containing doxycycline (Diet S3888, Bio-Serv; Frenchtown, NJ). After weaning, all mice were maintained on doxycycline chow until ~9–10 weeks of age; thereafter, they were transitioned to regular (no doxycycline) chow (Diet 8640, Harlan; Indianapolis, IN) for an additional 12 weeks. After switching to regular chow, the Cre transgene is expressed30, resulting in disruption of the floxed IR alleles (see supplementary figure 1). These mice are hereafter designated as postnatal-OIRKO.
Confirmatory genotyping was performed on all mice, using published procedures31. All mice were maintained in a 12-hour light-dark cycle, and provided ad libitum access to food and water throughout all studies, unless otherwise specified below. Individual body weights were measured weekly. Intraperitoneal (ip) glucose tolerance testing (ipGTT) was completed during the final week of the study8,32. For ipGTT, mice were weighed and then fasted for 4–5 hours with free access to water. Blood glucose and serum insulin levels were measured in the fasting state. Blood glucose measurements were obtained at 0, 15, 30, 60, and 120 minutes following an ip injection of glucose (1.5 g/kg body weight). Twelve weeks following removal of doxycycline from the diet, both male and female mice (mean age = 20.7 ± 0.2 weeks) from all three genotypes were then metabolically characterized. Specifically, body composition was analyzed by X-ray densitometry (DXA), as detailed below. Trunk blood was collected for physiological assessment of metabolic parameters and bone biomarkers. Pancreatic tissue was harvested at study end.
Dual-energy X-ray absorptiometry (DXA) analysis
Body composition was measured using the Lunar PIXImus2 (GE Lunar Corp.) and software version 2.10. Instrument performance was analyzed daily by scanning an aluminum/lucite phantom (TBMD = 0.0625 g/cm2, percentage fat = 10.9%). Mice were weighed immediately prior to DXA scan. Mice were anesthetized with isoflurane and placed in a prostrate position on the imaging tray as suggested by the manufacturer. Limbs were extended from the body and were taped to the imaging tray. Heads were excluded from all analyses by placing an exclusion region of interest over the head. All scans and analyses were performed by R.C.B.
Systemic biomarkers
Fasting or fed blood glucose (BG) was measured via glucometer (OneTouch Ultra 2 blood glucose monitoring system, Lifescan, Inc., Milpitas, CA). PINP (total procollagen type 1 N-terminal propeptide) was measured using the Rat/Mouse P1NP Competitive Enzyme Immunoassay (Immunodiagnostics Systems, Inc., Fountain Hills, AZ; #AC-33F1). RatLaps (C-terminal telopeptide I) and OPG (osteoprotegerin) were measured by rodent-specific ELISA (Immunodiagnostics Systems, Inc., Fountain Hills, AZ, AC-06F1; and RayBiotech Inc., Norcross, GA, ELM-OPG-1, respectively). Similarly, mouse osteocalcin components [carboxylated osteocalcin (cOC) and incompletely carboxylated or undercarboxylated osteocalcin (ucOC)] were measured by ELISA (MyBioSource.com, San Diego, CA; #MBS744268 and #MBS706251, respectively). Fasting insulin (measured at baseline during ipGTT), was quantified using a mouse ultrasensitive insulin ELISA (Crystal Chem USA, Elk Grove Village, IL; #90080).
Pancreatic islet histology
Islet morphometry
Islet area (μm2) and circularity were assessed by using Hematoxylin & Eosin stained sections of the pancreas (5 um thickness), from n = 3 mice from each group. Three sections per mouse (100 μm apart) were photographed with the AxioObserver Z1 inverted microscope (10x magnification) and analyzed with the ZEN 2.3 software (Carl Zeiss Microscopy, Jena, Germany).
Immunofluorescence
Pancreatic tissue was fixed overnight in 4% phosphate-buffered paraformaldehyde, paraffin-embedded and sectioned on a classical microtome (5 μm thickness). The sections were collected on Superfrost Plus coverslips, dewaxed in xylene, and rehydrated through a series of ethanol baths. After antigen retrieval (citrate buffer 10 mM, pH = 6), the sections were blocked with normal goat serum (Vector Laboratories, Burlingame, CA) and incubated overnight at 4 oC with the primary antibodies: mouse anti-Insulin (Sigma-Aldrich Corp., St. Louis, MO; 1:600) and rabbit anti-Glucagon (Abcam, Cambridge, MA; 1:400). The sections were than incubated at room temperature with a cocktail of secondary antibodies conjugated with Alexa594 and Alexa488 (Invitrogen Alexa Fluor dye-labeled secondary antibodies, Thermo Fisher Scientific, Waltham, MA; 1:200). Nuclei were stained with DAPI (Sigma-Aldrich Corp., St. Louis, MO). Negative controls were obtained by omitting the primary antibody. The sections were examined with the AxioObserver Z1 inverted microscope (Carl Zeiss Microscopy, Jena, Germany) equipped with an Axiocam 506 monochrome camera. Images were captured, recorded, and analyzed using the ZEN 2.3 Pro or ImageJ software.
Statistical analysis
Unless otherwise stated, results for individual parameters are reported as mean ± standard deviation (SD). A one-way Analysis of Variance (ANOVA) was used to determine differences between groups for each response variable, followed by the Tukey test to correct for multiple comparisons; statistically significant multiplicity-adjusted p values for each comparison were reported, and values were considered statistically significant at p ≤ 0.05. Where applicable, to determine the relationship between variables of interest, a Pearson correlation coefficient was computed between data sets, and the two-tailed p-value and R2 are reported.
References
Lee, N. K. et al. Endocrine regulation of energy metabolism by the skeleton. Cell, 130(3), 456–69, https://doi.org/10.1016/j.cell.2007.05.047 (2007).
Fulzele, K. et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell, 142(2), 309–19, https://doi.org/10.1016/j.cell.2010.06.002 (2010).
Ferron, M. et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell, 142(2), 296–308, https://doi.org/10.1016/j.cell.2010.06.003 (2010).
Andrews, N. A. Skeletal regulation of glucose metabolism: challenges in translation from mouse to man. IBMS BoneKEy, 10(353), https://doi.org/10.1038/bonekey.2013.87 (2013).
Manolagas, S. C. & Kronenberg, H. M. “Reproducibility of results in preclinical studies: a perspective from the bone field. J Bone Miner Res, 29(10), 2131–40, https://doi.org/10.1002/jbmr.2293 (2014).
Lambert, L. J. et al. Increased trabecular bone and improved biomechanics in an osteocalcin-null rat model created by CRISPR/Cas9 technology. Dis Model Mech, 9(10), 1169–1179, https://doi.org/10.1242/dmm.025247 (2016).
Pi, M. et al. Evidence for Osteocalcin Binding and Activation of GPRC6A in beta-Cells. Endocrinology, 157(5), 1866–80, https://doi.org/10.1210/en.2015-2010 (2016).
Thrailkill, K. et al. Loss of insulin receptor in osteoprogenitor cells impairs structural strength of bone. J Diabetes Res 2014, 703589, https://doi.org/10.1155/2014/703589 (2014).
Pi, M. Wu, Y. & Quarles, L. D. “GPRC6A mediates responses to osteocalcin in beta-cells in vitro and pancreas in vivo. J Bone Miner Res, 26(7), 1680–3, https://doi.org/10.1002/jbmr.390 (2011).
Huang, H. H. Harrington, S. & L. Stehno-Bittel, “The Flaws and Future of Islet Volume Measurements. Cell Transplant, 27(7), 1017–1026, https://doi.org/10.1177/0963689718779898 (2018).
Thrailkill, K. M. et al. Determinants of undercarboxylated and carboxylated osteocalcin concentrations in type 1 diabetes. Osteoporos Int, 23(6), 1799–806, https://doi.org/10.1007/s00198-011-1807-7 (2012).
Sabek, O. M. et al. Serum C-peptide and osteocalcin levels in children with recently diagnosed diabetes. Endocrinol Diabetes Metab, 3(1), e00104, https://doi.org/10.1002/edm2.104 (2020).
Kulkarni, R. N., et al. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell, 96(3), 329–39, https://doi.org/10.1016/s0092-8674(00)80546-2 (1999).
Michael, M. D. et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell, 6(1), 87–97, https://www.ncbi.nlm.nih.gov/pubmed/10949030 (2000).
Rached, M. T. et al. FoxO1 expression in osteoblasts regulates glucose homeostasis through regulation of osteocalcin in mice. J Clin Invest, 120(1), 357–68, https://doi.org/10.1172/JCI39901 (2010).
Iyer, S. et al. Deletion of FoxO1, 3, and 4 in Osteoblast Progenitors Attenuates the Loss of Cancellous Bone Mass in a Mouse Model of Type 1 Diabetes. J Bone Miner Res, 32(1), 60–69, https://doi.org/10.1002/jbmr.2934 (2017).
Pi, M. et al. GPRC6A null mice exhibit osteopenia, feminization and metabolic syndrome. PLoS One 3(12), e3858, https://doi.org/10.1371/journal.pone.0003858 (2008).
Jorgensen, C. V. et al. Metabolic and skeletal homeostasis are maintained in full locus GPRC6A knockout mice. Sci Rep, 9(1), 5995, https://doi.org/10.1038/s41598-019-41921-8 (2019).
S. Smajilovic et al. The L-alpha-amino acid receptor GPRC6A is expressed in the islets of Langerhans but is not involved in L-arginine-induced insulin release. Amino Acids, 44(2), 383–90, https://doi.org/10.1007/s00726-012-1341-8 (2013).
Clemmensen, C. et al. Increased susceptibility to diet-induced obesity in GPRC6A receptor knockout mice. J Endocrinol, 217(2), 151–60, https://doi.org/10.1530/JOE-12-0550 (2013).
Clemmensen, C. Jorgensen, C. V. Smajilovic, S. & H. Brauner-Osborne, “Robust GLP-1 secretion by basic L-amino acids does not require the GPRC6A receptor. Diabetes Obes Metab, 19(4), 599–603, https://doi.org/10.1111/dom.12845 (2017).
Goren, H. J. Kulkarni, R. N. & Kahn, C. R. Glucose homeostasis and tissue transcript content of insulin signaling intermediates in four inbred strains of mice: C57BL/6, C57BLKS/6, DBA/2, and 129X1. Endocrinology, 145(7), 3307–23, https://doi.org/10.1210/en.2003-1400 (2004).
Berglund, E. D. et al. Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes, 57(7), 1790–9, https://doi.org/10.2337/db07-1615 (2008).
Kido, Y. Philippe, N. Schaffer, A. A. & Accili, D. Genetic modifiers of the insulin resistance phenotype in mice. Diabetes, 49(4), 589–96, https://doi.org/10.2337/diabetes.49.4.589 (2000).
Kulkarni, R. N. et al. Impact of genetic background on development of hyperinsulinemia and diabetes in insulin receptor/insulin receptor substrate-1 double heterozygous mice. Diabetes, 52(6), 1528–34, https://doi.org/10.2337/diabetes.52.6.1528 (2003).
Heydemann, A. An Overview of Murine High Fat Diet as a Model for Type 2 Diabetes Mellitus. J Diabetes Res 2016, 2902351, https://doi.org/10.1155/2016/2902351 (2016).
Dallas, S. L. Xie, Y. Shiflett, L. A. & Ueki, Y. Mouse Cre Models for the Study of Bone Diseases. Curr Osteoporos Rep, 16(4), 466–477, https://doi.org/10.1007/s11914-018-0455-7 (2018).
F. Elefteriou and Yang, X. Genetic mouse models for bone studies–strengths and limitations. Bone, 49(6), 1242–54, https://doi.org/10.1016/j.bone.2011.08.021 (2011).
M. Gossen and Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proceedings of the National Academy of Sciences of the United States of America, 89(12), 5547–51, http://www.ncbi.nlm.nih.gov/pubmed/1319065 (1992).
Fowlkes, J. L. et al. Constitutive activation of MEK1 in osteoprogenitors increases strength of bone despite impairing mineralization. Bone, 130, 115106, https://doi.org/10.1016/j.bone.2019.115106 (2020).
Bruning, J. C. et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell, 2(5), 559–69, http://www.ncbi.nlm.nih.gov/pubmed/9844629 (1998).
Thrailkill, K. M. et al. Genetic ablation of SGLT2 function in mice impairs tissue mineral density but does not affect fracture resistance of bone. Bone, 133, 115254, https://doi.org/10.1016/j.bone.2020.115254 (2020).
Acknowledgements
The authors acknowledge and appreciate support for this work, provided by grants from the National Institutes of Health, R56DK084045 (to J.L.F.) and R21AR070620 (to K.M.T); as well as funding from the University of Kentucky Barnstable Brown Diabetes Center Research Endowment. The content is solely the responsibility of the authors and does not necessarily represent the official views of any of the listed funding agencies.
Author information
Authors and Affiliations
Contributions
J.L.F., K.M.T. and R.C.B., contributed to the design of the experiment. J.L.F., K.M.T., R.C.B., E.K., P.R., and I.P. researched data, contributed to discussion, wrote manuscript and reviewed/edited manuscript. J.L.F. and K.M.T. accept responsibility for the integrity of the data analysis. All authors have approved the final version of the submitted manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fowlkes, J.L., Clay Bunn, R., Kalaitzoglou, E. et al. Postnatal loss of the insulin receptor in osteoprogenitor cells does not impart a metabolic phenotype. Sci Rep 10, 8842 (2020). https://doi.org/10.1038/s41598-020-65717-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65717-3
- Springer Nature Limited