

Key Points
-
Cystinosis is a multi-systemic lysosomal storage disease caused by inactivating mutations in, or the absence of, the lysosomal membrane exporter for cystine, cystinosin; cystinosis is the main cause of hereditary renal Fanconi syndrome
-
Treatment with cysteamine efficiently depletes lysosomal cystine and delays progression to renal insufficiency; however, cysteamine does not reverse established renal Fanconi syndrome, indicating functions of cystinosin beyond cystine transport
-
Insights from mechanistic studies suggest that the pathological mechanisms of Fanconi syndrome in cystinosis are multifactorial, involving oxidative stress and impaired vesicular trafficking, autophagy, and mTORC1 and TFEB signalling
-
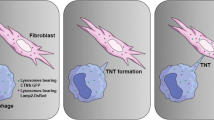
Haematopoietic stem cell (HSC) transplantation ameliorates renal Fanconi syndrome in cystinotic mice; HSCs differentiate into macrophages that transfer cystinosin-bearing lysosomes into proximal tubule cells via tunnelling nanotubes that cross the tubular basement membrane
-
Since tunnelling nanotubes contain donor-derived cytosol and carry all types of organelles, this mechanism should be generic and could be used to correct other genetic diseases that affect proximal tubule cells
Abstract
Cystinosis is an autosomal recessive metabolic disease that belongs to the family of lysosomal storage disorders. It is caused by a defect in the lysosomal cystine transporter, cystinosin, which results in an accumulation of cystine in all organs. Despite the ubiquitous expression of cystinosin, a renal Fanconi syndrome is often the first manifestation of cystinosis, usually presenting within the first year of life and characterized by the early and severe dysfunction of proximal tubule cells, highlighting the unique vulnerability of this cell type. The current therapy for cystinosis, cysteamine, facilitates lysosomal cystine clearance and greatly delays progression to kidney failure but is unable to correct the Fanconi syndrome. This Review summarizes decades of studies that have fostered a better understanding of the pathogenesis of the renal Fanconi syndrome associated with cystinosis. These studies have unraveled some of the early molecular changes that occur before the onset of tubular atrophy and identified a role for cystinosin beyond cystine transport, in endolysosomal trafficking and proteolysis, lysosomal clearance, autophagy and the regulation of energy balance. These studies have also led to the identification of new potential therapeutic targets and here, we outline the potential role of stem cell therapy for cystinosis and provide insights into the mechanism of haematopoietic stem cell-mediated kidney protection.







Similar content being viewed by others


References
Fanconi, G. Die nicht diabetischen glykosurien und hyperglykamiendes altem kindes. Jahrbuch Kinderheilkunde 133, 257–300 (1931).
De Toni, G. Remarks on the relations between renal rickets (renal dwarfism) and renal diabetes. Acta Paediatr. 16, 479–484 (1933).
Debre, R., Marie, J., Cleret, J. & Messimy, R. Rachitisme tardif coexistant avec une nephrite chronique et une glycosurie. Arch. Med. Enfants 37, 597–606 (1934).
Haffner, D. et al. Long-term outcome of paediatric patients with hereditary tubular disorders. Nephron 83, 250–260 (1999).
Gahl, W. A., Thoene, J. G. & Schneider, J. A. Cystinosis. N. Engl. J. Med. 347, 111–121 (2002).
Cohen, C. et al. Excellent long-term outcome of renal transplantation in cystinosis patients. Orphanet J. Rare Diseases 10, 90 (2015).
Gahl, W. A., Kuehl, E. M., Iwata, F., Lindblad, A. & Kaiser-Kupfer, M. I. Corneal crystals in nephropathic cystinosis: natural history and treatment with cysteamine eyedrops. Mol. Genet. Metab. 71, 100–120 (2000).
Brodin-Sartorius, A. et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 81, 179–189 (2012).
Langman, C. B. et al. Controversies and research agenda in nephropathic cystinosis: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 89, 1192–1203 (2016).
Nesterova, G. & Gahl, W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr. Nephrol. 23, 863–878 (2008).
Goldman, H., Scriver, C. R., Aaron, K., Delvin, E. & Canlas, Z. Adolescent cystinosis: comparisons with infantile and adult forms. Pediatrics 47, 979–988 (1971).
Cogan, D. G., Kuwabara, T., Kinoshita, J., Sheehan, L. & Merola, L. Cystinosis in an adult. J. Am. Med. Assoc. 164, 394–396 (1957).
Town, M. et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 18, 319–324. (1998).
Cherqui, S., Kalatzis, V., Trugnan, G. & Antignac, C. The targeting of cystinosin to the lysosomal membrane requires a tyrosine-based signal and a novel sorting motif. J. Biol. Chem. 276, 13314–13321 (2001).
Kalatzis, V., Cherqui, S., Antignac, C. & Gasnier, B. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. 20, 5940–5949. (2001).
Kalatzis, V. et al. The ocular anomalies in a cystinosis animal model mimic disease pathogenesis. Pediatr. Res. 62, 156–162 (2007).
Simpson, J. et al. Quantitative in vivo and ex vivo confocal microscopy analysis of corneal cystine crystals in the Ctns knockout mouse. Mol. Vision 17, 2212–2220 (2011).
Cherqui, S. et al. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol. Cell. Biol. 22, 7622–7632 (2002).
Cheung, W. W. et al. Muscle wasting and adipose tissue browning in infantile nephropathic cystinosis. J. Cachexia Sarcopenia Muscle 7, 152–164 (2016).
Gaide Chevronnay, H. P. et al. A mouse model suggests two mechanisms for thyroid alterations in infantile cystinosis: decreased thyroglobulin synthesis due to endoplasmic reticulum stress/unfolded protein response and impaired lysosomal processing. Endocrinology 6, 2349–2364 (2015).
Nevo, N. et al. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol. Dial Transplant 25, 1059–1066 (2010).
Mahoney, C. P. & Striker, G. E. Early development of the renal lesions in infantile cystinosis. Pediatr. Nephrol. 15, 50–56 (2000).
Yeagy, B. A. et al. Kidney preservation by bone marrow cell transplantation in hereditary nephropathy. Kidney Int. 79, 1198–1206 (2011).
Schneider, J. A. Approval of cysteamine for patients with cystinosis. Pediatr. Nephrol. 9, 254 (1995).
Thoene, J. G., Oshima, R. G., Crawhall, J. C., Olson, D. L. & Schneider, J. A. Cystinosis. Intracellular cystine depletion by aminothiols in vitro and in vivo. J. Clin. Invest. 58, 180–189 (1976).
Cherqui, S. Cysteamine therapy: a treatment for cystinosis, not a cure. Kidney Int. 81, 127–129 (2012).
Gahl, W. A., Balog, J. Z. & Kleta, R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann. Internal Med. 147, 242–250 (2007).
Emma, F. et al. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant 29, (Suppl 4) 87–94 (2014).
Schulman, J. D. & Schneider, J. A. Cystinosis and the Fanconi syndrome. Pediatr. Clin. North Amer. 23, 779–793 (1976).
Ivanova, E. A. et al. Cystinosin deficiency causes podocyte damage and loss associated with increased cell motility. Kidney Int. 89, 1037–1048 (2016).
Wilmer, M. J., Christensen, E. I., van den Heuvel, L. P., Monnens, L. A. & Levtchenko, E. N. Urinary protein excretion pattern and renal expression of megalin and cubilin in nephropathic cystinosis. Am. J. Kidney. Dis. 51, 893–903 (2008).
North American Pediatric Renal Trials and Collaborative Studies. NAPRTCS Annual Reports. NAPRTCS Online [online] https://web.emmes.com/study/ped/annlrept/annlrept.html, (2011).
Dufier, J. L., Dhermy, P., Gubler, M. C., Gagnadoux, M. F. & Broyer, M. Ocular changes in long-term evolution of infantile cystinosis. Ophthalm. Paediatr. Genet. 8, 131–137 (1987).
Tsilou, E., Zhou, M., Gahl, W., Sieving, P. C. & Chan, C. C. Ophthalmic manifestations and histopathology of infantile nephropathic cystinosis: report of a case and review of the literature. Survey Ophthalmol. 52, 97–105 (2007).
Gultekingil Keser, A., Topaloglu, R., Bilginer, Y. & Besbas, N. Long-term endocrinologic complications of cystinosis. Minerva Pediatr. 66, 123–130 (2014).
Besouw, M. T., Holewijn, S., Levtchenko, E. N. & Janssen, M. C. Non-invasive measurements of atherosclerosis in adult cystinosis patients. J. Inherited Metabol. Dis. 34, 811–818 (2011).
Ueda, M. et al. Coronary artery and other vascular calcifications in patients with cystinosis after kidney transplantation. Clin. J. Am. Soc. Nephrol. 1, 555–562 (2006).
Dixit, M. P. & Greifer, I. Nephropathic cystinosis associated with cardiomyopathy: a 27-year clinical follow-up. BMC Nephrol. 3, 8 (2002).
Broyer, M., Guillot, M., Gubler, M. C. & Habib, R. Infantile cystinosis: a reappraisal of early and late symptoms. Adv. Nephrol. Necker Hosp. 10, 137–166 (1981).
Klusmann, M., Van' t Hoff, W., Monsell, F. & Offiah, A. C. Progressive destructive bone changes in patients with cystinosis. Skeletal Radiol. http://dx.doi.org/10.1007/s00256-013-1735-z
Bacchetta, J. et al. Skeletal implications and management of cystinosis: three case reports and literature review. Bonekey Rep. 5, 828 (2016).
Ballantyne, A. O. & Trauner, D. A. Neurobehavioral consequences of a genetic metabolic disorder: visual processing deficits in infantile nephropathic cystinosis. Cogn. Behav. Neurol. 13, 254–263 (2000).
Scarvie, K. M., Ballantyne, A. O. & Trauner, D. A. Visuomotor performance in children with infantile nephropathic cystinosis. Percep. Mot. Skills 82, 67–75 (1996).
Trauner, D. A., Chase, C., Scheller, J., Katz, B. & Schneider, J. A. Neurologic and cognitive deficits in children with cystinosis. J. Pediatr. 112, 912–914 (1988).
Trauner, D. A., Spilkin, A. M., Williams, J. & Babchuck, L. Specific cognitive deficits in young children with cystinosis: evidence for an early effect of the cystinosin gene on neural function. J. Pediatr. 151, 192–196 (2007).
Trauner, D. A. et al. Neurological impairment in nephropathic cystinosis: motor coordination deficits. Pediatr. Nephrol. 25, 2061–2066 (2010).
Viltz, L. & Trauner, D. A. Effect of age at treatment on cognitive performance in patients with cystinosis. J. Pediatr. 163, 489–492 (2013).
Anikster, Y. et al. Pulmonary dysfunction in adults with nephropathic cystinosis. Chest 119, 394–401 (2001).
Sonies, B. C., Almajid, P., Kleta, R., Bernardini, I. & Gahl, W. A. Swallowing dysfunction in 101 patients with nephropathic cystinosis: benefit of long-term cysteamine therapy. Medicine 84, 137–146 (2005).
Levy, M. & Feingold, J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int. 58, 925–943 (2000).
Bois, E., Feingold, J., Frenay, P. & Briard, M.-L. Infantile cystinosis in France: genetics, incidence, geographic distribution. J. Med. Genet. 13, 434–438 (1976).
Cochat, P., Cordier, B., Lacôte, C. & Saïd, M.-H. in Cystinosis (ed. Broyer, M.) 28–35 (Elsevier, 1999).
DeBraekeleer, M. Hereditary disorders in Saguenay-Lac-St-Jean (Quebec, Canada). Hum. Hered. 41, 141–146 (1991).
Attard, M. et al. Severity of phenotype in cystinosis varies with mutations in the CTNS gene: predicted effect on the model of cystinosin. Hum. Mol. Genet. 8, 2507–2514 (1999).
Kalatzis, V. et al. Characterization of a putative founder mutation that accounts for the high incidence of cystinosis in Brittany. J. Am. Soc. Nephrol. 12, 2170–2174 (2001).
McGowan-Jordan, J. et al. Molecular Analysis of Cystinosis: Probable Irish Origin of the Most Common French Canadian Mutation. Eur. J. Hum. Genet. 7, 671–678 (1999).
Anikster, Y. et al. Identification and detection of the common 65-kb deletion breakpoint in the nephropathic cystinosis gene (CTNS). Mol. Genet. Metab. 66, 111–116 (1999).
Aldahmesh, M. A. et al. Characterization of CTNS mutations in Arab patients with cystinosis. Ophthalm. Genet. 30, 185–189 (2009).
Owen, E. P. et al. Common mutation causes cystinosis in the majority of black South African patients. Pediatr. Nephrol. 30, 595–601 (2015).
Shahkarami, S., Galehdari, H., Ahmadzadeh, A., Babaahmadi, M. & Pedram, M. The first molecular genetics analysis of individuals suffering from nephropatic cystinosis in the Southwestern Iran. Nefrologia 33, 308–315 (2013).
Soliman, N. A. et al. Mutational Spectrum of the CTNS Gene in Egyptian Patients with Nephropathic Cystinosis. JIMD reports 14, 87–97 (2014).
Aly, R., Makar, S., El Bakri, A. & Soliman, N. A. Neurocognitive functions and behavioral profiles in children with nephropathic cystinosis. Saudi J. Kidney Dis. Transpl. 25, 1224–1231 (2014).
Elmonem, M. A. et al. Lysosomal Storage Disorders in Egyptian Children. Indian J. Pediatr. 8, 805–813 (2016).
Mirdehghan, M., Ahmadzadeh, A., Bana-Behbahani, M., Motlagh, I. & Chomali, B. Infantile cystinosis. Indian Pediatr. 40, 21–24 (2003).
Kir'ianov, N. A., Bazhenov, E. L. & Stetsenko, E. V. [Cystinosis in an adult]. Arkh. Patol. 54, 34–36 (1992).
Tang, S., Danda, S., Zoleikhaeian, M., Simon, M. & Huang, T. An Indian boy with nephropathic cystinosis: a case report and molecular analysis of CTNS mutation. Genet. Test. Mol. Biomarkers 13, 435–438 (2009).
Yang, Y. J. et al. First report of CTNS mutations in a Chinese family with infantile cystinosis. ScientificWorldJournal 2015, 309410 (2015).
The Cystinosis Collaborative Research Group. Linkage of the gene for cystinosis to markers on the short arm of chromosome 17. Nat. Genet. 10, 246–248 (1995).
Taranta, A. et al. Identification and subcellular localization of a new cystinosin isoform. Am. J. Physiol. Renal Physiol. 294, F1101–1108 (2008).
Bellomo, F. et al. Carboxyl-Terminal SSLKG Motif of the Human Cystinosin-LKG Plays an Important Role in Plasma Membrane Sorting. PLoS ONE 11, e0154805 (2016).
Taranta, A. et al. Distribution of cystinosin-LKG in human tissues. Histochem. Cell Biol. 138, 351–363 (2012).
Andrzejewska, Z. et al. Lysosomal Targeting of Cystinosin Requires AP-3. Traffic 16, 712–726 (2015).
Kyttala, A., Ihrke, G., Vesa, J., Schell, M. J. & Luzio, J. P. Two motifs target Batten disease protein CLN3 to lysosomes in transfected nonneuronal and neuronal cells. Mol. Biol. Cell 15, 1313–1323 (2004).
Saudek, V. Cystinosin, MPDU1, SWEETs and KDELR belong to a well-defined protein family with putative function of cargo receptors involved in vesicle trafficking. PLoS ONE 7, e30876 (2012).
Ruivo, R. et al. Mechanism of proton/substrate coupling in the heptahelical lysosomal transporter cystinosin. Proc. Natl Acad. Sci. USA 109, E210–217 (2012).
Lee, Y., Nishizawa, T., Yamashita, K., Ishitani, R. & Nureki, O. Structural basis for the facilitative diffusion mechanism by SemiSWEET transporter. Nat. Commun. 6, 6112 (2015).
Jezegou, A. et al. Heptahelical protein PQLC2 is a lysosomal cationic amino acid exporter underlying the action of cysteamine in cystinosis therapy. Proc. Natl Acad. Sci. USA 109, E3434–3443 (2012).
Thamotharan, M., Lombardo, Y. B., Bawani, S. Z. & Adibi, S. A. An active mechanism for completion of the final stage of protein degradation in the liver, lysosomal transport of dipeptides. J. Biol. Chem. 272, 11786–11790 (1997).
Thoene, J. et al. In vitro correction of disorders of lysosomal transport by microvesicles derived from baculovirus-infected Spodoptera cells. Mol. Genet. Metab. 109, 77–85 (2013).
Leinekugel, P., Michel, S., Conzelmann, E. & Sandhoff, K. Quantitative Correlation between the Residual Activity of Beta-Hexosaminidase-a and Arylsulfatase-a and the Severity of the Resulting Lysosomal Storage Disease. Hum. Genet. 88, 513–523 (1992).
Kalatzis, V., Nevo, N., Cherqui, S., Gasnier, B. & Antignac, C. Molecular pathogenesis of cystinosis: effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Hum. Mol. Genet. 13, 1361–1371 (2004).
Thoene, J. G. & Lemons, R. M. Cystine accumulation in cystinotic fibroblasts from free and protein-linked cystine but not cysteine. Biochem. J. 208, 823–830 (1982).
Thoene, J. G. & Lemons, R. Modulation of the intracellular cystine content of cystinotic fibroblasts by extracellular albumin. Pediatr. Res. 14, 785–787 (1980).
Thoene, J. G., Oshima, R. G., Ritchie, D. G. & Schneider, J. A. Cystinotic fibroblasts accumulate cystine from intracellular protein degradation. Proc. Natl Acad. Sci. USA 74, 4505–4507 (1977).
Amsellem, S. et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J. Am. Soc. Nephrol. 21, 1859–1867 (2010).
Ovunc, B. et al. Exome sequencing reveals cubilin mutation as a single-gene cause of proteinuria. J. Am. Soc. Nephrol. 22, 1815–1820 (2011).
Oude Elferink, R. P., Harms, E., Strijland, A. & Tager, J. M. The intralysosomal pH in cultured human skin fibroblasts in relation to cystine accumulation in patients with cystinosis. Biochem. Biophys. Res. Commun. 116, 154–161 (1983).
Elmonem, M. A. et al. Cystinosis: a review. Orphanet J. Rare Diseases 11, 47 (2016).
Futter, C. E., Pearse, A., Hewlett, L. J. & Hopkins, C. R. Multivesicular endosomes containing internalized EGF-EGF receptor complexes mature and then fuse directly with lysosomes. J. Cell Biol. 132, 1011–1023 (1996).
Montgomery, R. R., Webster, P. & Mellman, I. Accumulation of indigestible substances reduces fusion competence of macrophage lysosomes. J. Immunol. 147, 3087–3095 (1991).
Gaide Chevronnay, H. P. et al. Time course of pathogenic and adaptation mechanisms in cystinotic mouse kidneys. J. Am. Soc. Nephrol. 25, 1256–1269 (2014).
Gubler, M. C., Lacoste, M., Sich, M. & Broyer, M. in Cystinosis Vol. 1st (ed. Broyer, M.) 42–48 (Elsevier, 1999).
Prencipe, G. et al. Inflammasome activation by cystine crystals: implications for the pathogenesis of cystinosis. J. Am. Soc. Nephrol. 25, 1163–1169 (2014).
Baum, M. The Fanconi syndrome of cystinosis: insights into the pathophysiology. Pediatr. Nephrol. 12, 492–497 (1998).
Coor, C., Salmon, R. F., Quigley, R., Marver, D. & Baum, M. Role of adenosine triphosphate (ATP) and NaK ATPase in the inhibition of proximal tubule transport with intracellular cystine loading. J. Clin. Invest. 87, 955–961 (1991).
Foreman, J. W. et al. Metabolic studies of rat renal tubule cells loaded with cystine: the cystine dimethylester model of cystinosis. J. Am. Soc. Nephrol. 6, 269–272 (1995).
Wilmer, M. J., van den Heuvel, L. P. & Levtchenko, E. N. The use of CDME in cystinosis research. Neurochem. Res. 33, 2373–2374 (2008).
Levtchenko, E. N. et al. Decreased intracellular ATP content and intact mitochondrial energy generating capacity in human cystinotic fibroblasts. Pediatr. Res. 59, 287–292 (2006).
Wilmer, M. J. et al. Mitochondrial complex V expression and activity in cystinotic fibroblasts. Pediatr. Res. 64, 495–497 (2008).
Taub, M. L., Springate, J. E. & Cutuli, F. Reduced phosphate transport in the renal proximal tubule cells in cystinosis is due to decreased expression of transporters rather than an energy defect. Biochem. Biophys. Res. Commun. 407, 355–359 (2011).
Taub, M. & Cutuli, F. Activation of AMP kinase plays a role in the increased apoptosis in the renal proximal tubule in cystinosis. Biochem. Biophys. Res. Commun. 426, 516–521 (2012).
Park, M., Helip-Wooley, A. & Thoene, J. Lysosomal cystine storage augments apoptosis in cultured human fibroblasts and renal tubular epithelial cells. J. Am. Soc. Nephrol. 13, 2878–2887 (2002).
Park, M. A., Pejovic, V., Kerisit, K. G., Junius, S. & Thoene, J. G. Increased apoptosis in cystinotic fibroblasts and renal proximal tubule epithelial cells results from cysteinylation of protein kinase Cdelta. J. Am. Soc. Nephrol. 17, 3167–3175 (2006).
Galarreta, C. I. et al. The swan-neck lesion: proximal tubular adaptation to oxidative stress in nephropathic cystinosis. Am. J. Physiol. Renal Physiol. 308, F1155–1166 (2015).
Sansanwal, P., Kambham, N. & Sarwal, M. M. Caspase-4 may play a role in loss of proximal tubules and renal injury in nephropathic cystinosis. Pediatr. Nephrol. 25, 105–109 (2010).
Chol, M., Nevo, N., Cherqui, S., Antignac, C. & Rustin, P. Glutathione precursors replenish decreased glutathione pool in cystinotic cell lines. Biochem. Biophys. Res. Commun. 324, 231–235 (2004).
Levtchenko, E. et al. Altered status of glutathione and its metabolites in cystinotic cells. Nephrol. Dial. Transplant. 20, 1828–1832 (2005).
Wilmer, M. J. et al. Elevated oxidized glutathione in cystinotic proximal tubular epithelial cells. Biochem. Biophys. Res. Commun. 337, 610–614 (2005).
Mannucci, L. et al. Impaired activity of the gamma-glutamyl cycle in nephropathic cystinosis fibroblasts. Pediatr. Res. 59, 332–335 (2006).
Rizzo, C. et al. Pyroglutamic aciduria and nephropathic cystinosis. J. Inherited Metabol. Dis. 22, 224–226 (1999).
Laube, G. F. et al. Glutathione depletion and increased apoptosis rate in human cystinotic proximal tubular cells. Pediatr. Nephrol. 21, 503–509 (2006).
Sakarcan, A., Timmons, C. & Baum, M. Intracellular distribution of cystine in cystine-loaded proximal tubules. Pediatr. Res. 35, 447–450 (1994).
Spear, G. S., Slusser, R. J., Tousimis, A. J., Taylor, C. G. & Schulman, J. D. Cystinosis. An ultrastructural and electron-probe study of the kidney with unusual findings. Arch. Pathol. 91, 206–221 (1971).
Pache de Faria Guimaraes, L. et al. N-Acetyl-cysteine is associated to renal function improvement in patients with nephropathic cystinosis. Pediatr. Nephrol. 29, 1097–1102 (2014).
Moldeus, P., Ormstad, K. & Reed, D. J. Turnover of cellular glutathione in isolated rat-kidney cells. Role of cystine and methionine. Eur. J. Biochem. 116, 13–16 (1981).
Meister, A., Anderson, M. E. & Hwang, O. Intracellular cysteine and glutathione delivery systems. J. Am. College Nutr. 5, 137–151 (1986).
Frey, I. M. et al. Profiling at mRNA, protein, and metabolite levels reveals alterations in renal amino acid handling and glutathione metabolism in kidney tissue of Pept2−/− mice. Physiol. Genom. 28, 301–310 (2007).
Ahmed, K., Dasgupta, P. & Khan, M. S. Cystine calculi: challenging group of stones. Postgrad. Med. J. 82, 799–801 (2006).
Kessler, A. et al. Antioxidant effect of cysteamine in brain cortex of young rats. Neurochem. Res. 33, 737–744 (2008).
Okamura, D. M. et al. Cysteamine modulates oxidative stress and blocks myofibroblast activity in CKD. J. Am. Soc. Nephrol. 25, 43–54 (2014).
Wilmer, M. J. et al. Cysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cells. Biochim. Biophys. Acta 1812, 643–651 (2011).
Raggi, C. et al. Dedifferentiation and aberrations of the endolysosomal compartment characterize the early stage of nephropathic cystinosis. Hum. Mol. Genet. 23, 2266–2278 (2014).
Ivanova, E. A. et al. Endo-lysosomal dysfunction in human proximal tubular epithelial cells deficient for lysosomal cystine transporter cystinosin. PLoS ONE 10, e0120998 (2015).
Christensen, E. I. et al. Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc. Natl Acad. Sci. USA 100, 8472–8477 (2003).
Vicinanza, M. et al. OCRL controls trafficking through early endosomes via PtdIns4,5P(2)-dependent regulation of endosomal actin. EMBO J. 30, 4970–4985 (2011).
Bandyopadhyay, D., Cyphersmith, A., Zapata, J. A., Kim, Y. J. & Payne, C. K. Lysosome transport as a function of lysosome diameter. PLoS ONE 9, e86847 (2014).
Li, X. et al. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 18, 404–417 (2016).
Ivanova, E. A. et al. Ca(2+) signalling in human proximal tubular epithelial cells deficient for cystinosin. Cell Calcium 60, 282–287 (2016).
Johnson, J. L. et al. Upregulation of the Rab27a-dependent trafficking and secretory mechanisms improves lysosomal transport, alleviates endoplasmic reticulum stress, and reduces lysosome overload in cystinosis. Mol. Cell. Biol. 33, 2950–2962 (2013).
Schulman, J. D., Bradley, K. H. & Seegmiller, J. E. Cystine: compartmentalization within lysosomes in cystinotic leukocytes. Science 166, 1152–1154 (1969).
Kooistra, T., Millard, P. C. & Lloyd, J. B. Role of thiols in degradation of proteins by cathepsins. Biochem. J. 204, 471–477 (1982).
Efeyan, A., Zoncu, R. & Sabatini, D. M. Amino acids and mTORC1: from lysosomes to disease. Trends Mol. Med. 18, 524–533 (2012).
Settembre, C., Fraldi, A., Medina, D. L. & Ballabio, A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296 (2013).
Napolitano, G. et al. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol. Med. 2, 158–174 (2015).
Sansanwal, P. et al. Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J. Am. Soc. Nephrol. 21, 272–283 (2010).
Fantus, D., Rogers, N. M., Grahammer, F., Huber, T. B. & Thomson, A. W. Roles of mTOR complexes in the kidney: implications for renal disease and transplantation. Nat. Rev. Nephrol. 12, 587–609 (2016).
Bar-Peled, L., Schweitzer, L. D., Zoncu, R. & Sabatini, D. M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208 (2012).
Sancak, Y. et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 (2010).
Laplante, M. & Sabatini, D. M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 126, 1713–1719 (2013).
Martina, J. A. & Puertollano, R. RRAG GTPases link nutrient availability to gene expression, autophagy and lysosomal biogenesis. Autophagy 9, 928–930 (2013).
Zoncu, R. et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683 (2011).
Andrzejewska, Z. et al. Cystinosin is a Component of the Vacuolar H+-ATPase-Ragulator-Rag Complex Controlling Mammalian Target of Rapamycin Complex 1 Signaling. J. Am. Soc. Nephrol. 27, 1678–1688 (2016).
Ivanova, E. A. et al. Altered mTOR signalling in nephropathic cystinosis. J. Inherited Metabol. Dis. 39, 457–464 (2016).
Gleixner, E. M. et al. V-ATPase/mTOR signaling regulates megalin-mediated apical endocytosis. Cell Rep. 8, 10–19 (2014).
Rega, L. R. et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 89, 862–873 (2016).
Platt, F. M., Boland, B. & van der Spoel, A. C. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J. Cell Biol. 199, 723–734 (2012).
Kaushik, S. & Cuervo, A. M. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 22, 407–417 (2012).
Kiffin, R., Christian, C., Knecht, E. & Cuervo, A. M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 15, 4829–4840 (2004).
Massey, A. C., Kaushik, S., Sovak, G., Kiffin, R. & Cuervo, A. M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl Acad. Sci. USA 103, 5805–5810 (2006).
Balmer, C. et al. Familial X-linked cardiomyopathy (Danon disease): diagnostic confirmation by mutation analysis of the LAMP2gene. Eur. J. Pediatr. 164, 509–514 (2005).
Fanin, M. et al. Generalized lysosome-associated membrane protein-2 defect explains multisystem clinical involvement and allows leukocyte diagnostic screening in Danon disease. Am. J. Pathol. 168, 1309–1320 (2006).
Horvath, J. et al. Identification of a novel LAMP2 mutation responsible for X-chromosomal dominant Danon disease. Neuropediatrics 34, 270–273 (2003).
Lima, W. R. et al. ZONAB promotes proliferation and represses differentiation of proximal tubule epithelial cells. J. Am. Soc. Nephrol. 21, 478–488 (2010).
Janssens, V. et al. in Fifth International Cystinosis Research Symposium (Univ. of California, 2016).
Chevalier, R. L. The proximal tubule in cystinosis: fight or flight? J. Am. Soc. Nephrol. 25, 1131–1132 (2014).
Chevalier, R. L. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am. J. Physiol. Renal Physiol. 311, F145–161 (2016).
Andrzejewska, Z. et al. Cystinosin is a Component of the Vacuolar H+-ATPase-Ragulator-Rag Complex Controlling Mammalian Target of Rapamycin Complex 1 Signaling. J. Am. Soc. Nephrol. 6, 1678–1688 (2015).
Dohil, R. et al. Long-term treatment of cystinosis in children with twice-daily cysteamine. J. Pediatr. 156, 823–827 (2010).
Gangoiti, J. A. et al. Pharmacokinetics of enteric-coated cysteamine bitartrate in healthy adults: a pilot study. Br. J. Clin. Pharmacol. 70, 376–382 (2010).
Dohil, R. & Cabrera, B. L. Treatment of cystinosis with delayed-release cysteamine: 6-year follow-up. Pediatr. Nephrol. 28, 507–510 (2013).
Dohil, R. et al. Twice-daily cysteamine bitartrate therapy for children with cystinosis. J. Pediatr. 156, 71–73 (2010).
Goldenberg, M. M. Pharmaceutical approval update. PT. 38, 323–324 (2013).
Langman, C. B. et al. Quality of life is improved and kidney function preserved in patients with nephropathic cystinosis treated for 2 years with delayed-release cysteamine bitartrate. J. Pediatr. 165, 528–533 (2014).
Langman, C. B. et al. A randomized controlled crossover trial with delayed-release cysteamine bitartrate in nephropathic cystinosis: effectiveness on white blood cell cystine levels and comparison of safety. Clin. J. Am. Soc. Nephrol. 7, 1112–1120 (2012).
Kaiser-Kupfer, M. I. et al. A randomized placebo-controlled trial of cysteamine eye drops in nephropathic cystinosis. Arch. Ophthalmol. 108, 689–693 (1990).
Kumar, A. & Bachhawat, A. K. A futile cycle, formed between two ATP-dependant gamma-glutamyl cycle enzymes, gamma-glutamyl cysteine synthetase and 5-oxoprolinase: the cause of cellular ATP depletion in nephrotic cystinosis? J. Biosci. 35, 21–25 (2010).
Anguiano, J. et al. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 9, 374–382 (2013).
McNeill, A. et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137, 1481–1495 (2014).
Medina, D. L. et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 21, 421–430 (2011).
Sardiello, M. et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009).
Dehay, B. et al. Pathogenic lysosomal depletion in Parkinson's disease. J. Neurosci. 30, 12535–12544 (2010).
Moskot, M. et al. The phytoestrogen genistein modulates lysosomal metabolism and transcription factor EB (TFEB) activation. J. Biol. Chem. 289, 17054–17069 (2014).
Piotrowska, E. et al. Genistein-mediated inhibition of glycosaminoglycan synthesis as a basis for gene expression-targeted isoflavone therapy for mucopolysaccharidoses. Eur. J. Hum. Genet. 14, 846–852 (2006).
Piotrowska, E. et al. Genistin-rich soy isoflavone extract in substrate reduction therapy for Sanfilippo syndrome: An open-label, pilot study in 10 pediatric patients. Curr. Ther. Res. Clin. Exp. 69, 166–179 (2008).
Lee, K. Y., Kim, J. R. & Choi, H. C. Genistein-induced LKB1-AMPK activation inhibits senescence of VSMC through autophagy induction. Vascul. Pharmacol. 81, 75–82 (2016).
Harrison, F. et al. Hematopoietic stem cell gene therapy for the multisystemic lysosomal storage disorder cystinosis. Mol. Ther. 21, 433–444 (2013).
Syres, K. et al. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood 114, 2542–2552 (2009).
Beck, M. Therapy for lysosomal storage disorders. IUBMB Life 62, 33–40 (2010).
Naphade, S. et al. Brief reports: lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells 33, 301–309 (2015).
Iglesias, D. M. et al. Stem cell microvesicles transfer cystinosin to human cystinotic cells and reduce cystine accumulation in vitro. PLoS ONE 7, e42840 (2012).
Onfelt, B. et al. Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J. Immunol. 177, 8476–8483 (2006).
Marzo, L., Gousset, K. & Zurzolo, C. Multifaceted roles of tunneling nanotubes in intercellular communication. Frontiers Physiol. 3, 72 (2012).
Sowinski, S., Alakoskela, J. M., Jolly, C. & Davis, D. M. Optimized methods for imaging membrane nanotubes between T cells and trafficking of HIV-1. Methods 53, 27–33 (2011).
Humphreys, B. D. Kidney injury, stem cells and regeneration. Curr. Opin. Nephrol. Hypertens. 23, 25–31 (2014).
Yeagy, B. A. & Cherqui, S. Kidney repair and stem cells: a complex and controversial process. Pediatr. Nephrol. 26, 1427–1434 (2011).
Abrahamson, D. R. & Leardkamolkarn, V. Development of kidney tubular basement membranes. Kidney Int. 39, 382–393 (1991).
Halfter, W. et al. Protein composition and biomechanical properties of in vivo-derived basement membranes. Cell Adhesion Migr. 7, 64–71 (2013).
Rocca, C. J. et al. Treatment of Inherited Eye Defects by Systemic Hematopoietic Stem Cell Transplantation. Investigative Ophthalmol. Visual Sci. 56, 7214–7223 (2015).
Gaide Chevronnay, H. P. et al. Hematopoietic stem cell transplantation can normalize thyroid function in a cystinosis mouse model. Endocrinology 56, 1363–1371 (2016).
Soos, T. J. et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 70, 591–596 (2006).
Biffi, A. et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341, 1233158 (2013).
Cartier, N. et al. Lentiviral hematopoietic cell gene therapy for X-linked adrenoleukodystrophy. Methods Enzymol. 507, 187–198 (2012).
DiGiusto, D. L. et al. Development of hematopoietic stem cell based gene therapy for HIV-1 infection: considerations for proof of concept studies and translation to standard medical practice. Viruses 5, 2898–2919 (2013).
Drakopoulou, E., Papanikolaou, E., Georgomanoli, M. & Anagnou, N. P. Towards more successful gene therapy clinical trials for beta-thalassemia. Curr. Mol. Med. 13, 1314–1330 (2013).
Zhang, L., Thrasher, A. J. & Gaspar, H. B. Current progress on gene therapy for primary immunodeficiencies. Gene Ther. 20, 963–969 (2013).
Pisoni, R. L., Acker, T. L., Lisowski, K. M., Lemons, R. M. & Thoene, J. G. A cysteine-specific lysosomal transport system provides a major route for the delivery of thiol to human fibroblast lysosomes: possible role in supporting lysosomal proteolysis. J. Cell Biol. 110, 327–335 (1990).
Lloyd, J. B. Disulphide reduction in lysosomes. The role of cysteine. Biochem. J. 237, 271–272 (1986).
Lloyd, J. B. Lysosomal handling of cystine residues: stoichiometry of cysteine involvement. Biochem. J. 286, 979–980 (1992).
Pisoni, R. L., Park, G. Y., Velilla, V. Q. & Thoene, J. G. Detection and characterization of a transport system mediating cysteamine entry into human fibroblast lysosomes. Specificity for aminoethylthiol and aminoethylsulfide derivatives. J. Biol. Chem. 270, 1179–1184 (1995).
Acknowledgements
S.C. and PJ.C. are funded by the Cystinosis Research Foundation, as well as NIH grants RO1-DK090058, R21-NS090066 and grant from the Sanford Stem Cell Clinical Center (to S.C.), and Belgian F.R.S./FNRS (to P.J.C.). We acknowledge Corinne Antignac, Imagine Institute, France, for her helpful comments while assembling this Review, Heloïse Gaide Chevronnay, CELL, de Duve Institute, Belgium, for her pivotal collaboration, and Patrick Van Der Smissen, CELL & PICT, de Duve Institute, Belgium, for kindly providing electron micrographs used in Fig. 2. We are also grateful to numerous colleagues who have provided so many insightful comments and contributive suggestions over the years.
Author information
Authors and Affiliations
Contributions
S.C. and P.J.C. contributed equally to researching data for the article, discussion of the content, and revising or editing the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Glossary
- Swan neck deformities
-
Tubular lesions that appear in microdissected nephrons as long atrophic tubules (the 'swan necks'), appending below the glomeruli (the 'swan heads').
- Founder mutations
-
Mutations that appear in the DNA of one or more individuals who are founders of a distinct population.
- Km
-
Concentration of half-maximal rate. For a transporter, the Km value is inversely related to its affinity for its cargo.
- Half-cystine
-
In biochemical assays, cystine values are reported as half-molecules because cystine was originally measured in reducing conditions (as cysteine), and cysteine is half a molecule of cystine. Cystine is now directly measured as cystine using a mass spectrometer but the values are still reported as half-cystine per mg of protein.
- Lysosomal fusion
-
Fusion of acidified late endosomes carrying endocytic cargo with resting lysosomes bearing concentrated enzymes, or fusion between resting overloaded lysosomes (then also called interlysosomal fusion).
- Lysosomal fission
-
Vesicular budding from endolysosomes (to regenerate late endosomes and resting lysosomes), or division of overloaded resting lysosomes into smaller ones to randomize residual content by interlysosomal fusion.
- Residual body
-
A resting lysosome, filled with undigestible content, that no longer engages in vesicular trafficking.
- Lysosomal defecation
-
Active exocytosis of overloaded lysosomes. This lysosomal clearing activity is stimulated by transcription factor EB.
- Lysosomal amino acid sensors
-
Component or interacting partner of the mTOR-complex machinery that informs mTOR kinase on starvation or amino acid abundance.
- Low molecular weight proteinuria
-
A commonly used term to describe tubular proteinuria due to defective proximal tubular endocytosis of plasma proteins selectively ultrafiltrated according to their size and/or sieving coefficient (thus excluding IgGs and other high molecular weight (MW) proteins), as opposed to non-selective glomerular leak. This terminology is convenient, but might be misleading since pure tubular proteinuria typically includes relatively high MW proteins such as transferrin (80 kDa) and even larger globular proteins.
- Cross-correction
-
Transfer of functional protein from normal cells to deficient cells.
- Pathogen spreading
-
Transmission of bacteria, virus or parasites from one cell to another. Bacteria and virus such as HIV have been shown to colonize cells using the tunnelling nanotube route.
Rights and permissions
About this article

Cite this article
Cherqui, S., Courtoy, P. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nat Rev Nephrol 13, 115–131 (2017). https://doi.org/10.1038/nrneph.2016.182
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrneph.2016.182
- Springer Nature Limited
This article is cited by
-
Disulfidptosis decoded: a journey through cell death mysteries, regulatory networks, disease paradigms and future directions
Biomarker Research (2024)
-
Addressing the psychosocial aspects of transition to adult care in patients with cystinosis
Pediatric Nephrology (2024)
-
Impact of compliance to oral cysteamine treatment on the costs of Kidney failure in patients with nephropathic cystinosis in the United Kingdom
BMC Nephrology (2023)
-
Evaluation of the efficacy of cystinosin supplementation through CTNS mRNA delivery in experimental models for cystinosis
Scientific Reports (2023)
-
Lysosomal cystine export regulates mTORC1 signaling to guide kidney epithelial cell fate specialization
Nature Communications (2023)