
Abstract
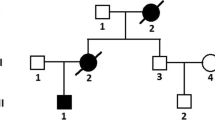
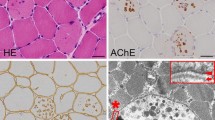
A boy presented at age 2.5 years with mild left ventricular hypertrophy and mild myopathy. Hypertrophic cardiomyopathy progressed relentlessly, leading to death at age 16 years shortly before planned heart transplantation. During the course of the disease, his mother developed severe dilated cardiomyopathy and died of its complications at 46 years of age. The combination of myopathy and cardiomyopathy, the biochemical and electron microscopy findings in a muscle biopsy, and the pedigree suggested Danon disease (MIM 300257), an X-linked lysosomal storage disorder caused by deficiency of lysosome-associated membrane protein-2 (LAMP2). The diagnosis was confirmed by the identification of a novel mutation, G138A, in the LAMP2gene, leading to the premature stop codon W46X. Conclusion:Early diagnosis of Danon disease is important for genetic counselling and timely cardiac transplantation, the only effective therapeutic option.





Similar content being viewed by others

Abbreviations
- LAMP2 :
-
lysosome-associated membrane protein-2
- SSCP :
-
single strand conformational polymorphism
References
Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB, Walter M, Li GH, Burgon PG, Maguire CT, Stapleton D, Schmitt JP, Guo XX, Pizard A, Kupershmidt S, Roden DM, Berul CI, Seidman CE, Sedman JG (2003) Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 107: 2850–2856
Baum H, Dodgson KS, Spencer B (1959) The assay of arylsulphatases A and B in human urine. Clin Chim Acta 4: 453–455
Bergia B, Sybers HD, Butler IJ (1986) Familial lethal cardiomyopathy with mental retardation and scapuloperoneal muscle dystrophy. J Neurol Neurosurg Psychiatry 49: 1423–1426
Beutler E (1975) Red cell metabolism. Grune and Stratton, New York, pp 42–45
Beutler E, Kuhl W, Matsumoto F, Pangalis G (1976) Acid hydrolases in leukocytes and platelets of normal subjects and in patients with Gaucher’s and Fabry’s disease. J Exp Med 143: 975–980
Bru P, Pellissier JF, Gatau-Pelanchon J, Faugère G, de Barsy T, Levy S, Gérard R (1988) Glycogenose lysosomiale cardio-musculaire de l’adulte sans deficit enzymatique connu. Arch Mal Coeur Vaiss 81: 109–114
Byrne E, Dennett X, Crotty B, Trounce I, Sands JM, Hawkins R, Hammond J, Anderson S, Haan EA, Pollard A (1986) Dominantly inherited cardioskeletal myopathy with lysosomal glycogen storage and normal acid maltase levels. Brain 109: 523–536
Danon MJ, Oh SJ, DiMauro S, Manaligod JR, Eastwood A, Naidu S, Schliselfeld LH (1981) Lysosomal glycogen storage disease with normal acid maltase. Neurology 31: 51–57
DeBakey ME, Bernowski R (1998) The DeBakey/NASA axial flow ventricular assist device. In: Akutsu T, Koyanagi H (eds) Heart replacement and artificial heart. Springer, Tokyo, pp 407–413
Dworzak F, Casazza F, Mora M, De Maria R, Gronda E, Baroldi G, Rimoldi M, Morandi L, Cornelio F (1994) Lysosomal glycogen storage with normal acid maltase: a familial study with successful heart transplant. Neuromuscul Disord 4: 243–247
Fukuda M (1994) Biogenesis of the lysosomal membrane. Subcell Biochem 22: 199–230
Galjaard H (1980) Genetic metabolic diseases. Early diagnosis and prenatal analysis. Elsevier, Amsterdam, pp 812–828
Hart ZH, Servidei S, Peterson PL, Chang CH, DiMauro S (1987) Cardiomyopathy, mental retardation and autophagic vacuolar myopathy. Neurology 37: 1065–1068
Hers HG (1964) Glycogen storage disease. In: Levine R, Luft R (eds) Advances in metabolic disorders, vol. I. Academic Press, New York London, pp 1–44
Horvath J, Ketelsen UP, Geibel-Zehender A, Boehm N, Olbrich H, Korinthenberg R, Omran H (2003) Identification of a novel LAMP2 mutation responsible for X-chromosomal dominant Danon disease. Neuropediatrics 34: 270–273
Huijing F (1974) Glycogen and enzymes of glycogen metabolism. In: Curtius HC, Roth M (eds) Clinical biochemistry, vol. II. W. de Gruyter, Berlin New York, pp 1208–1235
Krisman CR (1962) A method for the colorimetric estimation of glycogen with iodine. Anal Biochem 4: 17–23
Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, Sue CM, Yamamoto A, Murakami N, Shanske S, Byrne E, Bonilla E, Nonaka I, DiMauro S, Hirano M (2000) Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406: 906–910
Saftig P, Tanaka Y, Lüllmann-Rauch R, von Figura K (2001) Disease model: LAMP-2 enlightens Danon disease. Trends Mol Med 7: 37–39
Sugie K, Yamamoto A, Murayama K, Oh SJ, Takahashi M, Mora M, Riggs JE, Colomer J, Iturriaga C, Meloni A, Lamperti C, Saitoh S, Byrne E, DiMauro S, Nonaka I, Hirano M, Nishino I (2002) Clinicopathological features of genetically confirmed Danon disease. Neurology 58: 1773–1778
Tachi N, Tachi M, Sasaki K, Tomita H, Wakai S, Annaka S, Minami R, Tsurui S, Sugie H (1989) Glycogen storage disease with normal acid maltase: skeletal and cardiac muscles. Pediatr Neurol 5: 60–63
Takahashi M, Yamamoto A, Takano K, Sudo A, Wada T, Goto Y, Nishino I, Saitoh S (2002) Germline mosaicism of a novel mutation in lysosome-associated membrane protein-2 deficiency (Danon disease). Ann Neurol 52: 122–125
Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P (2000) Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406: 902–906
Tse HF, Shek TW, Tai YT, Lau YK, Ma L (1996) Case report: lysosomal glycogen storage disease with normal acid maltase: an unusual form of hypertrophic cardiomyopathy with rapidly progressive heart failure. Am J Med Sci 312: 182–186
Usuki F, Takenega S, Higuchi I, Kashio N, Nakagawa M, Osame M (1994) Morphologic findings in biopsied skeletal muscle and cultured fibroblasts from a female patient with Danon’s disease (lysosomal glycogen storage disease without acid maltase deficiency). J Neurol Sci 127: 54–60
Verloes A, Massin M, Lombet J, Grattagliano B, Soyeur D, Rigo J, Koulischer L, Van Hoof F (1997) Nosology of lysosomal glycogen storage diseases without in vitro acid maltase deficiency. Delineation of a neonatal form. Am J Med Genet 72: 135–142
Acknowledgement
We thank Dr. Max A. Spycher, Department of Pathology, Faculty of Medicine, University of Zurich, for his excellent electron microscopic evaluation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Balmer, C., Ballhausen, D., Bosshard, N.U. et al. Familial X-linked cardiomyopathy (Danon disease): diagnostic confirmation by mutation analysis of the LAMP2gene. Eur J Pediatr 164, 509–514 (2005). https://doi.org/10.1007/s00431-005-1678-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-005-1678-z