
Abstract
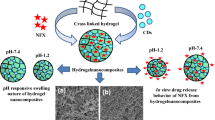
In this research, hydrogel biocomposites were prepared from whey protein isolate (WPI), reduced graphene oxide (rGO), and synthetic polymers in varied ratios. Their physicochemical properties were evaluated by FTIR, SEM, TGA, AFM, and TEM. FTIR spectra revealed significant peaks at 1167 cm−1 for C–O–C peak and at 1449 cm−1 for O–H bending for WPI and rGO, respectively. The hydrogels were loaded with proguanil hydrochloride and chloroquine diphosphate and in vitro release kinetics of individual drugs from the biocomposites were studied. The SEM images of the biocomposites after drug release confirmed that they are biodegradable. The drug release was controlled, pH-dependent which further confirmed that the hydrogels are pH-sensitive. The release of proguanil from the hydrogels was slow when compared to chloroquine, suggesting that the solubility of the drug influenced their rate of release. The drug release from the biocomposites fitted the Korsmeyer–Peppas model with n values for chloroquine between 0.46 and 0.49 at pH of 1.2 and between 0.72 and 1.41 at pH of 7.4. The n values for proguanil were between 0.66 and 0.83 at pH 1.2 and 0.85–0.92 at pH 7.4. The results obtained suggested that the biocomposites are potential systems that can be tailored for controlled delivery of bioactive agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hydrogels are referred to as smart systems with crosslinked polymeric network and swell in aqueous solution [1]. Some of them are biodegradable, non-toxic, and biocompatible, and exhibit porous network that can be tailored to control the rate of drug release [2]. The aforementioned features are dependent on the type of monomers employed in their preparation [2]. Hydrogels are used for targeted, controlled, and sustained drug delivery of bioactive agents such as anticancer, antimalaria, antibacterial drugs, etc., resulting in enhanced therapeutic effects when compared to the conventional administration of these drugs [3, 4].
Malaria is a chronic and serious disease that is prevalent in sub-Saharan Africa. In 2015, 450,000 malaria deaths worldwide were reported with over 92% occurring in Africa [5]. The currently used antimalarials suffer drug resistance, non-specificity, toxicity, poor bioavailability, and water solubility [6]. Proguanil hydrochloride (PG) is a synthetic derivative of pyrimidine and a biguanide organic compound. It is a prophylactic antimalarial [7], and overtime, it has been used in combination with other malaria drugs for many years [8]. It inhibits plasmodial dihydrofolate reductase (DHFR) via its active metabolite cycloguanil by its binding to the DHFR enzyme and inhibits the metabolism of folic acid [8]. Chloroquine diphosphate (CQ) is an aminoquinoline derivative used as a frontline treatment of malaria from the early 1950s [9] and it is believed to act by interfering with the detoxification of the byproducts of hemoglobin digestion taking place in the erythrocytes cell state of the plasmodium asexual life cycle [10, 11]. Despite the application of chloroquine and proguanil for the treatment of malaria, their application has been hampered by drug resistance.
Presently, there are few research reports on the design of hydrogels for the delivery of antimalarials. In one of our reports, soy protein-based hydrogels were prepared for dual delivery of antimalarials. The release profiles of both drugs were controlled, whereby both drugs can work over a different period of time [12]. Mavondo et al. developed asiatic and chloroquine-loaded pectin-based hydrogels for transdermal delivery in Plasmodium berghei-infected Sprague–Dawley rats with good therapeutic efficacy [13]. Simina et al. synthesized and characterized chitosan-based hydrogels for the delivery of quinine. Quinine formed temporary chelates in the hydrogels [14]. Dandekar et al. prepared hydrogel nanoparticles for loading of curcumin from a combination of hydroxyl propyl methyl cellulose and polyvinyl pyrrolidone. The hydrophilic nature of the formulation was exploited for enhanced absorption and prolonged rapid clearance of curcumin. In vivo antimalaria studies revealed significant antimalaria activity of the drug loaded hydrogel nanoparticles when compared to the free curcumin, suggesting that the formulation is a potential adjunct antimalarial therapy that can be used along with standard therapy [15].
In an earlier research report, we investigated the release mechanism of bioactive agent from reduced graphene oxide-based hydrogel biocomposites [16], as well; other researchers have utilized reduced graphene oxide-based biocomposites for other applications [17, 18]. In this manuscript, the focus is on the in vitro release study of antimalarials from the hydrogels with modified ratio of whey protein isolate and reduced graphene oxide. The hydrogels were characterized by FTIR, SEM, TGA, AFM, and TEM. In vitro drug release studies were performed at pH 1.2 and 7.4 simulating gastric juice and blood pH, respectively.
Experimental
Reagents
All chemicals used in this project were of analytical grades. Acrylamide (Am) was supplied by Sigma-Aldrich (Darmstadt, Germany). rGO was donated by Jayaramudu research group. Potassium persulfate (KPS), N,N′-methylenebisacrylamide (MBA), and the activator, N,N,N′,N′-tetramethylethylenediamine (TEMED) were supplied by Sigma-Aldrich (Darmstadt, Germany). Distilled water was used; the reagents and solvents used were purified by known laboratory procedures. Proguanil hydrochloride (PG) and chloroquine diphosphate (CQ) salt were purchased from Sigma-Aldrich, South Africa. The natural polymer, whey protein isolate (WPI), was supplied by Honeyville Food Products, Salt Lake City (Utah, USA). WPI contained 90% protein, 4% fat, 5% ash, and 1% of minor constituents.
Preparation of the thermally rGO-based hydrogel biocomposites
The hydrogel biocomposites were prepared by a modified method [16]. Table 1 is a summary of the composition of the hydrogels. The hydrogel biocomposites (GW2 and GW3) were prepared by dissolving WPI in sodium hydroxide solution, followed by the addition of acrylamide and MBA solution. The mixture was thoroughly stirred and a homogenous mixture was obtained before the addition of TEMED, rGO, and KPS, respectively (Table 1). The hydrogels were formed at a temperature between 30 and 50 °C. The hydrogel biocomposites were then soaked in distilled water overnight to get rid of unreacted amine, before drying at ambient temperature for 5 days.
GW1 and GW4 were prepared by similar procedure as shown above for GW2 and GW3. However, in these cases, the acrylamide was dissolved in MBA solution followed by the addition of TEMED and KPS for GW1 and addition of TEMED, rGO, and KPS for GW4, respectively. GW1 and GW4 were formed at a temperature between 30 and 50 °C and then soaked in distilled water overnight, before drying at ambient temperature for 5 days.
Drug loading of the hydrogels
1 mg of PG was dissolved in 40 mL of distilled water and 60 mg of the hydrogels were soaked separately in 10 mL of the drug solution overnight at ambient temperature. The hydrogel loaded with CQ was performed by dissolving 10 mg of CQ in 40 mL of distilled water. 60 mg of the hydrogels were soaked separately in 10 mL of the drug solution overnight at ambient temperature. The hydrogels were then removed from the drug solution, rinsed with distilled water to get rid of any excess drug present on the surface of the hydrogels, and allowed to dry at ambient temperature for 5 days. The percentage encapsulation efficiency of CQ and PG onto the hydrogels was determined spectrophotometrically. The remaining amount of the drug remaining in the solution after the hydrogel loading was assayed with UV spectrophotometer at wavelengths of 343 and 254 nm, respectively, for CQ and PG. The percentage encapsulation efficiency was calculated using Eq. (1) and amount of drug was calculated using Eq. (2):
The amount of drug loaded onto the hydrogels in mg:
where Wx is the dried hydrogel after loading with drug and Wy is the dry hydrogel before drug loading.
Physiochemical characterization of hydrogels
FTIR
FTIR spectroscopy was performed on (Perkin Elmer Spectrum 100 FTIR spectrometer, USA) in the range of 4000–500 cm−1. The sample was placed on the diamond sample surface of the machine. The number of scans used was 32 at a resolution of 4 cm−1. A scan of the background was taken before the sample was analyzed to act as a control. FTIR was used to evaluate the types of functional groups present in the hydrogel networks and the composition of the hydrogels, and to confirm the successful loading of the drugs to the hydrogel network.
SEM
To compare the surface morphology of the hydrogels, SEM analysis was performed at an accelerating voltage of 15 kV using (JEOL-JSM 7500F, Japan). The hydrogels were dried at room temperature and then sputtered with gold before SEM analysis.
TEM
The hydrogels were dispersed in deionized water and a drop of the solution was deposited onto copper grids and allowed to dry on a filter paper at room temperature for 15 min prior to TEM analysis. TEM analysis was performed to determine the morphology and particle size of the hydrogels using (JEM-1200EX, JEOL, Japan).
AFM
The surface properties of the hydrogels were evaluated using Atomic Force Spectroscopy (AFM) Digital Instruments Nanoscope, Veeco, MMAFMLN-AM (Multimode) instrument, USA. Flat samples of the gels were placed on the convex holder resting on the inner surface, underneath the microscope. The samples were scanned over lengths of 5 µm to give a surface area of 25 µm2. The experiment was performed at room temperature in a tapping mode using a probe designated as RTESPAW-300 (model). The scan rate was set to 0.50 Hz, the amplitude setpoint ranged from 1.27 to 1.440 V, and probe frequencies ranged from 280 to 310 kHz for all the analysis. Image processing such as roughness analysis, topography histogram images, surface skewness, average height, and surface kurtosis were done using the software package WSxM version 5.0.
XRD
X-ray diffraction was used to study the internal organization and crystalline nature of the hydrogels. It was performed on dried and finely grounded samples using PAnalytical X’Pert PRO diffractometer, The Netherlands (Cu Kα radiation, with a wavelength, λ = 0.1546 nm) running at 40 kV and 40 mA. The gels were packed into a hole of between 1 and 2 mm in diameter, in a piece of metal of ~ 1.5 mm thick. The metal piece was then mounted on a specimen holder, so that the X-ray beam can pass through.
TGA
This was performed on TA TGA Q500 (TA Instruments), USA, using between 5 and 10 mg of the hydrogel. TGA was used to evaluate the thermal stability of the hydrogel biocomposites. The analysis was performed at a heating rate of 10 °C/min under air and ramped up to 900 °C. The plot of the weight loss against temperature was achieved from the analysis.
Water uptake mechanism of the hydrogels
60 mg of the dry hydrogels were placed in 25 mL of buffer solutions (1.2 and 7.4) at ambient temperature. The water uptake of the hydrogels was evaluated by allowing the hydrogels to swell in selected buffer solutions until the equilibrium swelling was reached over a period of 24 h. They were then removed and blotted gently with blotting paper to remove the overloaded water on the surface and weighed. The swelling ratio at equilibrium (ER) was calculated from Eq. (3):
where Ws is the weight of the hydrogel at equilibrium and W t is the weight of the hydrogel before swelling. The swelling ratio (SR) of the hydrogel was evaluated by immersing dry hydrogel (50 mg) in (25 mL) of selected buffer solution (pH 7.4 and 10) at ambient temperature. After an interval of 30 min, the hydrogels were removed from the buffer solutions and blotted gently with blotting paper and weighed. SR was calculated from Eq. 4:
where M s is the weight of the hydrogel at time t and Md is the weight of the dried hydrogel before swelling.
The release kinetics of the drugs from the hydrogels was evaluated using Korsmeyer–Peppas model (Eq. 4), where Mt/M∞ = fraction of drug released, t = time of release, K = constant incorporating the structural geometric characteristics of the hydrogel, and n = release exponent. The plot of Log (Mt/M∞) against Log t was achieved with the experimental data according to Eq. 5.
The water uptake kinetics of the hydrogels were evaluated using Eq. 5:
where Mt and M∞ are the masses of the hydrogel at time t and equilibrium, respectively. K is the diffusion constant of water into the hydrogel matrix, and n is the diffusion exponent. n = 0.5 indicates case I, which is a perfect Fickian process, whereby the rate of network relaxation is faster than the rate of diffusion. When n = 1.0, it indicates a non-Fickian diffusion, whereby water transport is controlled and the rate of diffusion is faster than the network relaxation. When 0.5 < n < 1.0, it indicates that the rate of penetrant mobility and segmental relaxation is comparable.
The hydrogels diffusion coefficients were also calculated using Eq. 6:
where D, r, S, and t represent the diffusion coefficient, radius, fractional swelling of the hydrogels, and time, respectively. To investigate the diffusion coefficient of the hydrogels, a graph of S versus t1/2 were drawn and the diffusion coefficients were obtained from the slopes of these graphs.
In vitro drug release studies
The 60 mg hydrogels loaded with drug were placed in 50 mL of selected buffer solutions (pH 1.2 and pH 7.4). A shaker, BS-06 (Lab Companion) was used for the study at 100 rpm and at a temperature of 37 °C. The release profiles of CQ and PG from the hydrogels were obtained using UV–visible spectroscopy at wavelengths of 343 and 254 nm, respectively. The release study was performed over a period of 48 h by collecting 4 mL of the sample and replacing it with an equivalent amount of buffer solution. The cumulative drug release was calculated using Eq. 7:
where Io is the amount of drug release at time t and IF is the amount of drug loaded to the hydrogel.
Drug release kinetics were performed using different mathematical modeling drug release equations such as zero-order Eq. 8, Korsmeyer–Peppas Eq. 9, and Higuchi Eq. 10:
where Qt is the cumulative amount of drug release, Qo is the initial amount of drug, Ko is the zero-order release constant, and t is the time:
where Mt/M is the cumulative drug release, K is the release constant, t is the time, and n is the release exponent.
Q is the cumulative drug release, K is Higuchi release constant, and t is the time.
Results
FTIR
The FTIR spectrum for GW1 hydrogel exhibited absorption peaks for OH stretching at 3317 cm−1, NH stretching of amide at 3182 cm−1, C–H stretching at 2931 cm−1, C=O stretching of amide at 1648 cm−1, and CH2 bending at 1450 cm−1 (Fig. 1a). The FTIR spectrum of GW2 showed characteristic peaks for C–H stretch at 2944 cm−1 and C=C aromatic stretch at 1662 cm−1 and NH stretch of amide at 3175 cm−1 (Fig. 1a). The FTIR spectrum for GW3 exhibited absorption peaks for C-H sp3 stretch at 2938 cm−1 and C=C aromatic stretch at 1658 cm−1, OH stretch at 3345 cm−1, and NH stretch of amide at 3195 cm−1 (Fig. 1a). The FTIR spectrum for GW4 displayed absorption peaks for OH stretching at 3317 cm−1, CH stretching at 2927 cm−1, aand C=C stretching at 1653 cm−1, and OH bending of carboxylic acid from rGO was visible at 1445 and 1416 cm−1, respectively (Fig. 1a).

a FTIR spectra of GW1, GW2, GW3, and GW4. b FTIR spectra of GW3 loaded with CQ, CQ, acrylamide and whey protein isolate. c FTIR spectra of GW3 loaded with PG, PG, acrylamide, and whey protein isolate
CQ displayed N–H stretch at 3300 cm−1, C–N stretch at 1211 cm−1, C=C aromatic stretch at 1612 cm−1, and C–Cl peaks between 600 and 800 cm−1 (Fig. 1b). PG showed N–H stretch at 3292 cm−1, C=C stretch at 1532 cm−1, and C–Cl peaks between 600 and 800 cm−1 (Fig. 1c).
Hydrogels GW3PG and GW3CQ did not exhibit characteristic peaks for the loaded drug, confirming physical entrapment of CQ and PG into the hydrogel (Fig. 1b, c). Acrylamide FTIR spectrum revealed characteristic peaks at 3343 and 3156 cm−1 for N–H stretch for primary amide, at 1668 cm−1 for C=O stretch, and at 1610 cm−1 for C=C bending for alkene (Fig. 1b, c). The FTIR spectrum of WPI displayed characteristic peaks at 3279 cm−1 for N–H stretch, at 1645 cm−1 for C=O stretch, at 1537 cm−1 for N–H amide bending, and at 1226 cm−1 for C–O stretch (Fig. 1b, c). The FTIR spectrum of rGO showed peaks at 1727, 1550, and 1049 cm−1 for C=O, C=C, and C–O stretching, respectively.
SEM
Figure 2a–d depicts the SEM images of the hydrogels without the drugs. GW1 and GW2 displayed irregular and coarse morphologies (Fig. 2a, b). GW3 and GW4 exhibited distinguished rod-like morphology which can be attributed to the presence of rGO (Fig. 2c, d). GW3 loaded with PG exhibited irregular morphology (Fig. 2e, f). GW3 loaded with CQ exhibited rod-like morphology (Fig. 2g, h). GW3 loaded with CQ did not display rod-shaped morphology after drug release at pH 1.2 over a period of 48 h (Fig. 3a). GW3 loaded with CQ still displayed rod-shaped morphology after drug release at pH 7.4 over a period of 48 h (Fig. 3b).

a SEM image of GW1. b SEM image of GW2. c SEM image of GW3. d SEM image of GW4. e, f SEM images of GW2 hydrogel loaded with PG. g, h SEM images of GW3 hydrogel loaded with CQ

a SEM image of GW3 hydrogel after drug release at pH 1.2. b, c SEM images of GW3 hydrogel after drug release at pH 7.4
TEM and AFM
TEM images of GW3 are shown in Fig. 4a, b. AFM images of GW3CQ loaded with CQ and GW3PG loaded with PG are shown in Fig. 5a, b. The degree of roughness was reduced in the GW3 loaded with PG when compared to GW3 loaded with CQ. The WSxM software analysis of GW3 loaded with either PG or CQ are depicted in Table 2.

a, b TEM images of GW3

a AFM image of GW3 loaded with CQ. b AFM image of GW3 loaded with PG
XRD
XRD analysis was used to study the changes in the morphological structure of GW3, GW3 loaded with PG and GW3 loaded with CQ. GW3 loaded with either PG or CQ displayed broad peaks between Ɵ = 17o and 34o. (Fig. 6a) GW3 hydrogel showed significant peaks at Ɵ = 38o, 43o, and 49.7o.

a XRD diffractograms of GW3 before and after loading with drugs. b TGA thermograms of GW3 before and after loading with drugs
TGA
The thermal stability of GW3 after loading with the drugs and the free drugs were evaluated. The TGA thermograms of CQ exhibited two steps of weight loss (Fig. 6b). The first weight loss of 40% was between 200 and 380 °C, and the second weight loss was visible and rapid between 450 and 600 °C. GW3PG hydrogel loaded with PG exhibited the first weight loss of 12% between 180 and 240 °C, a second weight loss of 40% between 250 and 380 °C, a third weight loss of 12% was visible 390–440 °C, and a final weight loss of 28% was visible between 450 and 600 °C (Fig. 6b). GW3CQ hydrogel loaded with CQ exhibited the first weight loss of 22% between 210 and 320 °C, a second weight loss of 12% was visible between 250 and 400 °C, and the final weight loss of 28% was between 410 and 620 °C (Fig. 6b).
Water uptake mechanism of the hydrogels
The hydrogels were pH-sensitive with a significant degree of swelling at pH 7.4. The swelling capability of the hydrogel biocomposites was enhanced at pH 7.4 and at a temperature of 37 °C. GW1 and GW4 exhibited slow degree of swelling at 24 °C (Fig. 7a). GW4 containing only rGO exhibited low swelling ability at pH 7.4. At pH 1.2 and temperature of 24 °C, the hydrogels exhibited reduced degree of swelling (Fig. 7b). The swelling capacity of the hydrogels was enhanced at temperature of 37 °C when compared to 24 °C at pH 1.2 (Table 3). The swelling exponents n for the hydrogels were determined from the slope of the graph of ln (Mt/M∞) versus ln t for 60% swelling ratio. The n values were in the range of 0.38–1.10 with a coefficient of determination of 0.97–0.99 (Table 3). At pH 1.2 and temperature of 24 °C, the n values for GW2 and GW3 were 0.38 and 0.56, respectively. The n values for GW1 and GW4 were 0.64 and 0.81, respectively. At pH 7.4 and temperature of 24 °C, the n values for GW1–4 was 0.64, 0.86, 1.1, and 0.41, respectively (Table 3). The coefficient of determination was in a range of 0.99–0.97, indicating good linearity. At pH 1.2 and temperature of 24 °C, the n values for GW2 and GW3 were 0.38 and 0.56, respectively. GW1 and GW4 n values were 0.64 and 0.81. At pH 7.4 and temperature of 24 °C, the n values for GW1–4 were 0.64, 0.86, 1.1, and 0.41, respectively. The coefficient of determination was in the range of 0.99–0.97, indicating good linearity. At pH 1.2 and temperature of 37 °C, the n values for GW1, GW2, GW3, and GW4 were 0.50, 0.52, 0.49, and 0.52, respectively. At pH 7.4 and temperature of 37 °C, the n values for GW1–4 were 0.34, 0.48, 0.44, and 0.22, respectively (Table 3). The coefficient of determination was in the range of 0.99–0.97 indicating good linearity.

a Graph of water uptake capacity of the hydrogels at pH 7.4 (numbers 24 and 37 represent 24 and 37 °C, respectively). b Graph of water uptake capacity of the hydrogels at pH 1.2 (numbers 24 and 37 represent 24 and 37 °C, respectively). c Drug release profiles of CQ from GW1 to GW4 at pH 1.2. d Drug release profiles of CQ from GW1 to GW4 at pH 7.4. e Drug release profiles of PG from GW1 to GW4 at pH 1.2. f Drug release profiles of PG from GW1 to GW4 at pH 7.4
In vitro drug release
In vitro cumulative drug release of CQ and PG from GW1PG to GW4PG and GW1CQ–GW4CQ at pH 1.2 and 7.4 are depicted in Fig. 7c–f. The percentage encapsulation efficiency of proguanil hydrochloride onto GW1PG–GW4PG hydrogels was 88, 73, 72, and 90%, which relates to 0.88, 0.73, 0.72, and 0.9 mg, respectively. The percentage encapsulation efficiency of CQ onto GW1CQ–GW4CQ hydrogel was 53, 60, 60, and 52%, which relates to 5.3, 6, 6, and 5.2 mg, respectively. The release studies were performed over a period of 48 h and the amount of drug release at pH 7.4 was high. At pH 7.4: 53.5, 90.5, 95.9, and 50.7% of CQ were released over a period of 48 h from GW1CQ, GW2CQ, GW3CQ, and GW4CQ, respectively (Fig. 7d). At pH 1.2: 38.5, 17.8, 35.5, and 45% of PG were released over the same period from GW1PG, GW2PG, GW3PG, and GW4PG, respectively (Fig. 7c). At pH 1.2: 30, 17.6, 31, and 36% of CQ were released over the same period from GW1CQ, GW2CQ, GW3CQ, and GW4CQ, respectively (Fig. 7e). At pH 7.4: 46, 60.6, 69.5, and 40% of PG were released, respectively, over a period of 48 h from GW1PG, GW2PG, GW3PG and GW4PG, respectively (Fig. 7f). The release behaviours of the hydrogels were in agreement with their water sorption capacity. Using Korsmeyer–Peppas release equation, the release exponent n was between 0.38 and 0.49 for CQ at pH 1.2 and between 0.72 and 1.41 at pH 7.4 (Table 4). The release exponent was between 0.66 and 0.83 for PG at pH 1.2 and between 0.85 and 0.96 at pH 7.4 (Table 5). The coefficient of determination was between 0.98 and 0.99 signifying good linearity. The n values suggested that the release mechanism for CQ was pseudo-Fickian at pH 1.2 and super case II at pH 7.4. The n values suggested that the release mechanism for PG was anomalous at pH 1.2 and 7.4. Using zero-order release equation, the release of the drugs from the hydrogels did not follow zero-order release profile (Tables 4, 5).
Discussion
The percentage encapsulation of both drugs in the hydrogel network was high and is attributed to the water solubility of drugs. CQ water solubility was 50 mg/mL and PG water solubility was 156 mg/L. Similar results have been reported by other researchers in which percentage loading of hydrophilic drugs onto hydrogel network was enhanced [19,20,21]. The percentage encapsulation efficiency of PG for GW1PG–GW4PG hydrogels was 88, 73, 72, and 90%, respectively, while the percentage encapsulation efficiency of CQ onto GW1CQ–GW4CQ hydrogel was 53, 60, 60, and 52%, respectively. The FTIR spectrum (Fig. 1a) for GW1 hydrogel revealed characteristic peaks for OH stretching at 3317 cm−1, NH stretching of amide at 3182 cm−1, and C=O stretching of amide at 1648 cm−1, indicating the successful crosslinking of the acrylamide used for the preparation of the hydrogels [16]. The FTIR spectrum for GW2 and GW3 containing WPI showed C–O–C glycosidic absorption peak at 1167 cm−1 and C=O stretch for rGO at 1738 cm, confirming crosslinking of the WPI and rGO in the hydrogel network (Fig. 1a). The C=O stretch for rGO further confirmed the interaction of the rGO with the polymer in the hydrogel network. Some researchers reported similar findings for hydrogels containing rGO [16, 17]. The absence of the NH2 stretching which was conspicuous in pure acrylamide in the GW1–GW4 hydrogel further confirmed the successful crosslinking of acrylamide (Fig. 1a). Hydrogels GW3 loaded with either PG or CQ did not exhibit peaks for the drugs, confirming physical entrapment of CQ and PG into the hydrogel network (Fig. 1b, c). Apart from the FTIR, the hydrogels were also characterized by SEM and TEM analysis.
The surface morphology of GW3 and GW4 was rod-like, which is attributed to the interaction of rGO with the polymer in the hydrogel network (Fig. 2c, d). Similar findings were reported by Delbecq et al., and Sui et al., on hydrogels containing rGO [22, 23]. After drug loading, the hydrogels still retained the morphology (Fig. 2g, h). However, after drug release at pH 1.2, the rod-shaped morphology was not visible, suggesting degradation (Fig. 3a). Few rod-shaped features were visible in the hydrogels after drug release at pH 7.4. This finding suggests that the hydrogels degraded faster in acidic medium simulating gastric juice compared to the pH 7.4 simulating blood. TEM images of GW3 further revealed the interaction of rGO with the polymers in the hydrogel matrices (Fig. 4a, b). The average surface roughness (Ra) and mean-square-roughness (Rms) were obtained from the roughness analysis capacity of WS × M software [24]. Ra is the average distance of the roughness profile to the center plane of the surface profile, whereas Rms is the standard deviation from the mean surface plane. Both roughness parameters represent the maximum height identified within the observed area and do not reflect the actual topography of the hydrogels (Fig. 5a, b). The degree of roughness was reduced in the GW3PG when compared to GW3CQ and this is attributed to different levels of pores fillings within the polymeric matrix by drug loading (Table 2). The aforementioned findings were similar to those reported by Gonzalez-Meijome et al. [25].
The hydrogels were amorphous in nature, which were visible by their broad diffraction pattern. GW3PG and GW3CQ hydrogel loaded with either CQ or PG displayed broad peaks between Ɵ = 17o and 34o (Fig. 6a). GW3 hydrogel showed significant peaks at Ɵ = 38o, 43o, and 49.7o, respectively, which is due to oriented crystalline planes in the polymeric matrices. A similar finding was reported by Bastos et al. [26]. The intensity of the peaks at Ɵ = 38o, 43o, and 49.7o in GW3 was reduced in GW3PG and GW3CQ loaded with drugs and this is attributed to decrease in intermolecular interactions between the polymeric chains, which in turn decreases the degree of the crystallinity. However, a distinct peak was visible at Ɵ = 44o in all the hydrogels loaded with the drugs (Fig. 6a), suggesting a low degree of crystallinity of the hydrogel and less structural stability of the gels at the loaded concentrations and dispersion of the drug particle at a molecular level in the polymeric network. This observation is desirable, because it suggests enhanced degradation of the hydrogel after drug release.
The TGA thermograms of PG and CQ suggested that the drugs were not thermally stable. PG exhibited a curve, indicating a rapid rate of degradation (Fig. 6b). CQ exhibited two steps of weight loss. The weight loss of the free drugs was rapid, while the weight loss of the hydrogel loaded with the drug was steady. This observation suggested that the hydrogels enhanced the thermal stability of the drugs which is attributed to the high degree of crosslinking of the hydrogel and interaction between rGO and the polymeric network (Fig. 6b) [17].
The water uptake of the hydrogel is very important, because it influences the rate of release of drug from the hydrogel matrices. The hydrogels were pH-sensitive and their degree of swelling was very significant at pH 7.4 and at a temperature of 37 °C. At a temperature of 37 °C, the swelling ratio of the hydrogels was enhanced when compared to the swelling at a temperature of 24 °C. The enhanced swelling capability of the hydrogels was attributed to the destruction of hydrogen bonding between the polymer molecules, resulting in an increased in the mobility of the chain and network expansion. Similar findings were reported by other researchers [27, 28]. The presence of hydrophilic functionalities on the WPI in the polymer network also enhanced the swelling capacity of the hydrogels [29, 30]. At 24 °C, almost 30% of the total swelling capacities were observed in GW2 and GW3 in the first hour. GW2 and GW3 exhibited a high swelling ability over the period of 24 h at both temperatures, which is attributed to the presence of WPI within the gel matrix. GW1 and GW4 exhibited a slow degree of swelling at 24 °C and pH 7.4 (Fig. 7a). This observation suggests that the presence of WPI in the hydrogel matrices enhanced the swelling capacity of the hydrogels. WPI and rGO contain hydrophilic groups which contribute to the enhanced water sorption ability of the hydrogels. Some researchers reported similar findings in which the addition of rGO to hydrogel enhanced the swelling capacity of hydrogels [31, 32]. However, it is important to mention that the hydrogel GW4 containing only rGO exhibited low swelling ability at pH 7.4 and at a temperature of 24 °C, indicating suppressed swelling ability and suggesting good mechanical property that is not easily destroyed by swelling forces. However, further studies will be required to confirm the mechanical properties of these hydrogels. Similar findings were reported by Zhuang et al. for hydrogels prepared from alginate, synthetic polymers and rGO [33]. At pH 1.2, the hydrogels exhibited a reduced degree of swelling which is due to the protonation of carboxylate anions whereby anion–anion repulsive forces no longer exist (Fig. 7b). The swelling ability of the hydrogel containing rGO, i.e. GW4, was enhanced at acidic pH which is attributed to the low quantity of rGO dispersed in the hydrogel network, resulting in enhanced intermolecular interactions. A similar finding was reported by Huang et al. for hydrogels prepared from graphene oxide and polyacrylic-co-acrylamide [34]. At pH 7.4, the enhanced swelling capacity of the hydrogels was attributed to ionization of the carboxylate groups, thus resulting in the mutual repulsion between the carboxylate groups (Fig. 7a). The hydrogels were temperature sensitive (Table 3).
In vitro cumulative drug release of CQ and PG from hydrogels revealed that the hydrogels had the capacity to retain a substantial amount of drug in simulated gastric juice condition. Similar findings were reported by some researchers [35, 36]. The solubility of the drugs influenced their release rate from the hydrogels; Castro et al. [19] and Alhnan et al. reported similar findings [37]. The solubility of CQ in water is 50 mg/mL, while the solubility of PG in water is 156 mg/L. The swollen state of the hydrogel network contains a large amount of water which dissolves CQ which is highly soluble when compared to PG in water phase of the hydrogel, followed by the diffusion of CQ which was faster from the hydrogel when compared to PG. The drug release profiles of the hydrogels were found to follow Korsmeyer and Higuchi release models.
Conclusion
The aim of this research is to develop hydrogel biocomposites that are biodegradable for controlled release of bioactive agents. The biocomposites were characterized by FTIR, SEM, TEM, AFM, XRD, and TGA. Hydrogel biocomposites were prepared from a combination of thermally reduced graphene oxide (rGO) and natural and synthetic polymers followed by encapsulation of antimalarials. FTIR spectrum confirmed physical entrapment of the drugs to the hydrogel biocomposites. SEM analysis confirmed that the hydrogels are biodegradable and the rate of degradation was influenced by the pH. In vitro drug release was performed and the release of CQ was pseudo-Fickian at pH 1.2 and super case II at pH 7.4. The release mechanism of PG was anomalous at pH 1.2 and 7.4. The presence of WPI and rGO in the hydrogel network resulted in intermolecular interactions and influenced the drug release mechanisms. The results obtained suggested that the biocomposites are potential systems that can be tailored for controlled delivery of bioactive agents. However, more research is needed to evaluate the mechanical properties of the biocomposites.
References
Kopeček J (2007) Hydrogel biomaterials: a smart future? Biomaterials 28:5185–5192
Hoare TR, Kohane DS (2008) Hydrogels in drug delivery: progress and challenges. Polymer 49:1993–2007
Kioomars S, Heidari S, Malaekeh-Nikouei B, Shayani Rad M, Khameneh B, Mohajeri SA (2016) Ciprofloxacin-imprinted hydrogels for drug sustained release in aqueous media. Pharm Dev Technol 16:1–8
Wu H, Liu S, Xiao L, Dong X, Lu Q, Kaplan DL (2016) Injectable and pH-responsive silk nanofiber hydrogels for sustained anticancer drug delivery. ACS Appl Mater Interfaces 8:17118–17126
WHO (2015). World malaria report. http://www.who.int/malaria/publications/world-malaria-report-2015/report/en/. Accessed 15 Dec 2016
Sinha S, Medhi B, Sehgal R (2014) Challenges of drug-resistant malaria. Parasite 21:61
Irvine MH, Einarson A, Bozzo P (2011) Prophylactic use of antimalarials during pregnancy. Can Fam Physician 57:1279–1281
Abdel-Haleem FM, Saad M, Rizk MS (2016) Development of new potentiometric sensors for the determination of proguanil hydrochloride in serum and urine. Chin Chem Lett 27:857–863
Price RN, von Seidlein L, Valecha N, Nosten F, Baird JK, White NJ (2014) Global extent of chloroquine-resistant Plasmodium vivax: a systematic review and meta-analysis. Lancet Infect Dis 14:982–991
Ridley RG (2002) Medical need, scientific opportunity and the drive for antimalarial drugs. Nature 415:686–693
Sullivan DJ Jr, Matile H, Ridley RG, Goldberg DE (1998) A common mechanism for blockade of heme polymerization by antimalarial quinolines. J Biol Chem 273:31103–31107
Aderibigbe BA, Mhlwatika Z (2016) Dual release kinetics of antimalarials from soy protein isolate-carbopol-polyacrylamide based hydrogels. J Appl Polym Sci 133:43918
Mavondo GA, Tagumirwa MC (2016) Asiatic acid-pectin hydrogel matrix patch transdermal delivery system influences parasitaemia suppression and inflammation reduction in P. berghei murine malaria infected Sprague–Dawley rats. Asian Pac J Trop Med 9:1172–1180
Simina D, Kacso I, Popa A, Raita O, Bende A, Borodi G, Bratu I, Lazar MD (2012) Chitosan-based nanocarriers for antimalarials. AIP Conf Proc Am Inst Phys 1425:17
Dandekar PP, Jain R, Patil S, Dhumal R, Tiwari D, Sharma S, Vanage G, Patravale V (2010) Curcumin-loaded hydrogel nanoparticles: application in anti-malarial therapy and toxicological evaluation. J Pharm Sci 99:4992–5010
Aderibigbe BA, Owonubi SJ, Jayaramudu J, Sadiku ER, Ray SS (2015) Targeted drug delivery potential of hydrogel biocomposites containing partially and thermally reduced graphene oxide and natural polymers prepared via green process. Colloid Polym Sci 293:409–420
Jiao T, Guo H, Zhang Q, Peng Q, Tang Y, Yan X, Li B (2015) Reduced graphene oxide-based silver nanoparticle-containing composite hydrogel as highly efficient dye catalysts for wastewater treatment. Sci Rep 5:11873
Adhikari B, Biswas A, Banerjee A (2012) Graphene oxide-based hydrogels to make metal nanoparticle-containing reduced graphene oxide-based functional hybrid hydrogels. ACS Appl Mater Interface 4:5472–5482
Castro E, Mosquera V, Katime I (2012) Dual Drug Release of Triamterene and Aminophylline from Poly(N-Isopropylacrylamide) hydrogels. Nanomater Nanotechnol 1:11
Huang X, Lowe TL (2005) Biodegradable thermoresponsive hydrogels for aqueous encapsulation and controlled release of hydrophilic model drugs. Biomacromol 6:2131–2139
Wong RSH, Dodou K (2017) Effect of drug loading method and drug physicochemical properties on the material and drug release properties of poly (ethylene oxide) hydrogels for transdermal delivery. Polymers 9:29
Delbecq F, Endo H, Kono F, Kikuchi A, Kawai T (2013) Incorporation of graphene into photopolymerizable hydrogels of N-acyl glutanamides: rheological and swelling behavior study of soft nanocomposite materials. Polymer 54:1064–1071
Sui Z, Zhang X, Lei Y, Luo Y (2011) Easy and green synthesis of reduced graphite oxide-based hydrogels. Carbon 49:4314–4321
Horcas J, Fernandez R, Gomez-Rodriquez JM, Colchero J, Gomez-Herrero J, Baro AM (2007) A software for scanning probe microscopy and a tool for nanotechnology. Rev Sci Instrum 78:013705
González-Méijome JM, Lo´Pez-Alemany A, Almeida JB, Parafit MA (2008) Surface AFM microscopy of unworn and worn samples of silicon hydrogel contact lenses. J Biomed Mater Res Part B Appl Biomater 88:75–82
da Silva BD, do Pilar Goncalves M, de Andrade CT, de Lima Araújo KG, da Rocha Leão MH (2012) Microencapsulation of cashew apple (Anacardium Occidentale, L.) juice using a new chitosan-commercial bovine whey protein isolate system in spray drying. Food Bioprod Process 90:683–692
Gupta NV, Shivakumar HG (2012) Investigation of swelling behavior and mechanical properties of a ph-sensitive superporous hydrogel composite. Iran J Pharm Res 11:481–493
Deng KL, Zhong HB, Tian T, Gou YB, Li Q, Dong LR (2010) Drug release behavior of a pH/temperature sensitive calcium alginate/poly(N-acryloylglycine) bead with core-shelled structure. Express Polym Lett 4:773–780
Işıklan N, Küçükbalcı G (2016) Synthesis and characterization of pH-and temperature-sensitive materials based on alginate and poly (N-isopropylacrylamide/acrylic acid) for drug delivery. Polym Bull 73:1321–1342
Kajjari PB, Manjeshwar LS, Aminabhavi TM (2012) Novel pH-and temperature-responsive blend hydrogel microspheres of sodium alginate and PNIPAAm-g-GG for controlled release of isoniazid. AAPS Pharm Sci Tech 13:1147–1157
Zeng Y, Qiu L, Wang K, Yao J, Li D, Simon GP, Wang R, Wang H (2013) Significantly enhanced water flux in forward osmosis desalination with polymer-graphene composite hydrogels as a draw agent. RSC Adv 3:887–894
Kim S, Yoo Y, Kim H, Lee E, Lee JY (2015) Reduction of graphene oxide/alginate composite hydrogels for enhanced adsorption of hydrophobic compounds. Nanotechnol 26:405602
Zhuang Y, Yu F, Chen H, Zheng J, Ma J, Chen J (2016) Alginate/graphene double-network nanocomposite hydrogel beads with low-swelling, enhanced mechanical properties, and enhanced adsorption capacity. J Mater Chem A 4:10885–10892
Huang Y, Zeng M, Ren J, Wang J, Fan L, Xu Q (2012) Preparation and swelling properties of graphene oxide/poly (acrylic acid-co-acrylamide) super-absorbent hydrogel nanocomposites. Colloids Surf A Physicochem Eng Asp 401:97–106
Ye Y, Hu YA (2016) pH-sensitive injectable nanoparticle composite hydrogel for anticancer drug delivery. J Nanomater 2016:57
Byun E, Lee H (2014) Enhanced loading efficiency and sustained release of doxorubicin from hyaluronic acid/graphene oxide composite hydrogels by a mussel-inspired catecholamine. J Nanosci Nanotechnol 14:7395–7401
Alhnan M, Cosi D, Murdan S, Basit A (2012) Inhibiting the gastric burst release of drugs from enteric microparticles: the influence of drug molecular mass and solubility. J Pharm Sci 99:4576–4583
Acknowledgements
The financial assistance of National Research Foundation and Medical Research Council (Self-Initiated Research), South Africa towards this research are hereby acknowledged. The views and opinions expressed in this manuscript are those of the authors and not of MRC or NRF.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to urisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Owonubi, S.J., Aderibigbe, B.A., Mukwevho, E. et al. Characterization and in vitro release kinetics of antimalarials from whey protein-based hydrogel biocomposites. Int J Ind Chem 9, 39–52 (2018). https://doi.org/10.1007/s40090-018-0139-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40090-018-0139-2