
Abstract
A novel C17N Lycopodium alkaloid (LA), lycoplanine B (1), containing an unusual formyl group, along with two new LAs, lycoplanines C (2) and D (3), were isolated from the whole plant of Lycopodium complanatum. Their structures were elucidated by extensive NMR techniques, including 1D- and 2D-NMR experiments, as well as comparing their spectral data with those of the known analogues. A possible biogenetic pathway for 1 was also proposed.
Graphical Abstract

Similar content being viewed by others

1 Introduction
Lycopodium complanatum belonging to the family Lycopodiaceae, is mainly distributed in temperate and subtropical areas. This plant has been used as a folk medicine for the treatment of arthritic pain, quadriplegia, contusion, and blood stasis [1, 2]. Previous phytochemical investigations of this plant led to the isolation of a number of Lycopodium alkaloids (LAs) [3,4,5,6], which have attracted great interest from biogenetic [7, 8], synthetic [9,10,11], and biological [12, 13] points of view.
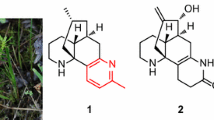
In our continuing efforts to find biogenetically interesting and structurally unique LAs, we reported a LA, lycoplanine A, posessing an intriguing tricyclic (6/9/5) ring skeleton from L. complanatum [14]. Further chemical investigation of the extracts of this plant resulted in the isolation of three new LAs, lycoplanines B-D (1–3), along with three known compounds lycopodine (4), lycopodine N-oxide (5), and des-N-methyl-α-obscurine (6). In current study, we describe the isolation, structure elucidation, and anti-AChE activity of 1–3 (Fig. 1), as well as a plausible biogenetic path for lycoplanine B (1).

Structures of compounds 1–5
2 Results and Discussion
The air-dried and powdered whole plant of L. complanatum was extracted with EtOH three times. The extract was partitioned between EtOAc and 1.0‰ HCl/H2O. The pH of the water-soluble portion was adjusted to pH 9 with saturated Na2CO3 aq. Then, it was extracted with CHCl3 to afford an alkaloidal extract. Further column chromatography over MCI gel, silica gel, Sephadex LH-20, and RP-18 led to the isolation of compounds 1 (5 mg), 2 (3 mg), 3 (6 mg), 4 (30 mg), and 5 (8 mg), and 6 (15 mg).
Lycoplanine B (1), \([\alpha]_{22.3}^{D}\) −166.85 (c 0.18, MeOH), white amorphous powder, gave rise to a quasi-molecular ion peak at m/z 274.1806 ([M+H]+) in the HRESIMS, which corresponded to the molecular formula C17H23NO2, and was further confirmed by its 13C NMR and DEPT analysis. The IR absorption bands indicated the existence of carbonyl (1703 cm−1), and olefinic functional (1637 cm−1) groups. The 13C NMR spectroscopic data of 1 (Table 1), with the aid of an HSQC NMR experiment, showed 17 carbons due to one methyl (δC 22.8), seven sp3 methylenes, one sp2 (δC 158.6) methine, four sp3 methines, one sp3 (δC 62.2) quaternary carbon, one sp2 (δC 113.2) quaternary carbon, one carbonyl group (δC 212.1), and one aldehyde group (δC 189.4), of which one sp3 quaternary carbon (δC 62.2) and one sp3 methylene carbon (δC 49.1) were attributed to those attached to a nitrogen atom.
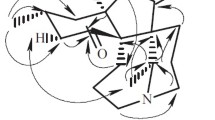
The gross structure of 1 was elucidated by analyses of 2D NMR data including 1H–1H COSY, HSQC, and HMBC spectra recorded in CD3OD (Fig. 2). The 1H-1H COSY spectrum of 1 revealed the presence of two fragments: a (C-3/C-4) and b (C-9/C-10/C-11/C-12/C-7/C-6, C-7/C-8/C-15/C-16 and C-15/C-14). The connection of C-4 and C-6 through the ketone group (C-5) as evidenced by the HMBC correlations from H-3, H-4, H-6, and H-7 to a ketone group (δC 212.1). The connection of C-4 and C-12 through quaternary carbon (C-13) as evidenced by the HMBC correlations of H-3 and H-4 with quaternary carbon (δC 62.2), and H-4 with C-12. What’s more, C-3 and C-17 was connected by sp2 quaternary carbon (C-2), which was also elucidated by the HMBC correlations from H-3 to C-1, C-2, and C-17. Meanwhile, HMBC cross-peaks of H-9 with C-1 and C-13 established the connections of C-1, C-9, and C-13 through a nitrogen atom. In addation, HMBC correlations from H-10 to C-11 and C-12, H-14 to C-4, C-12, C-13, C-15, and C-16 could be observed. Thus, the basic planar structure of 1 was determined.

Key 2D-NMR correlations for compound 1
The relative configuration of 1 was elucidated by the ROESY spectrum (Fig. 2) and biogenetic consideration of lycopodine-type alkaloid derivatives isolated from Lycopodiaceae family. The cross-peaks of H-12 with H-8a, H-15 with H-6a were observed in ROESY experiment indicating the β-orientation of H-12 and H-15. The correlation of H-4 with H-11b indicated the α-orientation of H-4. Thus, the structure of compound 1 was characterized as 2-formyl-1,2-dehydro-lycopodine.
Lycoplanine C (2), obtained as colorless oil, had a molecular formula of C16H23NO3 by the HRESIMS ion peak at m/z 278.1758 [M+H]+ (calcd for C16H24NO3, 278.1751) with six degrees of unsaturation. The IR absorption at 3414 cm−1 suggested the presence of hydroxy group. In agreement with its molecular formula, all the sixteen carbon signals were observed in the 13C NMR spectrum (Table 1), and were further classified by DEPT experiments into one methyl (δC 22.5), six methylenes, six methines (one olefinic at δC 123.2), and three quaternary carbons (one olefinic at δC 143.5, and one carbonyl at δC 210.1). The 1H NMR (Table 1) spectrum of 2 displayed one methyl (δH 0.84, d, J = 6.1 Hz) and one olefinic proton (δH 5.82, d, J = 4.5 Hz). Careful comparison of the NMR spectroscopic data of 2 with those of lycoposerramine K [15] suggested that they possessed the same carbon skeleton. The fragments of a C-1/C-2/C-3/C-4, b C-9/C-10/C-11, and c C-6/C-7/C-8/C-15(C-16)/C-14 (Fig. 3) determined by the 1H-1H COSY correlations (Fig. 3), in combination with a series of HMBC correlations (Fig. 3) of H-9 with C-1 and C-13, H-1 with C-9 and C-13, H-4 with C-5, C-12 and C-13, H-6 with C-4, C-5 and C-12, and H-7 with C-11 and C-12, further confirmed the above deduction. The only difference between 2 and lycoposerramine K was that 2 possessed an additional hydroxy group connected to C-10, as inferred from the HMBC correlations of H-9 and H-11 with C-10 (δC 64.3).

Key 2D-NMR correlations for compound 2
The relative configuration of 2 was assigned on the basis of ROESY experiment. The ROESY correlations (Fig. 3) of H-15 with H-6, H-11 with H-7, H-10 with H-14a, and H-4 with H-9b indicated that H-4 and H-7 were α-oriented, while H-6, H-10 and H-15 were β-oriented. Therefore, compound 2 was unambiguously established as 10α-hydroxy-lycoposerramine K.
Lycoplanine D (3) has the molecular formula C16H24N2O2 based on its HRESIMS (m/z 277.1914) ([M+H]+, calcd 277.1911) and 13C NMR spectroscopic data. The IR absorption bands indicated the existence of hydroxy (3495 cm−1), carbonyl (1698 cm−1), and olefinic functional (1657 cm−1) groups. The 13C NMR (Table 1) spectroscopic data of 3 showed the exitence of 16 signals classified as one methyl resonated at δC 22.3, eight methylenes, two methines, and five quaternary carbons (one amide, two olefinic carbons, and two sp3 quaternary carbons). Further 2D NMR analysis indicated that 3 should be a lycodine-type alkaloid with many similarities to that of des-N-methyl-α-obscurine [16], which was also isolated in present study. Differing from des-N-methyl-α-obscurine, the molecular formula of 3 contains one more oxygen atom. Meanwhile, the methine group (δC 44.5) at C-12 in des-N-methyl-α-obscurine was replaced by a sp3 quaternary carbon (δC 70.3) in 3. So, the H-12 in des-N-methyl-α-obscurine was replaced by a hydroxyl group in 3. The HMBC correlations of H-7, H-8, H-11, and H-14 with C-12 (δC 70.3) and C-13 (δC 60.5), further confirmed the above deduction. The relative configurations of 12-OH and H-15 were deduced to be β-oriented, according to the cross-peaks of H-11b/H-6b and H-15/H-6a in the ROESY spectrum. Therefore, compound 3 was determined as 12-hydroxy-des-N-methyl-α-obscurine.
Lycoplanine B (1), a novel C17N Lycopodium alkaloid, is the first example of Lycopodium alkaloids containing an unusual formyl group. A possible biogenetic pathway for 1 was also proposed as shown in Scheme 1. Compound 1 might be generated from lycopodine (4) [17], which was also isolated from the plant in current study. In brief, 4 underwent oxidation step to produce intermediate i. Intermediate i give ii through a aldol addition reaction. Intermediate ii could then underwent oxidation reaction to yield key intermediate iii. Then, intermediate iii accompanied by a isomerization behaviors of the double bond to get 1.

Plausible biogenetic formation of lycoplanine B (1)
Compounds 1–3 were also tested for AChE inhibitory activities using the Ellman method [18] reported previously (huperzine A as positive control, IC50 = 0.03 μM). Unfortunately, they showed no inhibitory activity (IC50 > 100 μM) after two repeated experiments.
3 Experimental
3.1 General
Optical rotations were measured with a Horiba SEPA-300 polarimeter (Horiba, Tokyo, Japan). UV spectra were obtained using a Shimadzu UV-2401A spectrophotometer (Shimadzu, Tokyo, Japan). IR spectra were obtained by a Tenor 27 spectrophotometer (Bruker Optics, Ettlingen, Germany) using KBr pellets. 1D and 2D NMR spectra were carried out on Bruker AM-400, DRX-500, or AVANCE III-600 spectrometers with TMS as an internal standard (Bruker, Karlsruhe, Germany). ESIMS and HRESIMS were run on an Agilent 6530 Q of spectrometer (Agilent, Palo Alto, CA, USA). Column chromatography (CC) was performed using MCI gel (CHP 20P, 75–150 m; Mitsubishi Chemical Corporation, Tokyo, Japan), Polyamide (PA, 80–100 mesh; Sinopharm Chemical Reagent Co., Ltd, Shanghai, China) and silica gel (100–200 or 200–300 mesh; Qingdao Haiyang Chemical Co., Ltd, Qingdao, China). Thin-layer chromatography (TLC) was conducted on silica gel plates GF254 (Qingdao Haiyang Chemical Co., Ltd, Qingdao, China). Fractions were monitored by TLC using various solvent systems, and spots were visualized by spraying improved Dragendorff’s reagent to the silica gel plates or by heating silica gel plates sprayed with 10% H2SO4 in EtOH.
3.2 Plant Material
The whole plants of Lycopodium complanatum were collected in Guizhou Province, People’s Republic of China, in September 2013. The sample was identified by Dr. Xiao Cheng. A voucher specimen (20130915) has been deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, People’s Republic of China.
3.3 Extraction and Isolation
The air-dried and powdered sample (30 kg) was extracted with 60% EtOH (24 h × 3), the extract was partitioned between EtOAc and 1.0‰ HCl/H2O. Water-soluble materials, which were adjusted to pH 10 with saturated Na2CO3 aq, were extracted with CHCl3 to give an alkaloidal extract (60 g). The alkaloidal extract was subjected to medium pressure liquid chromatography (MPLC) over RP-18 gel and eluted with MeOH/H2O (1:4–1:0) to yield five subfractions A-E. Fraction B (10 g) was chromatographed over silica gel CC (petroleumether/Me2CO, 8:1–1:1) to afford five subfractions, fractions B1-B6. Fraction B3 (3.5 g) was purified by silica gel CC (CHCl3/MeOH, 80:1–10:1) to obtain 1 (5 mg). Fraction C (18 g) was subjected to silica gel CC (petroleum ether/Me2CO, from 9:1 to 4:1) to give six subfractions, fractions C1-C6. Fraction C3 (5.4 g) was submitted to silica gel CC (CHCl3/MeOH, 50:1–10:1) to yield 2 (3 mg) and 4 (30 mg). Fraction D (11 g) was successively subjected to RP-18 (30% MeOH/H2O) to give three subfractions, fractions D1-D3. Fraction D3 (4.5 g) was further purified by a silica gel column chromatography (CHCl3/MeOH, 200:1) and Sephadex LH-20 to afford 3 (6 mg) and 5 (8 mg).
3.3.1 Lycoplanine B (1)
White amorphous powder; \([\alpha]_{22.3}^{D}\) −166.85 (c 0.18, MeOH); IR (KBr) υmax 1703, 1637, 1600, 1412 cm−1; UV (MeOH) λmax (log ε) 202 (3.80), 302 (4.36) nm; 1H- and 13C-NMR spectroscopic data, see Table 1; HRESIMS (pos.) m/z 274.1806 ([M+H]+) (calcd for C17H24NO2, 274.1802).
3.3.2 Lycoplanine C (2)
Colorless oil; \([\alpha]_{22.3}^{D}\) −50.28 (c 0.12, MeOH); IR (KBr) υmax 3414, 2925, 1711, 1454, 1043, cm−1; UV (MeOH) λmax (log ε) 203 (3.68) nm; 1H- and 13C-NMR spectroscopic data, see Table 1; HRESIMS (pos.) m/z 278.1758 ([M+H]+) (calcd for C16H24NO3, 278.1751).
3.3.3 Lycoplanine D (3)
White amorphous powder; \([\alpha]_{22.3}^{D}\) −26.58 (c 0.08, MeOH); IR (KBr) υmax 3495, 1657, 1384, 1196 cm−1; UV (MeOH) λmax (log ε) 201 (3.62), 253 (3.78) nm; 1H- and 13C-NMR spectroscopic data, see Table 1; HRESIMS (pos.) m/z 277.1914 ([M+H]+) (calcd for C16H25N2O2, 277.1911).
3.4 Acetylcholinesterase Inhibition Activity
Acetylcholinesterase (AChE) inhibitory activity of compounds 1–3 were assayed by the spectrophotometric method developed by Ellman et al. with slightly modification (Ellman et al. 1961). S-Acetylthiocholine iodide, S-butyrylthiocholine iodide,5,5′-dithio-bis-(2-nitrobenzoic) acid (DTNB, Ellman’s reagent), acetylcholinesterase derived from human erythrocytes were purchased from Sigma Chemical. Compounds 1–3 were dissolved in DMSO. The reaction mixture (totally 200 μL) containing phosphate buffer (pH 8.0), test compound (50 μM), and acetyl cholinesterase (0.02 U/mL), was incubated for 20 min (30 °C). Then, the reaction was initiated by the addition of 40 μL of solution containing DTNB (0.625 mM) and acetylthiocholine iodide (0.625 mM) for AChE inhibitory activity assay, respectively. The hydrolysis of acetylthiocholine was monitored at 405 nm every 30 s for 1 h. Tacrine was used as positive control with final concentration of 0.333 μM. All the reactions were performed in triplicate. The percentage inhibition was calculated as follows: % inhibition = (E − S)/E × 100 (E is the activity of the enzyme without test compound and S is the activity of enzyme with test compound).
References
X.C. Zhang, L.B. Zhang, Flora of China. Beijing: Science Press 6, 33–34 (2004)
Jiangsu New Medical College, Dictionary of the Tranditional Chinese Medicine (Shanghai Science and Technology Press, Shanghai, 1986), pp. 1138–1139
J. Kobayashi, Y. Hirasawa, N. Yoshida, H. Morita, J. Org. Chem. 66, 5901–5904 (2001)
X.D. Wu, J. He, G. Xu, L.Y. Peng, L.D. Song, Q.S. Zhao, Acta Bot Yunnan 31, 93–96 (2009)
K.I. Ishiuchi, T. Kubota, H. Ishiyama, S. Hayashi, T. Shibata, J. Kobayashi, Tetrahedron Lett. 52, 289–292 (2011)
J.T. Cheng, F. Liu, X.N. Li, X.D. Wu, L.B. Dong, L.Y. Peng, S.X. Huang, J. He, Q.S. Zhao, Org. Lett. 15, 2438–2441 (2013)
K.I. Ishiuchi, T. Kubota, Y. Mikami, Y. Obara, N. Nakahata, J. Kobayashi, Bioorg. Med. Chem. 15, 413–417 (2007)
K.I. Ishiuchi, T. Kubota, H. Ishiyama, S. Hayashi, T. Shibata, K. Mori, Y. Obara, N. Nakahata, J. Kobayashi, Bioorg. Med. Chem. 19, 749–753 (2011)
D.F. Fischer, R. Sarpong, J. Am. Chem. Soc. 132, 5926–5927 (2010)
Y. Nakamura, A.M. Burke, S. Kotani, J.W. Ziller, S.D. Rychnovsky, Org. Lett. 12, 72–75 (2010)
X. Cheng, S.P. Waters, Org. Lett. 12, 205–207 (2010)
J.S. Liu, Y.L. Zhu, C.M. Yu, Y.Z. Zhou, Y.Y. Han, F.W. Wu, B.F. Qi, Can. J. Chem. 64, 837–839 (1986)
M.G. Vallejo, M.G. Ortega, J.L. Cabrera, V.P. Carlini, S.R. de Barioglio, A.M. Agnese, J. Ethnopharmacol. 111, 685–687 (2007)
Z.J. Zhang, Y. Nian, Q.F. Zhu, X.N. Li, J. Su, J. Wu, J. Yang, Q.S. Zhao, Org. Lett. 19, 4668–4671 (2017)
H. Takayama, K. Katakawa, M. Kitajima, K. Yamaguchi, N. Aimi, Chem. Pharm. Bull. 51, 1163–1169 (2003)
W.A. Ayer, G.C. Kasitu, Can. J. Chem. 67, 1077–1086 (1989)
T.T. Nakashima, P.P. Singer, L.M. Browne, W.A. Ayer, Can. J. Chem. 53, 1936–1942 (1975)
G.L. Ellman, K.D. Courtney, V. Andres, R.M. Featherstone, Biochem. Pharmacol. 7, 88–95 (1961)
Acknowledgements
This work was financially supported by the NSFC-Joint Foundation of Yunnan Province (No. U1502223), the National Natural Science Foundation of China (Nos. 81773611 and 21602227), the Science and Technology Program of Yunnan province (Nos. 2016FB140 and 2015FB173).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, ZJ., Zhu, QF., Su, J. et al. Lycoplanines B-D, Three Lycopodium Alkaloids from Lycopodium complanatum. Nat. Prod. Bioprospect. 8, 177–182 (2018). https://doi.org/10.1007/s13659-018-0161-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-018-0161-2