
Abstract
Two rare lyconadin-type Lycopodium alkaloids, lyconadins G (1) and H (2), together with four known ones (3–6), were isolated from Lycopodium complanatum. The structures were determined on the basis of their spectroscopic analyses, and the absolute configuration of 1 was established by an X-ray crystallographic analysis. It is the first time to establish the absolute configuration of lyconadin-type Lycopodium alkaloid by an X-ray diffraction experiment. In addition, these findings may provide more information for the biosynthesis of lyconadins.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
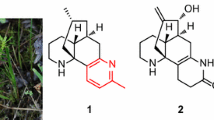
The Lycopodium alkaloids are a unique family of complex natural products that have garnered long-standing interest from chemists as challenging targets for total synthesis [1–5], due to their fascinating structure complexity and wide-ranging biological activities [6–8]. Of these, huperzine A (Hup A), isolated from the Chinese folk medicinal herb Qian Ceng Ta [whole plant of Huperzia serrata (Thunb.ex Murray) Trev.], is a promising agent to treat Alzheimer’s disease with highly specific and potent inhibitory activity against acetylcholinesterase (AChE) [9]. As a consequence, these structurally complex Lycopodium alkaloids have inspired many research groups to study the chemical constituents in the Lycopodium plants [10–13]. Lycopodium complanatum (L.) Holub, mainly distributed in the temperate and subtropical regions of the world [14], was used as a traditional Chinese herbal medicine for the treatment of arthritic pain, quadriplegia, and contusion [15]. Previously, lycospidine A, a unique C15N Lycopodium alkaloid possibly derived from proline instead of the lysine biosynthetically, was isolated from this species [16]. As a part of an ongoing research program to discover more Lycopodium alkaloids with fascinating structures and bioactivities serving as lead compounds for drug discovery [17, 18], two new rare lyconadins, termed lyconadins G and H (1 and 2), were isolated from L. complanatum (Fig. 1), together with four known ones, lyconadin A (3) [19] and lyconadins C–E (4–6) [20, 21]. To the best of our knowledge, although more than 300 Lycopodium alkaloids have been reported so far [22–25], only six ones belong to the lyconadin-type [19–21, 26]. In this paper, we describe the isolation and structure elucidation of lyconadins G (1) and H (2).

Structures of lyconadins G (1) and H (2)
2 Results and Discussion
The air-dried and powdered whole plant of L. complanatum was extracted with MeOH three times. The extract was partitioned between EtOAc and 1 % HCl/H2O. The pH of the water-soluble portion was adjusted to pH 9 with saturated Na2CO3 aq. Then, it was extracted with CHCl3 to afford an alkaloidal extract. Further column chromatography over MCI gel, silica gel, and semipreparative HPLC led to the isolation of compounds 1 (7 mg), 2 (2 mg), 3 (11 mg), 4 (8 mg), 5 (4 mg) and 6 (6 mg).
Lyconadin G (1), obtained as colorless blocks, was assigned to have a molecular formula of C16H20N2O by HR-EI-MS (m/z 256.1576, calcd 256.1576) and 13C NMR spectroscopic data (Table 1). The IR absorption implied the presence of amide carbonyl (1647 cm−1) functionality. The 13C NMR and DEPT spectra (Table 1) exhibited 16 carbon resonances, including four quaternary carbons (one carbonyl and three olefinic), seven methines [including three olefinic (δ C 120.7, 116.8, and 146.6) with corresponding protons as singlet signal at δ H 6.28 and two mutually coupled signals at δ H 6.34 and 7.30, respectively, in the 1H NMR spectrum], four methylenes, and one methyl. Based on the above evidence, the structure of 1 was determined to be similar to that of lyconadin C (4) [20]. Comparison of the NMR data of 1 and 4 suggested that the most obvious differences were the loss of two sp 3 carbons (one sp 3 methine carbon and one sp 3 methylene carbon) and the presence of one more trisubstituted double bond in 1. The 1H-1H COSY spectrum of 1 (Fig. 2) showed two partial structures a (C-2/C-3), b (C-9 to C-16 and C-8/C-15), implying that the additional double bond in 1 was located between C-6 and C-7. The HMBC correlations from H-15 and H2-8 to C-7 and from H-6 to C-4, C-5, and C-8 (Fig. 2) further supported the planar structure of 1 as shown in Fig. 2.

Key 1H-1H COSY and HMBC correlations of lyconadins G (1) and H (2)
In the ROESY spectrum of 1, the ROESY correlations of H-12/H-8b, H-12/H-14b suggested that they were in the same orientations. The fact that C-16 was in an equatorial position was deduced from the ROESY cross-peak of H-8b/H3-16. However, there were no solid proof which can be used to establish the relative configurations of C-10 and C-13. Fortunately, crystals suitable for single-crystal X-ray diffraction of 1 was obtained from CHCl3–MeOH. The structure of 1 was confirmed by single-crystal X-ray diffraction using the anomalous scattering of CuK radiation with a Flack parameter of 0.2(2) (Fig. 3). Its absolute configuration was unambiguously assigned as 10S, 12R, 13S, 15R.

The X-ray crystal structure of lyconadin G (1)
Lyconadin H (2), showed the pseudomolecular ion peak at m/z 259 [M + H]+ in the ESI–MS, and the molecular formula, C16H22N2O, was established by HR-EI-MS [m/z 258.1732 Calc. 258.1732]. The IR absorption implied the presence of amide carbonyl (1680 cm−1) functionality. The 13C NMR (Table 1) spectrum of 2 gave signals due to four quaternary carbons (one carbonyl, three olefinic), five methines (including one olefinic (δ C 123.1) with corresponding proton as a singlet signal at δ H 5.85 in the 1H NMR spectrum), six methylenes, and one methyl, implying that the structure of 2 was similar to that of lyconadin G (1). The sole difference was the loss of a 1,2-disubstituted double bond between C-2 and C-3, which was confirmed by the HMBC correlations of H2-2 and H2-3 with C-4, and of H2-3 with C-5, as well as the 1H-1H COSY correlation between H2-2 and H2-3.
The relative configuration of 2 was deduced from ROESY correlations as shown in computer-generated 3D drawing (Fig. 4). ROESY correlations of H-12/H-8b, H-12/H-14b and H-8b/H3-16 revealed they took the same orientations. A cis-fused ring junction between the cyclohexane ring and a piperidine ring (C-9–C-13 and N-9) was inferred from the coupling constant of 3 J H-13/H-14b value (4.3 Hz) and ROESY correlation of H-12/H-13 [19, 20]. ROESY cross-peaks of H-13/H-11a and H-13/H-9a indicated the chair form of the piperdine ring (C-9–C-13 and N-9). The relative configuration of H-10 was elucidated to be equatorial by the ROESY correlations of H-10/H-9a, and H-10/H-11a. Moreover, the specific rotation between 2 (+95.6) and 1 (+126.6) indicated they have the same absolute configuration. Accordingly, the structure of 2 was determined as shown in Fig. 4.

Key ROESY correlations of lyconadin H (2)
Among Lycopodium alkaloids, lyconadins are a unique family which contain intriguing tetra- or pentacyclic ring systems fused by the unique C-4–C-10 linkage and C-6–N-9 bond, and thus offer a new horizon for organic chemists [27–29]. Biogenetically, the discovery of two new compounds (1 and 2), together with four known lyconadins (3–6) from the title plant can provide some new insight into the biosynthesis of lyconadins [19, 20]. Possible biogenetic pathway for lyconadins G (1), H (2), and A (3) is proposed in Scheme 1. As illustrated in Scheme 1, the key intermediate A might arise from phlegmarane skeleton by C-4 and C-10 carbon bond formation and subsequent loss of H2O and oxidation to yield compounds 1 and 2. In addition, the double bond located between C-6 and C-7 in 1 could serve as a suitable electrophile in the process of the desired C-6–N-9 bond formation to produce lyconadin A (3). The potent biological activities of 1 and 2 against AChE using the improved Ellman method [30, 31] were also evaluated. However, neither of them showed obvious activity (IC50 > 100 µM).

Plausible biogenetic pathway for compounds 1 and 2
3 Experimental
3.1 General
Optical rotations were measured with a JASCO P-1020 polarimeter. UV spectra were recorded with a Shimadzu UV-2401A spectrophotometer. IR spectra were recorded on Bruker Tensor 27 spectrometer with a KBr disk. 1D and 2D NMR spectra were recorded on a Bruker Avance III 600 spectrometer. Chemical shifts were reported using TMS as the internal standard. ESI–MS were recorded on Shimadzu UPLC-IT-TOF-MS instrument and HREIMS spectra were measured with a Waters AutoSpec Premier P776 mass spectrometer. Column chromatography (CC) was performed on silica gel (90–200 µm; Qingdao Haiyang Chemical Co. Ltd., Qingdao, China), and MPLC was performed on a Lisui EZ Purify III System packed with RP-18 silica gel (40–63 μm, Merck, 71 Darmstadt, Germany) columns. Precoated silica gel GF254 plates (Qingdao Haiyang Chemical Co. Ltd.) were used for thin-layer chromatography (TLC). Preparative and semipreparative HPLC was performed on Shimadzu LC-8A equipped with a Shimadzu PRC-ODS (K) column and Agilent 1100 apparatus equipped with a Zorbax SB-C-18 75 (Agilent, 9.4 mm × 25 cm) column, respectively. Fractions were monitored by TLC and spots were visualized by Dragendorff’s reagent.
3.2 Plant Materials
The club moss L. complanatum was collected from Jinping County, Yunnan Province, People’s Republic of China in April, 2011. The plant was identified by Prof. Xiao Cheng (Kunming Institute of Botany, Chinese Academy of Sciences). And its voucher specimen (201104M) was deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, CAS.
3.3 Extraction and Isolation
The club moss L. complanatum (L.) Holub (100 kg) was chopped into sections and extracted with methanol under reflux for three times (4, 3, 3 h, respectively,). The resultant extract was partitioned between EtOAc and 1 % HCl/H2O solution to afford ethyl acetate and water soluble fractions, respectively. The water-soluble fractions was adjusted to pH 9 by saturated Na2CO3, and then extracted with chloroform to give an alkaloidal extract (128 g). The alkaloidal extract was subjected to a MCI gel CC (eluted with 20 % MeOH to 100 % MeOH) to afford fractions I-III. Fraction I (43.7 g) was further chromatographed over repeated silica gel columns (CHCl3/MeOH) to give subfractions Ia-Ic. Subfractions Ia was subjected to CC (SiO2, CHCl3/MeOH, gradient system) and finally purified by a C18 HPLC (MeOH/H2O/TFA, 20:80:0.1) to afford compounds 1 (7 mg) and 2 (2 mg). From subfraction Ic, compounds 3 (11 mg) and 4 (8 mg) were obtained after purified by repeated silica gel columns (CHCl3/MeOH) and semipreparative HPLC (MeOH/H2O/TFA, 18:82:0.1). Fraction II (35.0 g) was subjected to CC (SiO2, CHCl3/MeOH, gradient system) and repeated crystallization to give 5 (4 mg). Compound 6 (6 mg) was obtained after repeated chromatography and semipreparative HPLC (MeOH/H2O/TFA, 22:78:0.1) from fraction II.
3.3.1 Lyconadin G (1)
Colorless blocks; \({[{\rm \alpha}]}^{23.3}_{\rm D}\) + 126.6 (c = 0.2, MeOH); UV (MeOH) λ max (log ε) 206 (4.20), 252 (3.75) 345 (3.94) nm; IR (neat) ν max 3432, 2949, 2924, 2868, 1647, 1595, 1454, 1120 cm−1; for 1H, 13C NMR data see Table 1; HREIMS m/z 256.1576 (calcd for C16H20N2O, 256.1576).
3.3.2 Crystal Data for Lyconadin G (1)
C16H22N2O2 (C16H20N2O.H2O), MW = 274.36; orthorhombic, space group P212121; a = 8.2185(2) Å, b = 9.3774(2) Å, c = 18.2826(3) Å, α = β = γ = 90°, V = 1409.01(5) Å3, Z = 4, d = 1.293 g/cm3, crystal dimensions 0.20 × 0.25 × 0.75 mm were used for measurement on a Bruker APEX DUO with a graphite monochromator CuKα radiation. The total number of reflections measured was 6967, of which 2513 were observed, |F|2 ≥ 2σ|F|2. Final indices: R1 = 0.0337, wR2 = 0.0898 (w = 1/σ|F|2). Flack parameter = 0.2(2). The structure of 1 was solved by direct method SHELXS-97. Crystallographic data center (deposition number: CCDC 1507256).
3.3.3 Lyconadin H (2)
Colorless oil; [α] 23.3D + 95.6 (c = 0.1, MeOH); UV (MeOH) λ max (log ε) 223 (3.76), 291 (3.19) nm; IR (neat) ν max 3433, 2928, 1680, 1437, 1207, 1135, 801, 723 cm−1; for 1H, 13C NMR data see Table 1; HREIMS m/z 258.1732 (calcd for C16H22N2O, 258.1732).
3.4 Acetylcholinesterase Inhibitory Activity
AChE inhibitory activity of lyconadins G (1) and H (2) was assayed by the spectrophotometric method developed by Ellman with slightly modification. S-Acetylthiocholine iodide, S-butyrylthiocholine iodide, 5,5′-dithio-bis-(2-nitrobenzoic) acid (DTNB, Ellman’s reagent), AChE derived from human erythrocytes were purchased from Sigma Chemical. Compounds were dissolved in DMSO. The reaction mixture (totally 200 μL) containing phosphate buffer (pH 8.0), test compound (50 μM), and AChE (0.02 U/mL), was incubated for 20 min (30 °C). Then, the reaction was initiated by the addition of 40 μL of solution containing DTNB (0.625 mM) and acetylthiocholine iodide (0.625 mM) for AChE inhibitory activity assay, respectively. The hydrolysis of acetylthiocholine was monitored at 405 nm every 3 min for 1 h. Tacrine was used as positive control with final concentration of 0.333 μM. All the reactions were performed in triplicate. The percentage inhibition was calculated as follows: % inhibition = (E − S)/E × 100 (E is the activity of the enzyme without test compound and S is the activity of enzyme with test compound).
References
V. Bisai, R. Sarpong, Org. Lett. 12, 2551–2553 (2010)
S.M. Canham, D.J. France, L.E. Overman, J. Am. Chem. Soc. 132, 7876–7877 (2010)
H.M. Ge, L.D. Zhang, R.X. Tan, Z.J. Yao, J. Am. Chem. Soc. 134, 12323–12325 (2012)
T. Nishimura, A.K. Unni, S. Yokoshima, T. Fukuyama, J. Am. Chem. Soc. 135, 3243–3247 (2013)
S.Z. Jiang, T. Lei, K. Wei, Y.R. Yang, Org. Lett. 16, 5612–5615 (2014)
X. Ma, D.R. Gang, Nat. Prod. Rep. 21, 752–772 (2004)
Y. Hirasawa, J. Kobayashi, H. Morita, Heterocycles 77, 679–729 (2009)
M. Kitajima, H. Takayama, Top. Curr. Chem. 309, 1–31 (2012)
J.S. Liu, Y.L. Zhu, C.M. Yu, Y.Z. Zhou, Y.Y. Han, F.W. Wu, B.F. Qi, Can. J. Chem. 64, 837–839 (1986)
Y. Hirasawa, T. Tanaka, K. Koyama, H. Morita, Tetrahedron Lett. 50, 4816–4819 (2009)
K. Ishiuchi, S. Kodama, T. Kubota, S. Hayashi, T. Shibata, J. Kobayashi, Chem. Pharm. Bull. 57, 877–881 (2009)
F.W. Zhao, Q.Y. Sun, F.M. Yang, G.W. Hu, J.F. Luo, G.H. Tang, Y.H. Wang, C.L. Long, Org. Lett. 12, 3922–3925 (2010)
J. He, X.Q. Chen, M.M. Li, Y. Zhao, G. Xu, X. Cheng, L.Y. Peng, M.J. Xie, Y.T. Zheng, Y.P. Wang, Q.S. Zhao, Org. Lett. 11, 1397–1400 (2009)
S.G. Lu, G.F. Zhang, W.H. Su, R.X. Wang, C.X. Li, C.D. Xu, X.C. Deng, Y.Q. Duan, Y. Wang, B.B. Zhang, Pteridology (Higher Education Press, Beijing, 2007), pp. 45–47
X.D. Wu, J. He, G. Xu, L.Y. Peng, L.D. Song, Q.S. Zhao, Acta Bot Yunnanica. 31, 93–96 (2009)
J.T. Cheng, F. Liu, X.N. Li, X.D. Wu, L.B. Dong, L.Y. Peng, S.X. Huang, J. He, Q.S. Zhao, Org. Lett. 15, 2438–2441 (2013)
L.B. Dong, X. Gao, F. Liu, J. He, X.D. Wu, Y. Li, Q.S. Zhao, Org. Lett. 15, 3570–3573 (2013)
L.B. Dong, Y.N. Wu, S.Z. Jiang, X.D. Wu, J. He, Y.R. Yang, Q.S. Zhao, Org. Lett. 16, 2700–2703 (2014)
J. Kobayashi, Y. Hirasawa, N. Yoshida, H. Morita, J. Org. Chem. 66, 5901–5904 (2001)
K. Ishiuchi, T. Kubota, H. Ishiyama, S. Hayashi, T. Shibata, J. Kobayashi, Tetrahedron Lett. 52, 289–292 (2011)
K. Ishiuchi, T. Kubota, H. Ishiyama, S. Hayashi, T. Shibata, K. Mori, Y. Obara, N. Nakahata, J. Kobayashi, Bioorg. Med. Chem. 19, 749–753 (2011)
Z. Wang, J. Wu, N. Zhao, Y. Yang, Y. Chen, Nat. Prod. Res. 30, 241–245 (2016)
W.J. Meng, J. Xiong, W.X. Wang, H.Y. Zhang, H. Zeng, J.F. Hu, Tetrahedron Lett. 57, 3218–3221 (2016)
W.W. Jiang, Y.C. Liu, Z.J. Zhang, Y.C. Liu, J. He, J. Su, X. Cheng, L.Y. Peng, L.D. Shao, X.D. Wu, J.H. Yang, Q.S. Zhao, Fitoterapia 109, 155–161 (2016)
K. Ishiuchi, W.P. Jiang, Y. Fujiwara, J.B. Wu, S. Kitanaka, Bioorg. Med. Chem. Lett. 26, 2636–2640 (2016)
K. Ishiuchi, T. Kubota, T. Hoshino, Y. Obara, N. Nakahata, J. Kobayashi, Bioorg. Med. Chem. 14, 5995–6000 (2006)
Y. Yang, M. Dai, Synlett 25, 2093–2098 (2014)
S.P. West, A. Bisai, A.D. Lim, R.R. Narayan, R. Sarpong, J. Am. Chem. Soc. 131, 11187–11194 (2009)
D.C. Beshore, A.B. Smith, J. Am. Chem. Soc. 129, 4148–4149 (2007)
I.K. Rhee, N. Appels, B. Hofte, B. Karabatak, C. Erkelens, L.M. Stark, L.A. Flippin, R. Verpoorte, Biol. Pharm. Bull. 27, 1804–1809 (2004)
G.L. Ellman, K.D. Courtney, V. Andres Jr., R.M. Featherstone, Biochem. Pharmacol. 7, 88–95 (1961)
Acknowledgments
This work was financially supported by the NSFC-Joint Foundation of Yunnan Province (No. U1502223), the National Natural Science Foundation of China (No. 21402212), the Science and Technology Program of Yunnan Province (No. 2015FB173), and the CAS “Light of West China” Program and Youth Innovation Promotion Association CAS (X.-D. Wu).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cheng, JT., Zhang, ZJ., Li, XN. et al. Lyconadins G and H, Two Rare Lyconadin-Type Lycopodium Alkaloids from Lycopodium complanatum . Nat. Prod. Bioprospect. 6, 279–284 (2016). https://doi.org/10.1007/s13659-016-0111-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-016-0111-9