
Abstract
Fine-scale transition-iron-carbide precipitates in a lath martensitic microstructure of 4340 steel tempered at 200 °C for 1 h were examined via imaging and electron diffraction techniques with a transmission electron microscope. Region-to-region variations were eliminated by analyzing a small volume of material (about 0.03 μm3) at two tilt conditions. Geometric analyses showed that measured interplanar spacings compared favorably with accepted values from both epsilon-carbide and eta-carbide phases (within 1%), whereas measured interplanar angles were within 1−2% of accepted values. Centered-dark-field imaging identified precisely which reflections were produced from a single group of small precipitates (each about 10 nm in diameter). Consistent indexing schemes are provided for epsilon- and eta-carbide, including the proper angular relationship between the two tilt conditions. An interzonal angle of 17.9o ± 0.1o was determined for both candidate phases. The orientation relationship between the observed transition-iron-carbide phase and martensite was determined, and confirmed previous results reported for both carbide phases. Diffracted intensities of several reflections were estimated and compared favorably with those calculated from structure factors derived from idealized crystal structures of both transition-iron-carbide phases. All results are shown to be near-equally consistent with a hexagonal epsilon-carbide phase and an orthorhombic eta-carbide phase.
Similar content being viewed by others


Introduction
Medium-carbon alloy steels, such as 4340, are austenitized, quenched to form predominantly martensitic microstructures in comparatively thick sections, and tempered to improve toughness while maintaining reasonably high strength and hardness [1]. An important aspect of tempering is the reduction of carbon supersaturation of metastable as-quenched martensite. At low tempering temperatures (e.g., 200 °C), precipitates of transition-iron-carbide phases nucleate and grow, thereby removing carbon atoms from interstitial positions of the body-centered-tetragonal martensite lattice. As a result, the strained lattice evolves toward a lower-carbon version of martensite [2]. Oftentimes, a small portion of the high-temperature, parent austenite phase is retained as a metastable component of the martensitic microstructure.
Seminal investigations on the early stages of tempering of martensite resulted in the identification of a hexagonal iron-carbide phase, referred to as epsilon-carbide [3, 4], and an orthorhombic iron-carbide phase, referred to as eta-carbide [5]. In both cases, the chemical compositions are between Fe2C and Fe3C. These phases are transition phases with very different characteristics than orthorhombic cementite (Fe3C or θ-carbide), which tends to form at higher tempering temperatures and/or at longer times.
The design of steels for future applications will require a more detailed understanding of the evolution of microstructure during tempering. Several recent studies of advanced steels have provided keen insights on this topic using advanced characterization and modeling techniques to gain complementary results to those obtained from transmission electron microscopy [6–12]. However, careful examination of these recent additions to the literature reveals that some of the interpretations seem not to be in full agreement.
Partly in response to this state of affairs, a recent paper [13] presented and analyzed electron diffraction results from a highly symmetric zone axis of transition-iron-carbide precipitates within a martensitic matrix of 4340 steel. Key quantitative results from a single electron diffraction spot pattern (including interplanar spacing values, interplanar angles, and the orientation relationship) were shown to be consistent with both epsilon-carbide and eta-carbide, within estimated uncertainty ranges. Specifically, five slightly different unit cells were considered, and experimental results were compared with each of these (referred to in that paper as: \( \varepsilon_{\text{o}} \), \( \varepsilon \), \( \varepsilon^{\prime} \), \( \eta^{\prime} \), and \( \eta \)). Having established that these five unit cells can all be used to describe transition-iron-carbide precipitates in 4340 steel [13], a study of broader scope was conducted. An important aspect of the present work is that a single region was analyzed in detail, rather than analyses from many different regions contained in a single specimen or several specimens. Thus, variations in the carbon contents of the matrix martensite phase and precipitate phase (and possibly other variations) were presumed to have been minimized or eliminated. This paper presents results that initially will be compared with the originalFootnote 1 and most commonly cited descriptions of the hexagonal epsilon-carbide phase proposed by Jack [3, 4] and the orthorhombic eta-carbide phase identified by Hirotsu and Nagakura [5].
Experimental Procedures
Specimen material was received in the form of coupons of 4340 steel measuring 10 by 10 mm by about 1 mm (end-sliced pieces from Charpy V-notch specimens). The steel had been verified by the manufacturer to be within the 4340 chemical composition specification, with an approximate alloy composition (in mass pct.) of 0.4 C, 0.7 Mn, 0.3 Si, 1.7 Ni, 0.9 Cr, and 0.2 Mo. The thermal history of the Charpy specimen was reported as austenitized, quenched, and tempered at 200 °C for 1 h.
Steel pieces were ground to 0.1 mm, and several three-millimeter-diameter discs were punched from the thinned coupons. All remaining pieces proved too small for a reliable chemical analysis to verify the product composition. Thin discs were further ground to remove any surface oxide, and then they were electrochemically jet polished to perforation with a commercial twin-jet electropolishing system.
A transmission electron microscope, operated at 120 kV, was used to characterize several of the specimens, and two specimens were chosen for detailed analyses. Conventional bright-field (BF) and centered-dark-field (CDF) imaging techniques were employed and augmented with selected-area electron diffraction patterns. The camera constant was calibrated with a high-purity aluminum standard. Additionally, ring patterns from a thin surface iron oxide layer were also used as an internal calibration standard.
To maximize the reliability of the diffraction data, eccentricities of diffraction rings from a polycrystalline high-purity aluminum standard were determined. A method of correcting for eccentricity was employed [14] followed by recalibration of the camera constant. Based on this methodology and its application to numerous selected-area electron diffraction patterns from this microscope, the upper limits of uncertainties for interplanar spacings and angles were estimated to be ±0.003 nm and ±2o.
Results and Discussion
Microstructural Features
General Features
Figure 1 is an example of a typical lath martensitic microstructure from the 4340 steel chosen for analysis. The steel had been fully austenitized, quenched from the austenite phase field, and tempered for 1 h at 200 °C. A comparatively large lath (more than 10 μm in length and nearly 1 μm in width) of martensite extends almost vertically through this montage of bright-field (BF) transmission electron microscope images. Many smaller laths of a common physical orientation are aligned at a substantial angle with respect to the large lath, the smaller laths being typically up to 1 to 3 μm long and less than 0.5 μm wide. The majority of the smaller laths possess slight differences in orientation, giving the appearance of an interwoven or intertwined structure. Crystallographic factors responsible for this appearance have been described previously [15, 16].

Lath martensitic microstructure of 4340 steel. Tempered at 200 °C for 1 h
Within the large lath, complex contrast is observed, suggesting the presence of a variety of fine-scale microstructural features. Numerous, small iron-carbide precipitates are evident, e.g., in the circled region; similar precipitates are analyzed later in this paper. The precipitates show more than one direction of alignment, suggesting multiple variants of the same precipitate phase. The possibility of lower bainite appears to be ruled out since a single variant of internal carbide forms as compared with multiple carbide variants in tempered martensite [17–24]. The significant size difference between the large lath and adjacent smaller laths seems likely to be related to when, or more specifically, at what temperatures these laths formed during cooling [25–27].Footnote 2 The large lath, because of its size, is suggested to be one of the first laths of martensite to form within the parent grain of austenite. In addition, this lath seems to show iron-carbide precipitates which are larger than those in adjacent, smaller laths. Therefore, the larger lath is believed to have nucleated close to the martensite-start temperature upon cooling, thereby growing virtually unimpeded through a grain of austenite. Formation at comparatively high temperature also provides for a greater likelihood of autotempering during the quench [25–27]. Significant strain contrast is also evident within these martensite laths, which may be more pronounced in several of the smaller laths. These observations are consistent with volume accommodation effects associated with martensite formation [28, 29] and trapping of carbon within martensite that is relieved subsequently during tempering. Some twins were also observed, but Fig. 1 shows very little discernible evidence of this strain accommodation mechanism. Although the large lath appears to be nearly “continuous” within this field of view, a smaller lath seems to “cut through” the larger one toward the top of Fig. 1; the feature in the middle left (see arrow) is believed to be another small lath of a similar nature.
Previous transmission electron microscope studies of lath martensite have shown that the austenite-to-martensite phase transformation typically does not go to completion, and as a result, some of the parent austenite phase is retained as interlath films at room temperature [30–33]. In the present system, retained austenite was identified; however, it seemed to be present in a rather low quantity, estimated at a few volume percent. Figure 2 shows a BF/CDF (bright-field/centered-dark-field) pair of transmission electron microscope micrographs that reveals interlath films of austenite. The spot used to create the CDF image was determined to be from {200} planes of austenite. The films of retained austenite shown in Fig. 2 are on the order of 10 nm in thickness, consistent with previous research [33].

Thin films of retained austenite between laths of martensite; tempered at 200 °C for 1 h. a BF and b CDF transmission electron microscope images. Arrows indicate twins in a. Three white circles in b highlight groups of small transition-iron-carbide precipitates
The low magnification associated with Fig. 1 reveals iron-carbide precipitates that seem to be plate-shaped, rod-shaped, or some similarly anisotropic morphology. Tempering at 200 °C for an hour frequently does not result in the formation of cementite crystals (with plate-like morphology). However, numerous authors have shown that transition-iron-carbide precipitates nucleate and grow, rather than cementite, at low tempering temperatures [3–5, 13, 34–47]. These small precipitates tend to form along well-defined directions within the matrix martensite.
At noticeably higher magnification, these transition-iron-carbide precipitates are readily revealed, as shown in Figs. 3 and 4 (both figures are BF/CDF pairs of micrographs). The complex nature and very small size of the precipitates makes for difficult analysis of images as well as the electron diffraction spot patterns. The purpose of this paper was to investigate key crystallographic aspects of these iron-carbide precipitates and compare results with previous work [3–5].

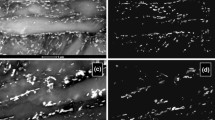
An array of transition-iron-carbide precipitates that appears to have segments of different crystallographic orientations. a BF and b CDF transmission electron microscope micrographs. Tempered at 200 °C for 1 h

An array of transition-iron-carbide precipitates that shows many nearly spherical precipitates. a BF and b CDF transmission electron microscope micrographs. Tempered at 200 °C for 1 h
The features shown by Figs. 1 through 4 are consistent with a typical low-temperature-tempered lath martensitic microstructure [15, 16]. Within this and similar specimens, there was no evidence of non-martensitic microconstituents, including proeutectoid/polygonal ferrite grains, pearlite colonies, or regions of upper or lower bainite. Thus, upon quenching, it is concluded that the parent austenite phase decomposed to a “fully” martensitic microstructure (with a very small amount of retained austenite). Although some amount of autotempering likely occurred during the quench, the majority of the transition-iron-carbide precipitate growth was presumed to have occurred during the tempering treatment. Crystallographic details of these precipitates are presented next.
Region Selected for Detailed Analysis
Figure 5a shows a montage of BF transmission electron microscope images from a different region of the same specimen described by Figs. 1, 2, 3, and 4. The largest lath has an apparent half-length estimated to be about 5 μm and an apparent width somewhat larger than 1 μm. This lath (and those nearby) is hypothesized to have formed close to the martensite-start temperature, as described for the large lath in Fig. 1, thereby explaining its large size (length and width). The extraordinarily large apparent width of these laths implies also that the lath width dimension is nearly perpendicular to the surface of this thin-foil specimen.

Lath martensite with transition-iron-carbide precipitates. Montage of a BF and b CDF transmission electron microscope micrographs. Spot used to form CDF image was shown to be a reflection from {102} ε or {121} η planes. Tempered at 200 °C for 1 h
Because of the size and orientation of the central laths in Fig. 5, and also the region’s proximity to the “edge” of the foil, numerous adjacent transition-iron-carbide precipitates of similar orientation were highly visible at various tilt conditions. The CDF images of the same region (Fig. 5b) were generated with a transition-iron-carbide precipitate reflection, and one or two precipitate variants are illuminated in this image. At this magnification and this diffracting condition, the transition-iron-carbide precipitates show frequently a continuous linear (or rod-like) nature. In some cases, a particulate nature is evident, but this latter description is more easily seen in Figs. 3 and 4.
Figure 5 is referred to and discussed throughout the remainder of this paper; Fig. 6 is a schematic illustration of Fig. 5 that identifies likely positions of low-angle lath boundaries and other features that are typical of a packet of lath martensite. The “tips” of laths labeled “A” and “B” in Fig. 6 are evident in Fig. 5a, and the majority of lath “B” is presumed to have been removed during specimen thinning. Other laths of similar crystallographic and geometrical orientation are denoted “C,” “D,” and “E” in Fig. 6. Dark regions at the periphery of Fig. 6 are presumed to be martensite laths of a different crystallographic orientation than that of the centralized laths, and their details are not shown.

Schematic diagram of key features of the lath martensitic microstructure shown in Fig. 6. Five laths from the same packet of martensite are labeled. In the gray areas, no features are indicated since this region is not of importance to this study. The circle indicates the region selected for detailed diffraction analyses
The majority of the small, bright features in the CDF image (Fig. 5b) are transition-iron-carbide precipitates. The seemingly linear nature of the precipitates is apparent and was confirmed by tilting the specimen in the microscope. These features always appeared as linear features, but never as plates. The arrays of these adjacent precipitates measured up to 200 nm in length with widths from 10 to 15 nm. Combined with observations from Figs. 3 and 4, individual, near-spherical or equiaxed precipitates measure 5 to 15 nm in diameter. These dimensions compare favorably with results reported previously [34, 35, 38–47]. Later in the paper, the directions of the linear precipitate arrays are shown to be consistent with <100> martensite directions (for the most part), and not <110> directions. This evidence lends further support to the assertion that the bright features shown in Fig. 5b are transition-iron-carbide precipitates, rather than cementite.
Two-Tilt Electron Diffraction Analysis
Diffraction at the 8°-tilt Condition
Selected-area electron diffraction patterns were generated at two different tilt conditions from the circled region illustrated in Fig. 6. The total volume of this region is estimated to be approximately 0.03 μm3. Using this limited volume of material for the complete analysis should minimize variations in diffraction data owing to variations in local chemical composition. Once this region had been chosen for detailed analyses, a near-eucentric position was established for the microscope stage, and tilting between the two conditions was accomplished with a single tilt axis (the main or α-tilt axis). Goniometer readings for these tilt conditions were estimated by the operator as 8° and 22°. Viewing the specimen holder from the end that is opposite the specimen itself, a clockwise rotation was used to go from the first to the second tilt condition.
The “as-recorded” selected-area electron diffraction pattern from the first tilt condition (the 8°-tilt condition) is shown in Fig. 7a. A second version of this pattern with labels for a zone of martensite spots and a zone of epsilon-carbide spots is shown in Fig. 7b, and an alternate indexing scheme of spots for eta-carbide is shown in Fig. 7c. A two-dimensional grid of solid lines (in Fig. 7b) represents the martensite phase, and spots are identified with black numbers of a comparatively large font size. The dashed line grids in Fig. 7b, c represent epsilon-carbide and eta-carbide, respectively, and reflections are labeled with smaller-font symbols (white numbers inside gray ovals).Footnote 3

Quantification of Fig. 7 was accomplished with standard procedures (and see [13, 48]). The camera constant (λL) was determined to be 3.50 nm·mm, where λ is the wavelength of the high-velocity electrons, and L is the camera length. Martensite was assumed to have the same crystal structure as body-centered-cubic iron, within experimental uncertainty. The lattice parameter of martensiteFootnote 4 was chosen as a = 0.2867 nm rather than using a measured value since peak broadening in x-ray diffraction was pronounced.
The lengths of important vectors in Fig. 7a were measured, and data are reported in Table 1. Calculated and accepted interplanar spacing (d hkl ) values are provided for martensite and also for both transition-iron-carbide phases. In all cases, the estimated upper limit of uncertainty (±0.003 nm) was not exceeded.
In Fig. 7b, the \( 2\bar{1}\bar{1} \)/\( \bar{2}11 \) martensite pair of spots lies horizontally within the figure.Footnote 5 This feature provides a convenient fiducial line for reporting angular measurements between diffracting plane normals. Angles between plane normals were measured (estimated to the nearest tenth of a degree), and a comparison with accepted values is shown in Table 2 for martensite and in Table 3 for both transition-iron-carbide phases. The deviations between measured and accepted angles are all within 1° for martensite. For the carbide phase, it was estimated that the \( 11\bar{2} \) g-vector of epsilon-carbide was 1° counterclockwise with respect to the \( 2\bar{1}\bar{1} \) g-vector of martensite, and this information is consistent with data in Table 3.
Data from Tables 1 through 3 were used to index spot patterns [48] from the matrix and precipitate phases, and results are shown in Fig. 7b, c.Footnote 6 From the indexed spot patterns, the zone axis of martensite was determined to be \( [102] \). For epsilon-carbide, it is \( [201] \) or \( [4\bar{2}\bar{2}3] \), and for eta-carbide, it is \( [\bar{1}11] \).
Diffraction at the 22°-tilt Condition
The “as-recorded” selected-area electron diffraction pattern from the 22°-tilt condition is shown in Fig. 8a. A second version of this pattern with labels for a zone of martensite spots and a zone of epsilon-carbide spots is shown in Fig. 8b, and similar data for eta-carbide is provided in Fig. 8c. A two-dimensional grid of solid lines in Fig. 8b represents the martensite phase, and spots are indexed with black numbers of comparatively large font. The dashed lines denote the carbide phase, and spots are indexed with white numbers of smaller font that are contained within gray ovals.

The lengths of important vectors in Fig. 8a were measured, and key data are reported in Table 4. With slightly different lens settings in the transmission electron microscope, the camera constant was determined to be 3.47 nm·mm for this pattern. Differences between calculated and accepted interplanar spacing values for martensite as well as both transition-iron-carbide phases did not exceed the estimated upper limit of ±0.003 nm. Angles were measured, and a comparison with accepted values is shown in Tables 5 and 6. Deviations did not exceed the estimated upper limit of ±2.0o.
Cumulatively, data in Tables 4, 5, and 6 provided the necessary information to index the spot patterns in an unambiguous manner [48], and results are provided in Figs. 8b, c. For martensite, the zone axis is \( [315] \); for epsilon-carbide, it is \( [312] \) or \( [5\bar{1}\bar{4}6] \); and for eta-carbide, it is \( [\bar{1}22] \).
Diffraction Summary
Comparison of measured and accepted values of interplanar spacings (Tables 1 and 4) reveals a mean relative error of 0.6% for eight martensite reflections, 1.2% for eight epsilon-carbide reflections, and 0.6% for eight eta-carbide diffraction spots. For both iron-carbide phases, a maximum difference of 0.003 nm was observed once. Concerning interplanar angles, the mean relative error for six measurements was 1.1% for martensite, and for seven measurements, the mean relative error was 1.5 and 1.3% for epsilon-carbide and eta-carbide, respectively. The maximum uncertainty of 2.0° was observed once for epsilon-carbide. As regards the identity of transition-iron-carbide phase based on this geometrical diffraction data, the agreement is slightly better for eta-carbide.
Since two zone axes were examined, and the correct link between them was established as part of the indexing process, a three-dimensional description of the reciprocal lattice of the transition-iron-carbide phase begins to emerge, which can be compared with the accepted reciprocal lattices of epsilon-carbide and eta-carbide (which seem to be nearly identical). The difference in the visually estimated goniometer settings for the two tilt conditions provides an angle of rotation equal to 14°. From the indexed diffraction patterns, the interzonal angle for the martensite phase is 10.67°. For epsilon-carbide and eta-carbide, the calculated interzonal angles are 17.95° and 17.84°, respectively. The average (for the matrix and one precipitate phase) is 14.3°. The interzonal angles for the transition-iron-carbide phase are assumed to be more accurate than for martensite since their pairs of diffracted intensities for +g hkl and −g hkl are very similar in Figs. 7a and 8a, whereas those for martensite are less so, e.g., notice the significant difference in the intensities of martensite spots \( \bar{2}\bar{1}1 \) and \( 21\bar{1} \) in Fig. 7a.
Diffraction from Magnetite
Thin steel specimens that are electropolished to perforation develop a surface oxide layer that forms very rapidly, and clear evidence of oxide layers on thin-foil transmission electron microscope specimens has been reported [13, 44, 50]. The oxide is nearly always identified as magnetite, Fe3O4. Oxide layers are an impediment to high-quality imaging, and the interpretation of spots in selected-area electron diffraction patterns from phases of interest can be confounded by spots from the oxide. Unfortunately, regions of thin-foil specimens that are very thin (i.e., at the edge of the perforation or the “hole”), which have a comparatively high volume ratio of oxide to steel, oftentimes provide the most visible diffraction spots from precipitates embedded in a dominant matrix phase as well as the best dark-field images. An analysis of some of the prominent oxide diffraction events in Fig. 7 is presented next since these spots are numerous and of reasonably high intensity. The proper interpretation of such spots helps to ensure that key information is not being ignored or misinterpreted.
Figure 9 shows evidence of a single zone of spots from an oxide phase as well as a ring pattern. All evidence is consistent with magnetite, and no other iron oxide phases were detected in this work. The zone axis for the spot pattern (Fig. 9a) from magnetite is \( [\bar{1}12] \); this pattern shows high-intensity spots in a rectangular array that surrounds the transmitted beam. Additionally, there is a row of prominent spots at about a 20° angle to a horizontal axis through the center of the pattern, which includes magnetite spots \( 1\bar{1}1 \), \( 2\bar{2}2 \), \( 3\bar{3}3 \), and \( 4\bar{4}4 \). Other than the rectangular array of oxide spots surrounding the transmitted beam, and the row of spots just described, many remaining spots that come from magnetite do not appear to be part of a periodic array.

Diffraction phenomena from iron oxide layers on the thin-foil steel specimen surfaces. a Spots from a single zone of iron oxide and b rings of diffracted intensity emanating from the transmitted beam (central spot within shadow of the pointer) and one ring centered about the \( 0\bar{2}0 \) martensite spot (owing to double diffraction). Many of the highest intensity iron oxide diffraction events are highlighted, but many of low intensity are not labeled
Other diffraction phenomena from the surface oxide layers are in the form of rings. The two most intense rings from magnetite are highlighted by black circles in Fig. 9b and labeled \( \{ 311\} \) and \( \{ 440\} \). In addition, the highest intensity martensite spots (especially \( 0\bar{2}0 \), \( 020 \), and \( \bar{2}\bar{1}1 \)) provide for significant sources of double diffraction. This statement is best appreciated by examining Fig. 7a that shows three faint-and-sporadic rings surrounding each of these spots. Note that a {311} ring of diffracted intensity surrounds the \( 0\bar{2}0_{\text{M}} \) spot (highlighted by a black circle near the top of Fig. 9b) and a second ring that surrounds the \( 020_{\text{M}} \) spot on the opposite side of the transmitted beam (in this case, without a black circle). Again, the labels are provided in Fig. 9b, whereas an unobstructed view of these features is more easily seen in Fig. 7a.
In some instances, the confounding effects of oxide diffraction are particularly complex. Examination of the \( \bar{1}\bar{3}1 \) oxide spot (labeled in Fig. 9a and seen unobstructed in Fig. 7a) shows an arc of intensity curved toward the transmitted beam as well as a second much-dimmer arc curved toward the \( 0\bar{2}0_{\text{M}} \) spot. This unusual intensity distribution results from a double diffraction effect produced by the \( 0\bar{2}0_{\text{M}} \) and \( 311 \) oxide spots that overlaps with the original \( \bar{1}\bar{3}1 \) oxide spot/ring combination. These and related features can explain many of the additional low-intensity spots throughout this pattern, as well as explaining the lack of an apparent periodicity of the remaining spots. In addition, there are a few additional spots of some significance that will be discussed later in this paper.
Centered-Dark-Field Imaging of Transition-Iron-Carbide Precipitates
The original electron diffraction spot pattern from the 8°-tilt condition (Fig. 7a) initially appears to be rather complex, although the analyses presented clearly show the presence of two-dimensional grids of spots produced by martensite, transition-iron-carbide precipitates, and surface oxide layers. The proper identification of each array of diffraction spots is aided by centered-dark-field imaging, especially in the case of the transition-iron-carbide precipitates. This section addresses this issue.
Figure 10a is a bright-field transmission electron microscope image from the largest lath in Fig. 5a. Figures 5, 6, and 10 have been rotated and positioned within the text so that each image has the same physical orientation. The large circle in Fig. 10a and the circle in Fig. 6 represent the same location within the large lath at “A.” All four parts of Fig. 10 were generated approximately 1 month earlier than Fig. 5. The specimen had been removed and reinserted in the microscope between these two sessions so the crystallographic relationship between the two sets of images (Figs. 5 and 10) was not available. However, because of the distinctive features of these laths and their proximity to the edge of the thin-foil specimen, many microstructural features were easily correlated between Figs. 5 and 10. The diffraction patterns (Figs. 7 and 8) in combination with Fig. 10 provided key quantitative information for this diffraction analysis. Finally, since Fig. 10 represents a small volume of material, Fig. 5 was included to provide a broader view of this portion of the specimen, thereby providing useful context.

Central region of martensite lath “A” in Fig. 5. Transmission electron microscope images: a BF, b CDF, c CDF, and d CDF. Tempered at 200 °C for 1 h. The small circle in each part of Fig. 10 shows the same group of transition-iron-carbide precipitates. The large circle in part (a) shows the region selected for diffraction (the same region circled in Fig. 6)
Figure 10b–d shows centered-dark-field transmission electron microscope images using different diffraction spots. In each case, the same transition-iron-carbide precipitates are illuminated, as exemplified by the small white circles in each image. Upon recording each centered-dark-field image, the x–y beam tilt readings were recorded, diagrams of the diffraction patterns were sketched (including which reflection was positioned on the optic axis), and an electron diffraction pattern was recorded on film along with the shadow of the objective aperture using a double exposure. This approach provided conclusive information as to which diffraction spot was used to produce each CDF image. Useful data are summarized in Fig. 11 and Table 7.

A montage of selected-area electron diffraction patterns at the 8°-tilt condition. Three reflections chosen for centered-dark-field images are highlighted by the shadow of the objective aperture. Labels “10(b)” and “(10(c)” were spots used to create Figs. 10b,c, respectively
Centered-dark-field images shown in Fig. 10b, c were generated at the 8°-tilt condition from spots labeled 10b, c in Fig. 11. Spot 10b is indexed as \( 0\bar{2}1_{\varepsilon } \) or as \( \bar{2}1\bar{2}_{\eta } \), and spot 10c is indexed as \( \bar{1}02_{\varepsilon } \) or as \( 12\bar{1}_{\eta } \), as indicated in Figs. 8 and 7, respectively. The spot labeled “ns” was also used to create a CDF image, but that image is not shown here as it subsequently was identified as a \( 220 \) magnetite reflection.
Comparison of Figs. 7b and 11 shows that spot 10c is included in the two-dimensional grid of indexed spots from the precipitate phase, as expected. Unfortunately, a slight complication arises upon realizing that spot 10b is not part of the same grid of spots. The arrow in Fig. 7b points to this latter spot, and this spot is also present in Fig. 8a. This spot is noticeably more intense in the latter figure, and it is included in the two-dimensional grid of spots from the precipitate phase (Fig. 8b).
Further comparison of Figs. 7 and 8 showed not only one, but several spots are evident in both patterns, i.e., at both tilt conditions. Most, though not all, of these “repeated” spots are from the magnetite phase (e.g., \( 1\bar{1}1 \), \( 2\bar{2}2 \)), and they are close to the horizontal axis (the approximate tilt axis between the two conditions). Specifically, the \( 2\bar{1}\bar{1} \)/\( \bar{2}11 \) pair of martensite spots is evident in both patterns, as is the \( 0\bar{2}1 \)/\( 02\bar{1}_{\varepsilon } \) or \( \bar{2}1\bar{2} \)/\( 2\bar{1}2_{\eta } \) pair. Evidence of the same spots in two spot patterns can be explained in association with three factors. First, the tilt angle between the patterns is only about 15°. Second, the “repeated” spots are aligned along the axis of tilt (or nearly so). Finally, the so-called rel-rod effect commonly occurs for diffraction spots because of the thin nature of the specimen [51, 52].
Figure 10d shows a third CDF image of the same transition-iron-carbide precipitates, although this image was generated at the 22°-tilt condition. In this case, the microscope camera system malfunctioned (unknown to the operator at the time), and a diffraction pattern with a shadow of the objective aperture was not recorded. However, since data were recorded and a schematic diagram drawn, as described above, it was still possible to provide convincing evidence of which reflection was used to produce the CDF image.
Based on the three spots recorded for the first tilt condition, an x–y beam tilt coordinate system was developed and added to Fig. 11. With only three points used to create this system, the uncertainty associated with properly locating spots based on x–y angles of tilt is estimated to be a few millimeters along x- and y-axes. This coordinate system was projected onto Fig. 8a, and the circled “dot” in this figure reveals the estimated position of the diffraction spot used to create Fig. 10d. The circle around this point represents the expected position and size of the objective aperture, indicating that Fig. 10d was produced from the high-intensity spot indexed as either \( \bar{1}11_{\varepsilon } \) or \( 210_{\eta } \).
Stereographic Representation of Diffraction Data
Figure 12 shows data from the indexed diffraction patterns in the form of stereographic projections. In the final stages of preparing Figs. 7 and 8, appropriate planes of a form were chosen so that Fig. 12 could be directly compared with the stereographic projections in Ref. [5].

Experimentally determined stereographic projections for a martensite and eta-carbide and b martensite and epsilon-carbide. Martensite labels include larger non-bold font and open circles. Iron-carbide labels include smaller bold font and filled circles
Hirotsu and Nagakura [5] list three relevant orientation relationships between a transition-iron-carbide phase and martensite, two of which are represented by their stereographic projections. For eta-carbide formed in martensite, they reported a ratio of lattice parameters for the martensite phase equal to c/a = 1.014 and referred to the partially supersaturated martensite as α”. Their orientation relationship was stated as:
and is shown by Fig. 13 of their paper [5]. Next, they listed the orientation relationship for epsilon-carbide in martensite as proposed by Jack [3, 4]:
\( \left[ {11.0} \right]_{\varepsilon } \) 5° from \( \left[ {100} \right]_{\alpha } \)This relationship is consistent with their Fig. 14. In addition, they listed a third similar relationship due to Pitsch and Schrader [53] as:
Comparison of the experimental results reported here (Figs. 7, 8, and 12) with the three “standard” orientation relationships listed above indicates good agreement with the first (Fig. 12a). Specifically, \( (110)_{\eta } // (010)_{\alpha } \) and \( [001]_{\eta } \) is about 1° away from \( [100]_{\alpha } \), which is within the often quoted uncertainty of such data plotted on stereograms.
Comparison of the experimentally determined orientation relationship between martensite and the transition-iron-carbide (now indexed as epsilon-carbide) with data from Jack [4] shows that \( (00.1)_{\varepsilon } \) is about 3° away from \( (011)_{\alpha } \) and \( [11.0]_{\varepsilon } \) is about 1° from \( [100]_{\alpha } \), rather than 5°, as reported by Jack [4]. The axis of rotation between Jack’s orientation relationship and the one reported here does not appear to be a simple one (i.e., of low index).
The orientation relationship reported by Pitsch and Schrader [53] indicates that \( [11.0]_{\varepsilon } //[100] _{\alpha } \), which is within a degree of that shown in Fig. 12b, but \( [\bar{1}1.1]_{\varepsilon } // [010]_{\alpha } \) is not consistent with the experimental one. The experimental relationship indicates that \( (\bar{1}12)_{\varepsilon } //[010] _{\alpha } \). It can be shown that \( (\bar{1}12)_{\varepsilon } // [\bar{5}56 ]_{\varepsilon } \) (within 0.02°). The angle between \( [\bar{5}56]_{\varepsilon } \) and \( [\bar{1}11]_{\varepsilon } \) is calculated to be 5.2°. Thus, the experimentally determined orientation relationship is about 5° away from that reported by Pitsch and Schrader [53] by a rotation about the near-parallel axes of \( \left[ {11.0} \right]_{\varepsilon } \) and \( \left[ {100} \right]_{\alpha } \).
The information presented in Fig. 12 also allows for further analysis of the direction of alignment of the transition-iron-carbide precipitates. To accomplish this task, Fig. 7 would require a counterclockwise rotation of 85° to align this diffraction pattern with the images shown as Fig. 10. Once accomplished, the direction of the linear aggregates of precipitates is nearly halfway between the \( \bar{2}\bar{1}1 \) and \( \bar{2}11 \) pair of martensite spots in Fig. 7b. This direction corresponds with the \( [\bar{2}01] \) martensite direction. As indicated by Fig. 12, the \( [\bar{1}00] \) martensite direction is about 27° directly beneath the plane of Fig. 7. This result supports the earlier claim that linear arrays of transition-iron-carbide precipitates are aligned consistent with <100> martensite directions, a result that has been reported in previous investigations [13, 35, 37, 39–42, 45–47].
Diffracted Intensities of Transition-Iron-Carbide Phases
To augment the information provided already, an attempt was made to address the issue of diffracted intensities. Analysis of intensities of spots in ordinary selected-area electron diffraction patterns is not typically recommended owing to a variety of phenomena, including the “rel-rod” effect [51, 52]. Nonetheless, such an analysis was conducted by Hirotsu and Nagakura [5], where their visual observations correlated well with their calculated values. Unfortunately, they included information for the eta-carbide phase only. Thus, for reflections from transition-iron-carbide precipitates in Figs. 7 and 8, visual estimates of relative diffracted intensity will be compared with calculated values. The latter values are generated from equations for structure factors for idealized crystal structures of both transition-iron-carbide precipitate phases.
Jack’s unit cell [3, 4] for epsilon-carbide can be described as hexagonal, with lattice parameters of a = 0.2752 nm, c = 0.4353 nm, iron atoms at \( 00\frac{1}{4} \) and \( \frac{2}{3}\frac{1}{3}\frac{3}{4} \), and a carbon atom at \( \frac{1}{3}\frac{2}{3}0 \). The structure factor is:
where values of hkl represent Miller indices of diffracting planes, and f Fe and f C are the atomic scattering factors for iron and carbon, respectively.
In the orthorhombic eta-carbide [5], iron atoms occupy equipoint 4 g with x = \( \frac{2}{3} \) and y = \( \frac{1}{4} \), and carbon atoms occupy equipoint 2a for space group P nnm . In this case, the structure factor is:
Notice that both expressions provide the stoichiometry of Fe2C, whereas several authors indicate a composition closer to Fe2.4C [4]. Variations in these expressions consistent with other stoichiometries have been considered by this author, but in this work only these two simple expressions will be examined. Since the unit cell of Jack [3, 4] has half the volume and half the number of atoms of the second cell, the first structure factor is multiplied by two prior to calculating \( \left| F \right|^{2} \).
As listed in the paper by Hirotsu and Nagakura [5], intensities were estimated visually (\( I_{\text{observed}} \)) and then compared with calculated values of the structure factor squared \( \left| {F_{\text{calculated}} } \right|^{2} \). The source of their values for atomic scattering factors appears not to have been provided in the paper. The values used in the present work come from the paper by Doyle and Turner [54] (and include a relativistic correction factor). Values of \( \left| {F_{\text{calculated}} } \right|^{2} \) for eta-carbide were calculated in the present work, and results were typically 20% lower than those reported by Hirotsu and Nagakura, suggesting that the procedure is effectively the same as theirs, but the source of scattering data likely was different than that used here.
Table 8 provides estimates of diffracted intensities for reflections of the transition-iron-carbide phase that were clearly seen within Fig. 7(a), i.e., spots that were not coincident or near-coincident with spots from other phases. The correct planar indices for epsilon-carbide and eta-carbide are both listed. The last two columns provide information from Hirotsu and Nagakura [5]. Table 9 presents similar information from Fig. 8a.
In general, estimated and calculated values in the present work compare fairly well. To provide a basis for comparison, assume that values of \( \left| {F_{hkl} } \right|^{2} \) above about 150 are very strong, between 150 to 100 are strong to moderate, between 100 to 50 are moderate to weak, and below 50 are weak or very weak. Of sixteen comparisons in Tables 8 and 9, thirteen are consistent with this approximate scale. Finally, the results for eta-carbide calculated here compare favorably (about 20% lower) with those calculated by Hirotsu and Nagakura [5].
Evidence of Cementite
The feature labeled “θ” in Figs. 10a, c–d, at a substantial angle with respect to the linear arrays of transition-iron-carbide precipitates, is a well-defined plate of cementite. Analysis suggests that the plate has a habit plane consistent with <110> martensite. More importantly, several spots in Fig. 7a and similar spots in Fig. 8a can be shown to be consistent with the cementite phase.
The \( \bar{1}\bar{3}1 \) magnetite spot (its identity indicated by Fig. 9) is in close proximity to cementite spots (best seen in Fig. 7a) where one spot is about 5 mm away (above and to the left) and a second spot is about 2 mm above and to the right. The first spot is consistent with an interplanar spacing value of 0.190 nm and is likely a \( 113 \) cementite reflection. The second spot has an interplanar spacing of 0.241 nm, likely making it a \( 112 \) cementite spot. The angle between these two reflections is 10.2°, while the accepted angle for cementite is 11.3°. Consistent with these measurements, \( {\mathbf{g}}_{001} \) of cementite is nearly parallel to \( {\mathbf{g}}_{{2\bar{1}\bar{1}}} \) of martensite, and \( {\mathbf{g}}_{110} \) of cementite is nearly parallel to \( {\mathbf{g}}_{{\bar{2}\bar{5}1}} \) of martensite. The resultant orientation relationship between cementite and martensite is within a few degrees of that due to Bagaryatskii [55, 56].
With this information in mind, the spot 4 mm to the left of spot “10(c) in Fig. 11 is indexed as \( \bar{1}\bar{1}\bar{2} \) θ. Spot “10(c)” has been indexed as either the \( \bar{1}02 \) ε or \( 12\bar{1}_{\eta } \) spot. The close proximity of these spots reveals why the cementite plate, labeled “θ”, is illuminated in Fig. 10c. Similarly, other cementite spots are present in these spot patterns, resulting in illumination of both iron-carbide phases, as shown vividly by Fig. 10d and even faintly in Fig. 10b.
Other regions within the thin-foil specimen showed evidence of several adjacent cementite plates, but in general the transition-iron-carbide precipitates were present in a much greater quantity. It is unclear whether cementite formed during tempering at 200 °C, or whether it formed upon quenching, i.e., autotempering.
Summary
A two-tilt crystallographic analysis of fine-scale transition-iron-carbide precipitates formed in martensite of 4340 steel that had been tempered at 200 °C for 1 h was completed. Bright-field and centered-dark-field imaging in a transmission electron microscope were used to complement a detailed selected-area electron diffraction analysis of patterns from two tilt conditions. Key information and conclusions are as follows:
-
1.
Transmission electron microscope imaging revealed approximately linear arrays of transition-iron-carbide precipitates measuring up to 200 nm in length, 10–15 nm in width, with individual precipitates about 5–15 nm in size. Linear arrays were aligned in a direction consistent with <100> martensite.
-
2.
Measured interplanar spacing values and interplanar angles compared well with accepted values for martensite, epsilon-carbide, and eta-carbide. Of the two iron-carbide phases, the latter compared slightly better with accepted values, but both were within the predicted range of experimental uncertainties.
-
3.
Centered-dark-field images from a small group of transition-iron-carbide precipitates were shown clearly to come from indexed electron diffraction spots produced by specific hkl planes. This information verifies the identity of several transition-iron-carbide diffraction events among many spots from other sources.
-
4.
The relationship between the two tilt conditions was determined precisely and associated with tilt angle of approximately 15°. Interzonal angles between indexed beam directions of the two patterns from both transition-iron-carbide phases compared to within 0.1°, providing additional support for the extremely similar crystallographic nature of epsilon-carbide and eta-carbide.
-
5.
Stereographic projections of experimental data were created and compared with “accepted” orientation relationships reported by Jack (epsilon-carbide) and Hirotsu and Nagakura (eta-carbide). The former relationship deviated from the experimental by nearly 5°, whereas the latter agreed within a degree or two.
-
6.
Structure factors derived from expected atom positions were used to calculate relative diffracted intensities. These values compared favorably with estimated diffracted intensities from selected-area electron diffraction patterns obtained from the transition-iron-carbide precipitates. This analysis indicates nearly identical results for the epsilon-carbide and eta-carbide precipitate phases.
-
7.
The data collected in this study compare favorably with the original crystallographic features (with a few minor adjustments) of epsilon-carbide and eta-carbide, as identified by Jack, and by Hirotsu and Nagakura, respectively. Distinction between these phases based on this and similar data seems unlikely.
-
8.
While transition-iron-carbide precipitates dominate the fine-scale microstructure of this material, the presence of a small amount of cementite was verified as well as a few percent of retained austenite.
Notes
This statement admits some minor changes since its early description circa 1950.
The significant difference is lath size is within the range observed in previous work [16].
Note that each diffraction spot labeled with Miller indices (i.e., hkl values) is associated with a vector that begins at the transmitted beam and ends at the center of the spot. The vector is generally referred to as g hkl , its magnitude is the reciprocal of d hkl , and the scaling factor of the diffraction “image” (which changes as does lens strength) is λL, which is the camera constant.
Microstructural features consistent with lath martensite were ubiquitous within the thin-foil specimens. However, numerous fine-scale transition-iron-carbide precipitates were also evident, and their formation removes a substantial amount of carbon from the previously supersaturated martensite. If 0.05 mass pct of carbon remains in martensite, the difference between a and c lattice parameters of martensite is less than 0.001 nm. Hence, the c/a ratio is presumed to be nearly equal to one, martensite matches body-centered-cubic iron, and [100], [010], and [001] “martensite” directions are indistinguishable.
Cullity [49] states that “the Miller indices of a reflecting plane hkl, written without parentheses, stand for the reflected beam from the plane (hkl).”
References
G. Krauss, Principles of Heat Treatment of Steel (American Society for Metals, Metals Park, OH, 1980), pp. 187–227
C.S. Roberts, B.L. Averbach, M. Cohen, The mechanism and kinetics of the first stage of tempering. Trans. ASM 45, 576–604 (1953)
K.H. Jack, Results of further x-ray structural investigations of the iron-carbon and iron-nitrogen systems and of related interstitial alloys. Acta Cryst. 3, 392–394 (1950)
K.H. Jack, Structural transformations in the tempering of high-carbon martensitic steels. J. Iron Steel Inst. 169, 26–36 (1951)
Y. Hirotsu, S. Nagakura, Crystal structure and morphology of the carbide precipitated from martensitic high carbon steel during the first stage of tempering. Acta Met. 20, 645–655 (1972)
P.V. Morra, A.J. Böttger, E.J. Mittemeijer, Decomposition of iron-based martensite: a kinematic analysis by means of differential scanning calorimetry and dilatometry. J. Therm. Anal. Calor. 64, 905–914 (2001)
M. Perez, C. Sidoroff, A. Vincent, C. Esnouf, Microstructural evolution of martensitic 100Cr6 bearing steel during tempering: from thermoelectric power measurements to the prediction of dimensional changes. Acta Mater. 57, 3170–3181 (2009)
J.H. Jang, I.G. Kim, H.K.D.H. Bhadeshia, ε-carbide in alloy steels: first-principles assessment. Scr. Mater. 63, 121–123 (2010)
A.T.W. Barrow, J.-H. Kang, P.E.J. Rivera-Díaz-del-Castillo, The ε → η → θ transition in 100Cr6 and its effect on mechanical properties. Acta Mater. 60, 2805–2815 (2012)
W. Song, J. von Appen, P. Choi, R. Dronskowski, D. Raabe, W. Bleck, Atomic-scale investigations of ε and θ precipitates in bainite in 100Cr6 bearing steel by atom probe tomography and ab initio calculations. Acta Mater. 61, 7582–7590 (2013)
D.T. Pierce, D.R. Coughlin, D.L. Williamson, K.D. Clarke, A.J. Clarke, J.G. Speer, E. De Moor, Characterization of transition carbides in quench and partitioned steel microstructures by Mössbauer spectroscopy and complementary techniques. Acta Mater. 90, 417–430 (2015)
W.S. Choi, J. Lee, B.C. De Cooman, Internal-friction analysis of dislocation-interstitial carbon interactions in press-hardened 22MnB5 steel. Mat. Sci. Eng. A 639, 439–447 (2015)
S.W. Thompson, Structural characteristics of transition-iron-carbide precipitates formed during the first stage of tempering in 4340 steel. Mater. Charact. 106, 452–462 (2015)
G.C. Capitani, P. Oleynikov, S. Hovmöller, M. Mellini, A practical method to detect and correct for lens distortion in the TEM. Ultramicroscopy 106, 66–74 (2006)
A.R. Marder, G. Krauss, The formation of low-carbon martensite in Fe-C alloys. Trans. ASM 62, 957–964 (1969)
C.A. Apple, R.N. Caron, G. Krauss, Packet microstructure in Fe-0.2 pct C martensite. Metall. Trans. 5, 593–599 (1974)
R.F. Hehemann: The Bainite Transformation. in: Phase Transformations. (American Society for Metals, Metals Park, OH, 1970), pp. 397–432
Report of Committee E-4 on Metallography: Electron microstructure of bainite in steel, Proceedings, ASTM 52:543-596 (1952)
H. Modin, S. Modin, Pearlite and bainite structures in a eutectoid carbon steel. Jernkont. Ann. 139, 480–515 (1955)
D.H. Huang, G. Thomas, Metallography of bainitic transformation in silicon containing steels. Metall. Trans. 8A, 1661–1674 (1977)
G.Y. Lai, On the precipitation of epsilon-carbide in lower bainite. Metall. Trans. 6A, 1469–1471 (1975)
R.W.K. Honeycombe, Steels: Microstructure and Properties (Edward Arnold (Publishers) Ltd and American Society for Metals, Metals Park, OH, 1981), p. 109
H.K.D.H. Bahdeshia, Bainite in Steels (The Institute of Materials, London, 1992), p. 66
G. Spanos, The fine structure and formation mechanism of lower bainite. Metall. Mater. Trans. 25A, 1967–1980 (1994)
B. Hutchinson, J. Hagström, O. Karlsson, D. Lindell, M. Tornberg, F. Lindberg, M. Thuvander, Microstructures and hardness of as-quenched martensites (0.1–0.5%C). Acta Mater. 59, 5845–5858 (2011)
C. Lerchbacher, S. Zinner, H. Leitner, Atom probe study of the carbon distribution in a hardened martensitic hot-work tool steel X38CrMoV5-1. Micron 43, 818–826 (2012)
L. Morsdorf, C.C. Tasan, D. Ponge, D. Raabe, 3D structural and atomic-scale analysis of lath martensite: effect of the transformation sequence. Acta Mat. 95, 366–377 (2015)
G. Krauss, Martensite in steel: strength and structure. Mater. Sci. Eng. A273–275, 40–57 (1999)
B.P.J. Sandvik, C.M. Wayman, Characteristics of lath martensite: part I. Crystallographic and substructural features. Metall. Trans. 14A, 809–822 (1983)
B.V.N. Rao, J.Y. Koo, G. Thomas: in Proceedings Electron Microscopy Society of America, 33rd Annual Meeting, Claitors Publishing, Baton Rouge, p. 30 (1975)
G. Thomas, Retained austenite and tempered martensite embrittlement. Metall. Trans. 9A, 439–450 (1978)
N.C. Law, P.F. Howell, D.V. Edmonds, Structure of lath martensite and occurrence of retained austenite in as-quenched Fe-V-C low-alloy steels. Met. Sci. 13, 507–515 (1979)
D.H. Sherman, S.M. Cross, S. Kim, F. Grandjean, G.J. Long, M.K. Miller, Characterization of the carbon and retained austenite distributions in martensitic medium carbon, high silicon steel. Metall. Mat. Trans. A 38A, 1698–1711 (2007)
M.G.H. Wells, An electron transmission study of the tempering of martensite in an Fe-Ni-C alloy. Acta Met. 12, 389–399 (1964)
E. Tekin, P.M. Kelly, A Study of the Tempering of Steel Using Transmission Electron Microscopy, in Precipitation from Iron-Base Alloys, ed. by G.R. Speich, J.B. Clark (Gordon and Breach, New York, 1965), pp. 173–221
C.J. Barton, The tempering of a low-carbon internally twinned martensite. Acta. Met. 17, 1085–1093 (1969)
S. Murphy, J.A. Whiteman, The precipitation of epsilon-carbide in twinned martensite. Metall. Trans. 1, 843–848 (1970)
K. Shimizu, H. Okamoto, High voltage electron microscopy study of the metastable iron carbide in a eutectoid Fe-C alloy. Trans. JIM 15, 193–199 (1974)
Y. Hirotsu, Y. Itakura, K.C. Su, S. Nagakura, Electron microscopy and diffraction study of the carbide precipitated from martensitic low and high nickel steels at the first stage of tempering. Trans. JIM 17, 503–513 (1976)
D.L. Williamson, K. Nakazawa, G. Krauss, A study of the early stages of tempering of an Fe-1.2 pct alloy. Metall. Trans. A 10A, 1351–1363 (1979)
Y. Tanaka, K. Shimizu, Carbide formation upon tempering at low temperatures in Fe-Mn-C alloys. Trans. Jap. Inst. Met. 22, 779–788 (1981)
K.A. Taylor, G.B. Olson, M. Cohen, J.B. Vander Sande, Carbide precipitation during stage I tempering of Fe-Ni-C martensites. Metall. Trans. 20A, 2749–2765 (1989)
O.N.C. Uwakweh, J.-M.R. Génin, J.-F. Silvain, Electron microscopy study of the aging and first stage of tempering of high-carbon Fe-C martensite. Metall. Trans. 22A, 797–806 (1991)
Y. Ohmori, I. Tamura, Epsilon carbide precipitation during tempering of plain carbon martensite. Metall. Trans. A 23A, 2737–2751 (1992)
S.S. Babu, K. Hono, T. Sakurai, Atom probe field ion microscopy study of the partitioning of substitutional elements during tempering of a low-alloy steel martensite. Metall. Trans. A 25A, 499–508 (1994)
W.J. Nam, C.S. Lee, D.Y. Ban, Mat. Sci. Eng. A289, 8–17 (2000)
A.J. Clarke, M.K. Miller, R.D. Field, D.R. Coughlin, P.J. Gibbs, K.D. Clarke, D.J. Alexander, P.A. Powers, P.A. Papin, G. Krauss, Atomic and nanoscale chemical and structural changes in quenched and tempered 4340 steel. Acta Mater. 77, 17–27 (2014)
J.W. Edington, Practical Electron Microscopy in Materials Science (Van Nostrand Reinhold, New York, 1976), pp. 44–47
B.D. Cullity, Elements of x-ray Diffraction, 2nd edn. (MA, Addison-Wesley, Reading, 1978), p. 87
S.H. Chen, J.W. Morris, Electron microscopy study of the passivating layer on iron-nickel martensite. Metall. Trans. 8A, 19–26 (1977)
P.B. Hirsch, A. Howie, R.B. Nicholson, D.W. Pashley, M.J. Whelan, Electron Microscopy of Thin Crystals (Butterworths, Washington D.C., 1965), pp. 44–47
D.B. Williams, C.B. Carter, Transmission Electron Microscopy: A Textbook for Materials Science (Plenum Press, New York, 1996), p. 196
W. Pitsch, A. Schrader, Die ausscheidungs form des ε-karbids im ferrit und im martensit beim anlassen. Arch. Eisenhütt. 29, 715–721 (1958)
P.A. Doyle, P.S. Turner, Relativistic Hartree-Fock x-ray and electron scattering factors. Acta Cryst. A24, 390–397 (1968)
K.W. Andrews, D.J. Dyson, S.R. Keown, Interpretation of Electron Diffraction Patterns, 2nd edn. (Plenum, New York, 1971), p. 195
Y.A. Bagaryatsi, Veroyatnue mechanezm raspada martenseeta (Likely mechanism for the tempering of martensite). Dokl. Akad. Nauk SSSR 73, 1161–1164 (1950)
Acknowledgments
Support from the Advanced Steel Processing and Products Research Center is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Thompson, S.W. A Two-Tilt Analysis of Electron Diffraction Patterns from Transition-Iron-Carbide Precipitates Formed During Tempering of 4340 Steel. Metallogr. Microstruct. Anal. 5, 367–383 (2016). https://doi.org/10.1007/s13632-016-0302-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13632-016-0302-0