
Abstract
We have previously shown that liquid microjunction surface sampling of dried blood spots coupled with high resolution top-down mass spectrometry may be used for screening of common hemoglobin variants HbS, HbC, and HbD. In order to test the robustness of the approach, we have applied the approach to unknown hemoglobin variants. Six neonatal dried blood spot samples that had been identified as variants, but which could not be diagnosed by current screening methods, were analyzed by direct surface sampling top-down mass spectrometry. Both collision-induced dissociation and electron transfer dissociation mass spectrometry were employed. Four of the samples were identified as β-chain variants: two were heterozygous Hb D-Iran, one was heterozygous Hb Headington, and one was heterozygous Hb J-Baltimore. The fifth sample was identified as the α-chain variant heterozygous Hb Phnom Penh. Analysis of the sixth sample suggested that it did not in fact contain a variant. Adoption of the approach in the clinic would require speed in both data collection and interpretation. To address that issue, we have compared manual data analysis with freely available data analysis software (ProsightPTM). The results demonstrate the power of top-down proteomics for hemoglobin variant analysis in newborn samples.
Similar content being viewed by others


1 Introduction
Hemoglobin (Hb) is a tetramer consisting of four polypeptide chains. Normal adult hemoglobin (HbA), dominant in the blood from 3 to 6 months after birth, comprises two α and two β globin chains (α2β2) of 141 and 146 amino acids residues, respectively, with a total molecular weight of ~68,000 Da. The fetus’ reliance on an oxygen supply from the mother accounts for the presence of the higher oxygen affinity fetal hemoglobin (HbF) α2γ2 in newborns. The γ chains are homologous to the β chain, differing by 39 out of 146 residues [1]. Hemoglobinopathies, variants of Hb, are the most common single gene recessive disorder with approximately 269 million carriers worldwide [2]. Hemoglobinopathies are classified into two types: thalassemias, which are characterized by a reduction in the synthesis of the globin chains, and structural hemoglobin variants which are caused by a point mutation in the globin gene typically leading to a single amino acid substitution in the globin chain. There are over 1000 Hb variants classified [3] and in the majority of cases the variants are not clinically significant. The most well-known clinically significant structural Hb variant is the sickle variant (HbS) (β6 Glu→Val Δm –29.97 Da) [4]. Whereas heterozygous inheritance of the sickle variant (HbAS) results in mild complications, the phenotypic consequence of inheriting the HbS variant in homozygous form is sickle cell disease (SCD), the symptoms of which are severe [5]. Patients with SCD experience reduced lifespans [6]. Rapid diagnosis during newborn blood screening programs, which allows for the implementation of suitable healthcare strategies, has been credited with the ability to virtually eradicate childhood mortality in SCD and SCD-related disorders [7, 8]. Newborn screening programs typically also screen for the structural variants HbC (β6 Glu→Lys Δm–0.9476 Da), HbD (β121 Glu→Gln Δm –0.9840 Da), HbE (β26 Glu→Lys Δm –0.9476 Da), and HbO-Arab (β121 Glu→Lys Δm –0.9476 Da). In the heterozygous or homozygous state these variants are benign but become clinically significant when co-inherited with HbS, resulting in sickling disorders [9, 10]. In addition, screening may detect the presence of other variants that cannot be identified. Generally, these variants will be benign but if the variant is dominant, it will be clinically significant in the heterozygous state. Additionally, these variants may be significant if co-inherited with HbS.
In the UK, the newborn screening program screens ~700,000 neonates each year for five inherited disorders [11]. Current screening methods for hemoglobinopathies involve collection of blood samples via heel prick onto standard NHS dried blood spot (DBS) cards from neonates between 5 and 8 days old. Samples are left to dry at room temperature before being sent to a newborn screening laboratory. The first step in the screening process involves punching out small discs (~3 mm in diameter) from the dried blood card and eluting the sample for around 30 min [12]. The resolubilized samples are analyzed by cation exchange high-performance liquid chromatography (ceHPLC) and/or isoelectric focusing (IEF) [13]. Both methods involve lengthy sample preparation and are presumptive rather than absolute (i.e., diagnosis of Hb variants is not possible by these methods). Absolute determination of an Hb variant can only be determined by second-line testing, via DNA analysis (sequencing) or mass spectrometry [14], carried out in a separate laboratory.
Mass spectrometry-based approaches for identifying hemoglobin variants have been developed both for whole blood and for samples resolubilized from dried blood spots [15–21]. Typically, mass analysis of tryptic peptides of the hemoglobin is performed. That is, a bottom-up approach is utilized. For example, Daniel et al. developed a multiple reaction monitoring method for the analysis of tryptic digests of whole blood and latterly to blood eluted from dried blood spots [15, 18]. An alternative approach involves mass analysis of intact globin chains followed by MS/MS of tryptic peptides [20, 21] .
In contrast, the top-down approach [22, 23], in which intact proteins are analyzed, negates the need for lengthy proteolysis. Top-down mass spectrometry also addresses the problems of ‘information loss’ inherent in bottom-up approaches [24]. That is, there is no loss of regions of the protein due to poor ionization, or connectivity between substitutions and/or modifications, or confusion due to the presence of isomeric peptides. We contend, therefore, in terms of identifying unknown hemoglobin variants, top-down analysis of the globin chains is the most suitable approach.
We recently demonstrated that liquid microjunction (LMJ) surface sampling [25] of newborn dried blood spots coupled with high resolution mass spectrometry can be applied to the screening of Hb variants, HbS, HbC, and HbD [26]. The method involves no sample preparation and the results are unambiguous. Here, we optimize the approach to maximize protein sequence coverage following collision-induced dissociation (CID) and electron transfer dissociation (ETD) mass spectrometry with the aim of diagnosing unknown variants. The method was applied to six variant samples, which could not be diagnosed by the standard screening methods. Five of the six were diagnosed unambiguously. The sixth was either misassigned as a variant by the standard screening methods, or was an isomeric substitution (Ile→Leu or vice versa).
Our results demonstrate the robustness of the direct surface sampling top-down proteomics approach and confirm that the approach is applicable both for screening and diagnosis. All data were analyzed manually, with the attendant time costs, constituting a bottle-neck in the workflow and highlighting the need for data analysis software specifically for the diagnosis of Hb variants from top-down MS/MS data. Therefore, the freely available web-based top-down protein characterization software Prosight PTM [27] was investigated as an alternative to manual data analysis.
2 Experimental
2.1 Materials
Normal adult (HbA) DBS specimens were collected via finger prick on to standard NHS blood spot (Guthrie) cards, Ahlstrom grade 226 filter paper (ID Biological Systems, Greenville, SC, USA). Anonymized dried blood spots from normal neonates (FA), and unknown variants (FAV), acquired from newborn patients between 5 and 8 days, were supplied by Birmingham Children’s Hospital in accordance with the Code of Practice for the Retention and Storage of Residual Spots [28]. All samples have previously undergone analysis by the standard NHS screening protocols using ceHPLC and IEF. For the FAV samples, screening suggested the presence of an Hb variant but could not determine its identity. Samples were stored at -20 °C, with 1 g desiccant packets (Whatman International Ltd., Kent, UK). The electrospray solution consisted of methanol (J.T. Baker, Deventer, The Netherlands) and water (J.T Baker) (48.5:48.5), with 3 % formic acid (Sigma-Aldrich Company Ltd., Dorset, UK).
2.2 Surface Sampling
DBS were placed onto the Advion LESA (Liquid Extraction Surface Analysis) universal plate adapter (Advion, Ithaca, NY, USA) and an image of the DBS was acquired using an Epson Perfection V300 photo scanner. Using the LESA Points software (Advion), the precise location of the DBS to be sampled was selected using the scanned image. The universal plate adapter was placed into the Triversa Nanomate chip-based electrospray device (Advion, Ithaca, NY, USA), coupled to the ThermoFisher Scientific Orbitrap Velos ETD (ThermoFisher Scientific, Bremen, Germany).
Sample analysis was performed using the LESA feature of the Triversa Neonate ChipSoft Manager software. This platform was used to set the parameters for the surface sampling routine based on robotic arm movements (X,Y,Z) of the Nanomate probe. The routine begins with the Nanomate probe collecting a conductive tip, which is used for sample recovery and delivery, from the tip rack. The tip is then moved to solvent well containing the electrospray solution and collects 7 μL of the solution. The robotic arm relocates to a predetermined position above the surface of the DBS and the tip is lowered towards the surface of the DBS leaving a gap of 1.6 mm between the sample surface and tip. Six μL of the electrospray solution is dispensed onto the DBS forming the sample surface to tip liquid microjunction (LMJ). The LMJ is maintained for 5 s, and then 5 μL is reaspirated and introduced via nanospray to the mass spectrometer.
2.3 Mass Spectrometry
The sample was introduced at flow rate of ~80 nL/min, with a gas pressure of 0.5 psi, a tip voltage of 1.75 kV and a capillary temperature of 250 °C. MS data was collected in selected ion monitoring (SIM) mode (m/z 1055–1090) at a resolution of 100,000 at m/z 400 in the Orbitrap. Each scan comprises 30 co-added microscans. SIM-mode mass spectra shown comprise between 1 and 3 scans (acquired for ~2 min). Automatic gain control (AGC) target was 1 × 106 with a maximum fill time of 2 s. CID was performed in the ion trap and the fragment ions detected in the orbitrap with a resolution of 100,000 at m/z 400. The isolation width was 8–10 Th. The AGC target for CID was 1 × 106 with a maximum fill time of 2 s and CID experiments were performed with helium gas at normalized collision energy between 20 % and 40 % as discussed in the text. Each CID scan comprises 30 co-added microscans. CID MS/MS spectra shown comprise 3 co-added scans (acquired for ~3 min). ETD was performed in the ion trap and the fragments detected in the orbitrap with a resolution of 100,000 at m/z 400. The isolation width was 10 Th. The AGC target for ETD was 1 × 106 with a maximum fill time of 2 s. The reagent ion (fluoranthene) AGC target was 1 × 105 with a maximum fill time of 1 s. ETD activation time was 20 ms with a supplemental activation energy of 20 %. Each ETD scan comprises 30 co-added microscans. ETD MS/MS spectra shown comprise ~3 co-added scans (acquired for ~3 min).
Data were analyzed using Xcalibur 2.10 software (ThermoFisher Scientific). For the SIM mode mass spectra, the Xtract program was used to calculate monoisotopic masses (44 % fit factor, 25 % remainder, S/N threshold 2.1). CID and ETD MS/MS spectra were analyzed manually resulting in de novo identification of the site and nature of amino acid substitution/insertion. That information was manually searched against the HbVar database (http://globin.bx.psu.edu/hbvar/menu.html) enabling Hb variant identification. Experimentally measured fragment m/z values were compared with theoretical m/z values for the variants. Theoretical m/z values for comparison were calculated in Protein Prospector (http://prospector.ucsf.edu/prospector/mshome.htm). Manual fragment assignments for all the hemoglobin species studied here are given in Supplemental File 1. For data analysis using ProSightPTM 1.0, CID and ETD MS/MS spectra were deconvoluted using the Xtract program (44 % fit factor, 25 % remainder, S/N threshold 2.1). Xtracted fragment masses were searched against the (manually determined) globin/variant sequence, mass error = 10 ppm, in ProSightPTM 1.0.
3 Results
3.1 Protein Sequence Coverage
Previously we showed, in the analysis of a neonatal heterozygous HbS dried blood spot (DBS) sample, that collision-induced dissociation of [M + 15H]15+ ions resulted in a sequence coverage of 39 % for the β-chain and 32 % for the HbS variant [26]. Clearly, top-down diagnosis of unknown variants requires the maximum sequence coverage possible. CID of [M + 15H]15+ ions of the β-chain from normal adult routinely gives a mean sequence coverage of 44 %, however we found that reducing the normalized collision energy by 5 % (from 35 % to 30 %) dramatically improves sequence coverage to a highly reproducible 68 %, see Supplemental Figure 1. If electron transfer dissociation (ETD) is performed the combined sequence coverage (CID + ETD) is 81 %. Neonate DBS samples analyzed with the optimized CID parameters gave average protein sequence coverage of 63 %. This slightly lower value may be due to the lower abundance of β chain in neonate samples. (HbA comprises 25 % of the total Hb content [13]).
3.2 Diagnosis of Unknown Variants
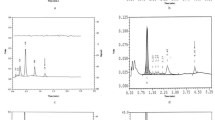
Six unknown neonate samples (FAV1-6) were analyzed by LMJ surface sampling. The samples had been identified as variants by ceHPLC and IEF; however, the nature of the variant had not been diagnosed. Figure 1a shows the SIM-mode mass spectrum obtained from sample FAV1. At first glance, the mass spectrum appears identical to that expected for a normal neonatal sample [26]. Peaks corresponding to [M + 14H]14+ ions of the α-chain and [M + 15H]15+ ions of the γ-chains (γA and γG) are present. However, the peak at m/z 1058 corresponds to a measured mass MWmeas 15856.3072 Da, suggesting the presence of a variant with a -1 Da mass shift. (The calculated mass of the β-chain is 15857.2497 Da). The ions centered at m/z 1058 were selected and subjected to CID. The resulting fragments were compared with those observed for CID of [M + 15H]15+ ions of the β-chain. The fragment peaks observed in the CID mass spectrum of FAV1 and their assignments are shown in Supplemental Table 1. The sample was identified as heterozygous Hb D-Iran. Hb D-Iran is caused by an amino acid substitution a position 22 on the β chain, glutamic acid to glutamine, causing a mass shift Δm 0.9840 Da [29]. Figure 1b shows an expanded m/z region containing the b 3+21 fragment ions of the β-chain (monoisotopic m/z calc 759.0742) (top) and the equivalent region for the variant (bottom). Figure 1c shows the expanded m/z region containing the b 3+22 fragment ions of the β-chain (monoisotopic m/z calc 802.0884) (top) and the equivalent region for the variant (bottom). Whereas the regions containing the b21 fragments are identical, the regions containing the b22 fragments are not. Two sets of peaks are observed for the variant sample: One set corresponds to the isotope distribution of the b 3+22 fragment of the β-chain (monoisotopic m/z meas 802.0865), confirming the heterozygous nature of the variant. The second set corresponds to the b 3+22 fragment of Hb D-Iran (m/z meas 801.7623, m/z calc 801.7604, Δ 2.4 ppm). Similar observations are made for the b 3+23 fragment ions shown in Figure 1d. The protein sequence coverage obtained was 54 %, see Figure 1e. It should be noted, however, that as this sample is heterozygous and the mass shift is <1 Da, both the normal β-chain and the variant ions were selected for CID, an unavoidable consequence of instrumental resolution limitations and overlapping isotopic distributions. Fifteen of the observed fragments are unique to Hb D-Iran, whereas the remaining 62 are common to both.

(a) Selected ion monitoring mode mass spectrum of unknown variant FAV1. Expanded m/z region showing peaks corresponding to the isotopic distributions of (b) b 3+21 fragments (m/z calc 759.0742) observed following CID of 15+ ions of β-chain (top) and variant Hb D-Iran (bottom); (c) b 3+22 fragments observed following CID of 15+ ions of β chain (m/z calc 802.0884) (top) and variant Hb D-Iran (m/z calc 801.7604) (bottom); (d) b 3+23 fragments following CID of 15+ ions β chain ions (m/z calc 835.1112) (top) and Hb D-Iran variant ions (m/z calc 834.7832) (bottom). (e) Protein sequence coverage obtained following CID of the Hb D-Iran variant (54 %)
Clearly, manual analysis of top-down mass spectrometry data for the diagnosis of hemoglobin variants is time-consuming and is a potential barrier to adoption in the clinical laboratory. Ideally, data collection would be followed by automated searching against a hemoglobin variant database. To test that approach, we searched the CID MS/MS data for FAV1 against the Hb D-Iran sequence using ProSightPTM 1.0. The aim was to mimic a search against a dedicated Hb variant database. The protein sequence coverage for the Hb D-Iran variant chain generated using ProSightPTM was 43 % (see Supplemental Figure 2a). The source of the discrepancy in sequence coverage was investigated further. For example, fragment ions b 4+32 and b 4+33 were assigned manually but were not identified by ProSightPTM (see Supplemental Figure 2b and c). For these fragments, the Xtract program had failed to distinguish the presence of two overlapping isotope distributions, one from the β-chain and one from the Hb D-Iran variant. Despite the difference in sequence coverage, the site of the substitution was identified and the variant could be unequivocally diagnosed.
A second unknown variant, FAV2, was also identified as heterozygous Hb D-Iran. Again, because of the small mass difference, both the variant and the β-chain were selected for CID. The fragment peaks observed in the CID mass spectrum of FAV2 and their assignments are shown in Supplemental Table 2. The protein sequence coverage obtained was 57 %. Again, the data were searched against the Hb D-Iran sequence using ProSightPTM and gave a coverage of 56 %, and unequivocal diagnosis. A study reviewing screening results over a 10 year period in the North Thames health region showed that Hb D variants are the third most common variants with a carrier incidence rate of 1 in 631. Hb D-Iran is the second most prevalent of the Hb D variants after Hb D Punjab (Los Angeles) [12]. Heterozygous Hb D-Iran is clinically benign.
Figure 2a shows the SIM-mode mass spectrum obtained for variant FAV3. In addition to the peaks corresponding to the α, β, and γ chains, a peak is observed at m/z 1063 (MWmeas 15926.24 Da) which corresponds to a β-chain mass shift Δm + 69.06 Da (i.e., substitution of a serine for an arginine residue). The presence of both the β-chain and the variant confirm the sample as heterozygous. The variant ions were isolated and fragmented by CID and ETD. The fragment peaks observed in the CID mass spectrum and their assignments are shown in Supplemental Table 3. The β-chain contains five serine residues: Ser9, Ser44, Ser49, Ser72, Ser89. Two Ser→Arg variants are known: Hb Headington (Ser72→Arg) and Hb Vanderbilt (Ser89→Arg). The protein sequence coverage observed following CID was 46 % and is summarised in Figure 2b. (Note, unlike Hb D-Iran, there is no interference from β-chain fragments as the larger mass shift makes it possible to fully isolate the variant precursor ions). Potential substitution of Ser9, Ser44 and Ser49 are ruled out by the presence of the b11 through b49 fragments at the expected masses. The nature of the variant (Hb Vanderbilt or Hb Headington) could not be confirmed by the CID data due to the lack of fragments in the central region of the protein. The protein sequence coverage observed following ETD was 34 %, see Figure 2c. The fragment peaks and their assignments are summarised in Supplemental Table 4. Hb Vanderbilt is ruled out by the presence of fragments z61-63, z67-70 and z74 at the expected masses. The variant is confirmed as Hb Headington [30] by the presence of the c + H72 6+ fragment (m/zmeas 1309.0297, m/z calc 1309.0339, Δ 3.2 ppm) and the z 7+77 fragment (m/z meas 1208.7861, m/z calc 1208.7754, Δ 8.9 ppm), see Figure 2d and e. The combined protein sequence coverage (CID + ETD) was 63 %. The ETD data were searched against the Hb Headington sequence using ProSightPTM, which returned a coverage of 20 %. The variant was assigned on the basis of the c 6+77 fragment ion. That fragment was not assigned in the manual analysis due to the very low signal-to-noise of the monoisotopic peak.

(a) Selected ion monitoring mode mass spectrum of an unknown variant FAV3. Protein sequence coverage observed following (b) CID (46 %) and (c) ETD (34 %) of V3. Site of substitution (Ser72→Arg) is shown in both but can only be confirmed by ETD. * Indicates fragments unique to ETD. Expanded m/z region showing peaks corresponding to (d) c + H 6+72 (m/z calc 1309.0339) and (e) z 7+77 (m/z calc 1208.7754) fragment ions following ETD of 15+ ions of V3 and the equivalent regions (above) from the ETD of 15+ ions of β chain
Figure 3a shows the SIM-mode mass spectrum obtained for unknown variant FAV4. An additional peak is observed at m/z 1062, corresponding to a β-chain variant with mass shift Δm +58.01 Da (MWmeas 15915.27). A mass shift of +58.01 Da could either result from a Gly→Asp substitution or an Ala→Glu substitution. The β-chain contains thirteen glycine residues and fifteen alanine residues, offering numerous possibilities for variation. The ions at m/z 1062 were isolated and subjected to CID. The fragment peaks observed in the CID mass spectrum and their assignments are shown in Supplemental Table 5. The protein sequence coverage, see Figure 3b, was 28 %, the lowest of all the variants studied. Nevertheless, the fragmentation observed narrows down the region of substitution to between Gly16 and Glu22: The b 2+15 fragment is observed at the expected mass m/z meas 831.9470 (m/z calc 831.9461, Δ 1.1 ppm) while the b 3+22 fragment is observed with the +58.01 mass shift, m/z meas 821.4248 (m/z calc 821.4236 Δ –1.7 ppm), see Figure 3c and d. The only glycine or alanine residue in this region is Gly16. Thus the variant corresponds to a Gly16→Asp substitution (i.e., Hb J-Baltimore) [31]. The ProsightPTM search of the data against the Hb J-Baltimore sequence gave a sequence coverage of 19 %. Again the reduced coverage appears to be the result of incomplete deconvolution by the Xtract program. Nevertheless, the variant could be diagnosed unambiguously.

(a) Selected ion monitoring mode mass spectra spectrum of an unknown variant FAV4. (b) Protein sequence coverage observed following CID of V4 (Hb J-Baltimore), 28 %. Expanded m/z region showing peaks corresponding to isotopic distributions of (c) b 4+15 fragments (m/z calc 831.9461) observed following CID of 15+ ions of β-chain (top) and variant Hb J-Baltimore (bottom); (d) b 3+22 fragments following CID of 15+ ions of variant Hb J-Baltimore (m/z calc 821.4248), (bottom) and the equivalent m/z region from the CID mass spectrum of 15+ ions of β-chain (top)
The SIM-mode mass spectrum obtained for the variant FAV5 is shown in Figure 4a. Peaks corresponding to 15+ ions of β and γ chains and 14+ ions of α-chains are observed, confirming the variant is heterozygous. In addition, a peak corresponding to 14+ ions was observed at m/z 1090 suggesting the presence of an α-chain variant. β variants are more common than α variants: ~60 % are β variants [3]. In addition, β variants are more abundant because only two β globin chains are inherited (one per chromosome) by an individual compared with four α globin chains, therefore inheritance of an β variant makes up 50 % of the Hb content whereas an α variant would be 25 % [32, 33]. The measured mass of the α-chain was MWmeas 15116.95 Da (MWcalc 15116.89 Da) and the measured mass of the variant was 15229.88 Da, a mass shift Δm of +113 Da. That mass shift does not correspond to any possible amino acid substitution, however it does correspond to insertion of an isoleucine or leucine residue within the sequence. The α-chain and the variant were each selected for CID. The fragments observed for the α-chain are shown in Supplemental Table 6. The α-chain sequence coverage was 54 %. The CID fragments observed for the variant are shown in Supplemental Table 7. The variant was identified as Hb Phnom Penh which arises through insertion of an isoleucine between position 117 and 118 on the α-chain coded by the α 1 gene [34]. The protein sequence coverage was 46 % and is summarised in Figure 4b. Fragments y3 through y24 were observed at the expected masses. Figure 4c shows an expanded m/z region containing the y 3+25 of the Hb Phnom Penh variant (m/z meas 902.1746, m/z calc 902.1726 Δ 2.2 ppm) (i.e., the fragment adjacent to the insertion, and the corresponding region from the CID of the α-chain). Another example, the y 4+48 fragment (m/z meas 1305.4803, m/z calc 1305.4768 Δ 2.7 ppm), is shown in Figure 4d. Bakhtiar et al. [35] showed that sequence coverage of the α-chain can be improved by fragmentation of multiple charge states, and that may also be the case here. Nevertheless the coverage obtained for the 14+ charge state was more than sufficient to identify the variant. The ProsightPTM search of the data against the Hb Phnom Penh sequence gave a sequence coverage of 44 % and unequivocal diagnosis.

(a) Selected ion monitoring mode mass spectrum of an unknown variant FAV5. (b) Protein sequence coverage observed following CID of V5 (Hb Phnom Penh), 46 %. Expanded m/z regions showing peaks corresponding to isotopic distributions of (c) y 3+25 fragments (m/z calc 902.1726) and (d) y 4+48 fragments (m/z calc 1305.4768) observed following CID of 14+ ions of variant V5 (Hb Phnom Penh) (bottom) and the equivalent m/z regions from the CID mass spectrum of 14+ ions of α-chain (top)
The SIM-mode mass spectrum for the final unknown variant FAV6 is shown in Supplemental Figure 3. The mass spectrum does not reveal the presence of any additional peaks. The measured masses of the α, β, Gγ and Aγ chains were MWmeas 15117.8211 (MWcalc 15117.8924 Δ -4.7 ppm), MWmeas 15858.3217 (MWcalc 15858.2570 Δ 4.1 ppm), MWmeas 15986.3329 (MWcalc 15986.2626 Δ 4.4 ppm), MWmeas 16000.2849 (MWcalc 16000.2782 Δ 0.4 ppm), respectively. We therefore performed CID of the peaks centered at m/z 1059 (β-chains), m/z 1068 (γ-chains) and m/z 1081 (α-chains). The CID protein sequence coverages were 46 % (β-chain), 45 % (α-chain), 43 % (Gγ-chain) and 32 % (Aγ-chain). Fragments observed are detailed in Supplemental Tables 8–11.The ETD protein sequence coverages were 17 % (β-chain), 44 % (α-chain), 28 % (Gγ-chain) and 15 % (Aγ-chain). Fragments observed are detailed in Supplemental Tables 12–15. No erroneous fragment peaks were observed for any of the globin chains suggesting that either the sample had been incorrectly identified as a variant in the screening process or contains an isomeric substitution (i.e., Leu→Ile or vice versa). (It is worth noting that no such substitution is currently described in the HbVar database [3].) It was not possible to reconfirm the screening results because the sample was anonymized prior to mass spectrometry analysis. The estimated false positive rate in screening is ~1 % [36] and documented sources of error include administration errors or unfocused/ merged bands in IEF [37].
4 Discussion
We have shown that direct surface sampling of newborn dried blood spots coupled with top down mass spectrometry may be used for the diagnosis of unknown hemoglobin variants. The identified variants are summarised in Table 1. Six samples which had been identified as variants by ceHPLC and IEF, but which could not be diagnosed by those methods, were analyzed. Two of the samples were unambiguously determined to be heterozygous Hb D-Iran following CID of the overlapping β-chain and variant peaks. Mass analysis of the third sample revealed the presence of a Ser→Arg β-chain variant. CID of the variant narrowed the possibilities to Hb Headington or Hb Vanderbilt. ETD confirmed the variant as Hb Headington. Mass analysis of the fourth variant suggested either a Gly→Asp or Ala→Gln β-chain substitution. CID confirmed the variant as Hb J-Baltimore. The fifth sample was confirmed as a rare α-variant with a mass shift of +113 Da. CID revealed it to be the insertion variant Hb Phnom Penh. The mass analysis, CID and ETD of the sixth sample do not indicate the presence of a variant. Either the sample had been incorrectly identified as a variant by the ceHPLC and IEF (the false positive rate in screening is estimated as ~1 % [36]) or contained an isomeric substitution (Ile→Leu or vice versa).
We postulate that the top-down proteomics approach could be employed within a newborn screening program, combining screening and diagnosis on a single platform. Efficient application of the approach would require improvements to Xtract program and the use or development of software capable of searching top-down MS/MS data (CID, ETD) against a dedicated hemoglobin variant database. We have used ProSightPTM to search against specific variant sequences to illustrate such an approach. In all cases, the automated search identified the unique fragment ions, allowing variant diagnosis, albeit with reduced overall protein sequence coverage.
References
Adachi, K., Zhao, Y., Yamaguchi, T., Surrey, S.: Assembly of γ- with α-globin chains to form human fetal hemoglobin in vitro and in vivo. J. Biochem. 275, 12424–12429 (2000)
Trent, R.J.A.: Diagnosis of the hemoglobinopathies. Clin. Biochem. Rev. 27, 27–38 (2006)
Giardine, B., van Baal, S., Kaimakis, P., Riemer, C., Miller, W., Samara, M., Kollia, P., Anagnou, N.P., Chui, D.H.K., Wajcman, H., Hardison, R.C., Patrinos, G.P.: HbVar database of human hemoglobin variants and thalassemia mutations: 2007 update. Hum. Mutat 28, 1–10 (2007)
Ingram, V.M.: Abnormal human hemoglobins. III the chemical difference between normal and sickle hemoglobins. Biochim. Biophys. Acta 36, 402–411 (1959)
Embury, S.H.: The clinical pathophysiology of sickle cell disease. Annu. Rev. Med. 37, 361–376 (1986)
Scott, R.B.: Health care priority and sickle cell anemia. J. Am. Med. Assoc. 214, 731–734 (1970)
Davis, H., Schoendorf, K.C., Gergen, P.J., Moore, R.M.: National trends in the mortality of children with sickle cell disease, 1968 throught 1992. Am. J. Public Health 87, 1317–1322 (1997)
Telfer, P., Coen, P., Chakravorty, S., Wilkey, O., Evans, J., Newell, H., Smalling, B., Amos, R., Stephens, A., Rogers, D., Kirkham, F.: Clinical outcomes in children with sickle cell disease living in England: A neonatal cohort in East London. Haematologica 92(7), 905–912 (2007)
Black, A.J., Condon, P.I., Gompels, B.M., Green, R.L., Huntsman, R.G., Jenkins, G.C.: Sickle-cell hemoglobin C disease in London. J. Clin. Pathol. 25, 49–55 (1972)
River, G.L., Robbins, A.B., Schwartz, S.O.: S-C hemoglobin: A clinical study. Blood 18, 385–416 (1961)
Streetly, A., Latinovic, R., Hall, K., Henthorn, J.: Implementation of universal newborn bloodspot screening for sickle cell disease and other clinically significant hemoglobinopathies in England: Screening results for 2005–2007. J. Clin. Pathol. 62(1), 26–30 (2009)
Almeida, A.M., Henthorn, J.S., Davies, S.C.: Neonatal screening for haemoglobinopathies: The results of a 10-year programme in an English Health Region. Br. J. Haematol. 112(1), 32–35 (2001)
Ryan, K., Bain, B.J., Worthington, D., James, J., Plews, D., Mason, A., Roper, D., Rees, D.C., De La Salle, B., Streetly, A.: Significant haemoglobinopathies: Guidelines for screening and diagnosis. Br. J. Haematol. 149(1), 35–49 (2010)
Old, J.M.: Screening and genetic diagnosis of hemoglobin disorders. Blood Rev. 17(1), 43–53 (2003)
Daniel, Y.A., Turner, C., Haynes, R.M., Hunt, B.J., Dalton, R.N.: Rapid and specific detection of clinically significant haemoglobinopathies using electrospray mass spectrometry–mass spectrometry. Br. J. Haematol. 130(4), 635–643 (2005)
Boemer, F., Cornet, Y., Libioulle, C., Segers, K., Bours, V., Schoos, R.: 3-years experience review of neonatal screening for hemoglobin disorders using tandem mass spectrometry. Clin. Chim. Acta 412(15/16), 1476–1479 (2011)
Williams, J.P., Creese, A.J., Roper, D.R., Green, B.N., Cooper, H.J.: Hot electron capture dissociation distinguishes leucine from isoleucine in a novel hemoglobin variant, Hb askew, β54(D5)Val→Ile. J. Am. Soc. Mass Spectrom. 20(9), 1707–1713 (2009)
Daniel, Y.A., Turner, C., Haynes, R.M., Hunt, B.J., Dalton, R.N.: Quantification of hemoglobin A2 by tandem mass spectrometry. Clin. Chem. 53(8), 1448–1454 (2007)
Rai, D.K., Griffiths, W.J., Landon, B., Wild, B.J., Alvelius, G., Green, B.N.: Accurate mass measurement by electrospray ionisation quadrupole mass spectrometry: Detection of variants differing by <6 Da from normal in human hemoglobin heterozygotes. Anal. Chem. 75(9), 1978–1982 (2003)
Wild, B.J., Green, B.N., Cooper, E.K., Lalloz, M.R.A., Erten, S., Stephens, A.D., Layton, D.M.: Rapid identification of hemoglobin varints by electrospray ionization mass spectrometry. Blood Cells Mol. Dis. 27, 691–704 (2001)
Wild, B.J., Green, B.N., Stephens, A.D.: The potential of electrospray ionization mass spectrometry for the diagnosis of hemoglobin variants found in newborn screening. Blood Cells Mol. Dis. 33, 308–317 (2004)
Kelleher, N.L., Lin, H.Y., Valaskovic, G.A., Aaserud, D.J., Fridriksson, E.K., McLafferty, F.W.: Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J. Am. Chem. Soc. 121, 806–812 (1999)
Reid, G.E., McLuckey, S.A.: ‘Top down’ protein characterization via tandem mass spectrometry. J. Mass Spectrom. 37(7), 663–675 (2002)
Cui, W., Rohrs, H.W., Gross, M.L.: Top-down mass spectrometry: Recent developments, applications and perspectives. Analyst 136, 3854–3864 (2011)
Van Berkel, G.J., Kertesz, V., Koeplinger, K.A., Vavrek, M., Kong, A.-N.T.: Liquid microjunction surface sampling probe electrospray mass spectrometry for detection of drugs and metabolites in thin tissue sections. J. Mass Spectrom. 43(4), 500–508 (2008)
Edwards, R.L., Creese, A.J., Baumert, M., Griffiths, P., Bunch, J., Cooper, H.J.: Hemoglobin variant analysis via direct surface sampling of dried blood spots coupled with high-resolution mass spectrometry. Anal. Chem. 83(6), 2265–2270 (2011)
LeDuc, R.D., Taylor, G.K., Kim, Y.-B., Januszyk, T.E., Bynum, L.H., Sola, J.V., Garavelli, J.S., Kelleher, N.L.: ProSight PTM: An integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res. 32(Suppl 2), W340–W345 (2004)
Newborn Blood Spot Screening in the UK- Polices and Standards. (2005)
Rahbar, S.: Hemoglobin D Iran: β222 glutamic acid→glutamine (B4). Br. J. Haematol. 24(1), 31–35 (1973)
Rochette, J., Barnetson, R., Kiger, L., Kister, J., Littlewood, T.J., Webster, R., Poyart, C., Thein, S.L.: Association of a novel high oxygen-affinity haemoglobin-variant with delta-δ thalassemia. Br. J. Haematol. 86(1), 118–124 (1994)
Vandenesch, F., Baklouti, F., Francina, A., Vianeyliaud, C., Bertrand, A., Ledevehat, C., Delauney, J.: Hemoglobin J-Baltimore (β-16(A13)Gly-Asp) – interference with the assay of HBA1C. Clin. Chim. Acta 168(2), 121–128 (1987)
Fucharoen, S., Winichagoon, P.: Thalassemia and abnormal hemoglobin. Int. J. Hematol. 76, 83–89 (2002)
Lang, A., Lorkin, P.A.: Genetics of human hemoglobins. Br. Med. Bull. 32(3), 239–245 (1976)
Wajcman, H., Prehu, M.O., Prehu, C., Blouquit, Y., Prome, D., Galacteros, F.: Hemoglobin Phnom Penh [α 117Phe(H1)-Ile-α 118Thr(H2)]: Evidence for a hotspot for insertion of residues in the third exon of the α 1-globin gene. Hum. Mutat. S1, S20–S22 (1998)
Bakhtiar, R., Wu, Q., Hofstadler, S.A., Smith, R.D.: Charge state specific facile gas-phase cleavage of Asp75-Met76 peptide bond in the α-chain of human apohemoglobin probed by electrospray ionization mass spectrometry. Biol. Mass Spectrom. 23(11), 707–710 (1994)
NHS Sickle Cell and Thalassemia Screening Programme: Data Report: 2008/09. Informing Policy and Improving Quality. (2008–2009)
Papadea, C., Eckman, J.R., Kuenhert, R.S., Platt, A.F.: Comparison of liquid and dried blood for neonatal hemoglobinopathy screening—laboratory and programmatic issues. Pediatrics 93(3), 427–432 (1994)
Acknowledgments
R.L.E. received an EPSRC-funded studentship. The Advion Triversa Nanomate and ThermoFisher Orbitrap mass spectrometer used in this research were funded through the Birmingham Science City Translational Medicine: Experimental Medicine Network of Excellence Project, with support from Advantage West Midlands (AWM). The authors thank Mark Baumert for technical discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Edwards, R.L., Griffiths, P., Bunch, J. et al. Top-Down Proteomics and Direct Surface Sampling of Neonatal Dried Blood Spots: Diagnosis of Unknown Hemoglobin Variants. J. Am. Soc. Mass Spectrom. 23, 1921–1930 (2012). https://doi.org/10.1007/s13361-012-0477-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-012-0477-9