
Abstract
Therapeutic options for multiple sclerosis (MS) have significantly increased over the last few years. T lymphocytes are considered to play a central role in initiating and perpetuating the pathological immune response. Currently approved therapies for MS target T lymphocytes, either in an unspecific manner or directly by interference with specific T-cell pathways. While the concept of “T-cell-specific therapy” implies specificity and selectivity, currently approved approaches come from a general shaping of the immune system towards anti-inflammatory immune responses by non-T-cell-selective immune suppression or immune modulation (e.g., interferons—immune modulation approach) to a depletion of immune cell populations involving T cells (e.g., anti-CD52, alemtuzumab—immune selective depletion approach), or a selective inhibition of distinct molecular pathways in order to sequester leucocytes (e.g., natalizumab—leukocyte sequestration approach). This review will highlight the rationale and results of different T-cell-directed therapeutic approaches coming from basic animal experiments to clinical trials. We will first discuss the pathophysiological rationale for targeting T lymphocytes in MS leading to currently approved treatments acting on T lymphocytes. Furthermore, we will disuss previous promising concepts that have failed to show efficacy in clinical trials or were halted as a result of unexpected adverse events. Learning from the discrepancies between expectations and failures in practical outcomes helps to optimize future research approaches and clinical study designs. As our current view of MS pathogenesis and patient needs is rapidly evolving, novel therapeutic approaches targeting T lymphocytes will also be discussed, including specific molecular interventions such as cytokine-directed treatments or strategies enhancing immunoregulatory mechanisms. Based on clinical experience and novel pathophysiological approaches, T-cell-based strategies will remain a pillarstone of MS therapy.
Similar content being viewed by others


Introduction
In the last few decades, treatment strategies for multiple sclerosis (MS) have undergone radical change. Approximately 25 years ago, interferon (IFN)-β and glatiramer acetate revolutionized the field of MS therapy as, for the first time, it was possible to suppress new relapses, thereby improving the disease course and long-term disability of patients with MS. These so called disease-modifying drugs have broad immune-modulatory effects, including effects on T-cell activation, shifting of antigen-specific responses towards a helper T cell 2 (Th2)/regulatory T cell (Treg) pattern, as well as on blood–brain barrier (BBB) transmigration and local inflammatory events within the central nervous system (CNS). However, their therapeutic potential is limited as they reduce the annual relapse rate (ARR) by about 30–40 % [1, 2]. With the approval of the monoclonal antibody (mAb) natalizumab, a new era of selective therapeutic approaches has entered the field of neuroimmunotherapy. Novel emerging therapies are often sophisticated mAb approaches based on a growing understanding of the underlying molecular signatures and immunepathophysiology of MS. The most common treatment strategies include a targeting of cell surface markers and receptors, an antagonization of soluble signal molecules, or a depletion of specific immune cell populations. In this review, we will first discuss the available data arguing for a key role of T lymphocytes in the pathophysiology of MS. We will include a brief summary of currently approved drugs targeting T lymphocytes and discuss previous promising concepts that have failed expectations in clinical trials, as well as novel, attractive T-cell strategies currently under development.
Pathophysiological Rationale for Targeting T Cells in MS
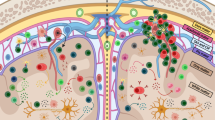
MS is a prototypic chronic autoinflammatory disease of the CNS initiated by a breakdown of peripheral immune tolerance to as yet unidentified antigens in genetically predisposed individuals [3]. The pathogenesis of MS is complex and involves nearly all cell types of the adaptive and innate immune system. T cells have traditionally been a key target of MS research and their assumed central role for MS pathology is based on data from extensive studies not only in animal models (which biased research and pathogenetic concepts towards Th cells), but also from MS lesions, genetic susceptibility, and, finally, the mode of action of approved immunotherapies [4]. MS has therefore long been considered the “classical” prototype of a T-cell-mediated autoimmune disorder. This view still holds true, even in light of growing evidence of an additional involvement of B cells, cytokines, innate immune cells, or immune system-independent neurodegenerative mechanisms. Abundant evidence could be acquired in favor of a central role of T lymphocytes in MS pathology. Inflammatory lesions in the CNS of patients with MS contain considerable numbers of both CD4+ and CD8+ T cells [5]. Interestingly, active MS lesions in some patients have a predominance of CD8+ T cells and clonal expansion of CD8+ T cells but not CD4+ T cells [6]. In contrast, murine experimental autoimmune encephalomyelitis (EAE) lesions are dominated by CD4+ T lymphocytes [7, 8]. Adoptive transfer of both Th1- and Th17-polarized cells from mice with EAE symptoms could transfer disease to recipient animals and depletion of CD4+ T cells from mice with EAE symptoms led to suppression of disease symptoms [9–11]. Data from different genome-wide susceptibility screens have now identified > 150 genes and nearly all of them are associated with the immune system [12]. Furthermore, the largest group of genes is involved in antigen presentation to T cells or in T-cell pathways themselves [13, 14]. An integrated analysis of genome-wide association studies with DNAse hypersensitivity sites in 112 cell types showed that Th1, Th17, and CD8+ T cells are those in which MS-associated genes are most active [15]. In a study with patients with clinically isolated syndrome, gene analysis was performed in naïve CD4+ T cells at baseline and 1 year after developing a second relapse, that is, clinically definite MS [16]. Ninety-two percent of patients who converted to clinically definite MS displayed a decreased expression of the transcription regulator Tob1, a key regulator of T-cell quiescence. These results were confirmed in the EAE model, demonstrating that loss of T-cell quiescence is associated with inflammatory relapses in MS [17]. From a clinician’s point of view, currently approved therapies used in MS include the targeting of T lymphocytes but are not selective or specific for T cells. They do this either directly (e.g., natalizumab, alemtuzumab, fingolimod) or at least affect them among other immune cells in therapy concepts with pleiotropic effects on the immune system (e.g., glatiramer acetate, IFN-β [18, 19]). However, none of these are T-cell-selective or antigen-specific.
Based on the pathophysiological concept and results, numerous different strategies for T-cell-based immunotherapy have been proposed previously and some of them have either proved their usefulness in daily clinical practice or are currently being evaluated in clinical trials. Extensive preclinical research on basic pathological mechanisms of T-cell-mediated CNS autoimmunity facilitates the advancement of preclinical research findings towards meaningful clinical approaches for patients with MS.
Approved or Late-stage Preclinical Therapies Targeting T Cells in MS
Various strategies of immune intervention have been considered for attenuating disease activity and progression in MS. In the last 2 decades, there has been considerable progress in immune therapy for MS [20]. While the first generation of drugs (IFN-β, glatiramer acetate) acts via pleiotropic effects on the immune system, a deeper understanding of pathological immune processes has led to highly specific and targeted novel therapeutic approaches. At present, all approved therapies for MS have an influence on functional properties of T lymphocytes (see Table 1). In the current review, the focus is on mAb therapies conceptually directed at specific T-cell pathways (natalizumab, alemtuzumab, daclizumab). We will not discuss the mode of action of other therapies also affecting T cells (including IFN, glatiramer acetate, mitoxantrone, fingolimod, teriflunomide, and dimethylfumarate; Table 1).
Natalizumab
Natalizumab is a humanized mAB direct against α4-integrin molecules preventing leukocyte migration across the BBB [35, 36]. The main mode of action in MS is the inhibition of the molecular interaction between α4β1-integrin [very late antigen 4 (VLA-4)] on T lymphocytes and vascular cell adhesion protein 1 on endothelial cells. The disruption of this process antagonizes a key molecular interaction essential for leukocyte adhesion and necessary for efficient migration across the BBB. This leads to a reduction of infiltrating immune cells and lower levels of CNS inflammation, as has been confirmed using animal models [37–39]. Natalizumab showed efficacy in 2 double-blind, placebo-controlled, multicenter phase III studies against placebo and IFN-β1a (AFFIRM and SENTINEL), leading to its approval nearly 10 years ago. Based on the results of these clinical trials, clinical experience, and phase IV studies, natalizumab appears to be one of the most potent drugs currently available for the management of MS. The major drawback of this potent therapy is the rare reactivation of latent JC virus (JCV), leading to progressive multifocal leukoencephalopathy (PML). PML is an often fatal inflammation of the brain caused by a viral infection of oligodendrocytes and neurons accompanied by an immune reaction against infected cells, leading to permanent neurological damage. Risk evaluation tools have been developed to determine the individual risk of developing PML [40, 41]. Classicaly, 3 risk factors have been established: anti-JCV antibody status, duration of treatment with natalizumab of > 2 years, and prior treatment with immunosuppressive therapies such as mitoxantrone. Currently, 2 novel independent approaches are being evaluated that have been proposed as additional risk stratification parameters: anti-JCV antibody titers and L-selectin (CD62L) expression levels [42–44]. While the association of PML and natalizumab is well established, a small number of cases have also been reported for patients treated with dimethylfumarate and fingolimod [45, 46]. Further studies on risk stratification for PML under different treatment options are clearly recommended.
In summary, natalizumab has proven to be a highly effective treatment against inflammatory activity in patients with MS. Natalizumab reduces leukocyte entry with a certain differentiation between distinct immune cell subsets. It has become clear that lymphocytes depend on different trafficking routes into the CNS, each of them characterized by a different set of endothelial or epithelial cells with a unique expression of (adhesion) molecules. Treatment with natalizumab changes the CNS inflammatory milieu rather than completely disrupting any immune surveillance in the CNS [47]. In this context, it was shown that long-term treatment with natalizumab leads to an upregulation of P-selectin glycoprotein ligand-1 on T cells, enhancing their rolling capacity over endothelial cells. Under these circumstances, a subpopulation of T cells expressing melanoma cell adhesion molecule is able to adhere to endothelial cells independently of the VLA-4 pathway [48]. In vivo, this results in high quantities of melanoma cell adhesion molecule-expressing T cells (mainly Th17 cells) in the cerebrospinal fluid (CSF) of patients treated with natalizumab. Obviously, these cells are unable to trigger relapses on their own as entry of Th1 cells depending on VLA-4 is blocked by natalizumab. However, another important aspect is the observation of reoccurrence of disease activity in patients with MS after cessation of natalizumab treatment, which can be partially explained by the mechanism of action: as relapse-triggering Th17 cells are already located within the CNS, the removal of the VLA-4 blockade initiates a strong recruitment of Th1 cells, enhancing relapse activity. Up to 50 % of patients suffer from significant clinical detoriation after de-escalation of natalizumab therapy and optimal clinical management is still a matter of discussion [49–51].
The unique situation in MS therapy is the availability of a very potent drug that exerts a clear risk in a subgroup of patients. As a consequence, a clear need for a personalized risk-management and risk-mitigation strategies has been requested, and natalizumab has already shown some paradigmatic approaches for individual risk stratification efforts, alredy partially useful in clinical practice [41]. The need for personalized risk management will become even more crucial in future, owing to other novel therapeutic approaches in patients with MS. It is very important to note that the experience with natalizumab has tought important lessons for the underlying pathogenesis of various other autoimmune inflammatory CNS disorders. Specificallly, Th1-driven MS pathology might be effectively treated with blockade of VLA-4, but Th17-driven pathology will probably not respond. Indeed, natalizumab has been reported to exacerbate disease symptoms in neuromyelitis optica spectrum disorders (NMOSD) [52, 53]. Th17 cells and B cells have been strongly implicated in the pathogenesis of NMOSD and natalizumab causes not only an enrichment of Th17 cells in the CSF, but also a redistribution of lymphocyte subsets in the periphery. Levels of immature B cells and plasma cells are elevated in the blood of patients treated with natalizumab [53]. All together, the lack of clinical treatment response to natalizumab in NMOSD underlines both the heterogenic pathophysiology of MS and NMOSD and the need to develop therapies targeting different CNS entry pathways.
Furthermore, second-generation approaches targeting VLA-4 are underway, which might present an improved risk profile for PML. A phase II trial showed efficacy in 88 patients treated with the CD49d antisense oligonucleotide ATL1102 [54]. Other pharmacological strategies are derived from important tissue-specific differences of α4-integrins. While α4β1 integrin mediates the migration of T lymphocytes into the CNS, bone marrow, and skin via adhesion to vascular cell adhesion protein 1, α4β7 integrin preferentially regulates the migration of T lymphocytes into the gut via adhesion to mucosal adressin cell adhesion molecule 1. In 2014, the α4β7 integrin-specific antibody vedolizumab was approved for Crohn disease and ulcerative colitis [55]. Treatment with vedolizumab is expected to provide gut-specific immunosuppression without systemic effects. Indeed, no cases of PML have so far been reported for vedolizumab. However, a putative role for α4β7 integrin inhibitors in MS seems unlikely as this pathway has so far not been shown to be involved in experimental models [56]. In agreement with this, vedolizumab did not affect experimental autoimmune encephalomyelitis in nonhuman primates [57], and, in healthy volunteers, it did not influence trafficking of CD4+ and CD8+ T cells into the CNS.
Alemtuzumab
CD52 is a cell-surface glycoprotein with largely unknown biological functions that is predominantly expressed on T and B lymphocytes and, to a lesser degree, on monocytes, macrophages, and eosinophil granulocytes but not on their respective hematopoietic precursors [58]. Targeting of CD52 by alemtuzumab selectively depletes CD52-bearing immune cells rapidly after infusion by antibody-dependent and complement-dependent cytolysis [32, 59, 60]. The slow repopulation of these cell populations from hematological stem cells is believed to reset and rebalance immunological responses, mediating long-term beneficial therapeutic effects [33, 61]. While the half-life of alemtuzumab itself is only a few days, the slow repopulation of immune populations follows a distinct temporal and probably spatial pattern. Monocytes recover to baseline levels after 3 months, as do B lymphocytes, which show a further increase and overshoot to approximately 150 % of baseline levels after 12 months [58, 62]. CD8+ T lymphocytes reach starting levels after 31 months, while CD4+ T cells need around 60 months for complete restauration. The repopulation of the immune system is accompanied by a long-term reduction of MS-related inflammatory responses. Recovered immune cells show a bias towards a tolerogenic phenotype with elevated numbers of regulatory T lymphocytes, a shift towards Th2 responses, increased levels of immunoregulatory cytokines [transforming growth factor-β, interleukin (IL)-10] and lower levels of Th1 and Th17 cytokines (IFN-γ, IL-12, and IL-17) and higher expression levels of co-inhibitory receptor molecues (programmed cell death protein 1, lymphocyte-activation gene 3) on T lymphocytes [63, 64]. Autoreactive T-cell clones were reduced after immune cell reconstitution and T cells displayed an increased T-cell receptor (TCR) diversity [61, 65]. The migratory capacity of T cells was shown to be reduced after alemtuzumab treatment, leading to a lower migration of T cells into the CNS. These data demonstrate that alemtuzumab treatment goes beyond a simple immune cell depletion approach. Instead, alemtuzumab influences qualititative functional properties of immune cells, promoting a rebalancing of immune tolerance networks in MS.
In phase II (CAMMS223) and phase III (CARE-MS I and CARE-MS II) clinical trials, alemtuzumab has shown high efficacy on clinical and radiological disease outcome parameters in replasing–remitting MS (RRMS). The randomized, rater-blinded, phase III clinical trials demonstrated superiority of alemtuzumab over IFN-β1a concerning clinical disease activity measured according to ARR and radiological parameters [58]. A significant difference in the sustained accumulation of confirmed disability was only observed for CARE-MS II but not for CARE-MS I. This result is most likely owing to the unexpectedly low rate of disability progression in the IFN-β1a group, indicating an underpowered clinical trial (11 % instead of the expected 20 %, based on the CAMMS223 trial and historical data). Novel readout parameters showed that significantly more patients were free of any clinical and magnetic resonance imaging (MRI) disease activity (no evidence of disease activity) in the alemtuzumab compared with the IFN-β1a group. Preliminary data from extension studies from patients completing the phase II and III trials demonstrate substained effects on relapse rate, disability, and MRI measures [66]. No evidence of disease activity was observed in 50–60 % of patients in years 3 and 4 after starting alemtuzumab, and a substantial proportion of patients have a stable or improved expanded disability status scale score over 3–4 years (data presented at the European Committee for Treatment and Research in Multiple Sclerosis congress 2014 and the American Academy of Neurology congress 2015). The 4-year follow-ups support the notion that alemtuzumab leads to a sustained “immune reprogramming” after immune cell reconstitution as retreatment rates were only 36 % and 32 % (CARE-MS I and II, respectively) over 3 years since the initial 2 treatment courses. Only 2–5 % of patients received other disease-modifying drugs in years 3 and 4.
In summary, alemtuzumab treatment aims at a sustained reprogramming of the immune system and avaible clinical data underline the strong effect of this new therapy for patients with MS. Detailed studies on the subtype composition and functional properties of both the T- and B-lymphocyte compartment after repopulation will provide further valuable information on the exact mode of action of alemtuzumab after cell depletion in the future. Frequent and considerable adverse events are the major drawback of alemtuzumab treatment. Infusion-associated reactions and mild-to-moderate infections occur regularly but can, in almost all cases, be safely controlled by routine clinical measures. Opportunistic infections in patients treated with alemtuzumab have been reported in single cases before. Of note, a first report of listeria meningitis in the CAMMS223 trial was followed by 2 cases in patients with MS, occurring immediately after the first cycle of alemtuzumab infusions [67]. Therefore, physicians and patients should be aware of the possibility of rare opportunistic infections following alemtuzumab treatment. In contrast, treatment with alemtuzumab is associated with secondarily induced autoimmune phenomens. When summarizing experience from all clinical trials, thyroid autoimmunity occurred in 30–40 % of patients with a range of onset from 6 to 61 months, and a peak in year 3, after alemtuzumab [68, 69]. Idiopathic thrombocytopenic purpura can be found in 1–3 % of patients whith the index patients having succumbed to a lethal intracranial hemorrhage. Additionally, 4 cases of glomerulonephritis (0.3 % of patients) and single cases of autoimmune neutropenia, hemolytic anemia, and type 1 diabetes have been reported [70, 71]. The development of secondary autoimmune phenomens seems to have no impact on the efficacy of alemtuzumab itself. These findings demonstrate that strict and continuous monitoring in all patients receiving alemtuzumab is indispensable for years after treatment induction. The underlying mechanisms of secondary autoimmune phenomens are not completely understood, and different hypotheses have been proposed. An incomplete depletion of few autoreactive T lymphocytes might favor their expansion, while a delayed T-cell recovery favors an uncontrolled reoccurrence of B lymphocytes. Altered cytokine levels (e.g., IL-21 or IL-7) have also been proposed to be involved. Clearly, future studies are necessary to identify biomarker signatures indicative for individual risks of post-treatment autoimmune responses, as well as to identify treatments for the occurrence of secondary autoimmune reactions under alemtuzumab [72, 73].
Recently, a phase I study was initiated with a next-generation anti-CD52 mAb (GZ402668, NCT02282826; Table 2) in patients with progressive MS, underlining ongoing research interest in this direction.
Daclizumab
Daclizumab is a humanized IgG antibody targeting CD25 and thereby preventing the binding of IL-2 to its receptor, which is highly expressed on activated T lymphocytes and regulatory CD4+CD25+FoxP3+ T lymphocytes [74]. Daclizumab does not trigger cell depletion or interfere with CD25 receptor signaling itself. Interestingly, the mode of action of daclizumab is only partially explained by a direct inhibition of T-cell activation or autocrine T-cell expansion, and only a modest reduction of CD4+ and CD8+ T-cell populations can be observed [75]. Higher levels of available IL-2 support an expansion of cells that constitutively express the intermediate affinity IL-2 receptor, such as CD56bright natural killer (NK) cells [76], a distinct cell population with immunoregulatory properties suppressing antigen-specific responses. Therapy response in clinical studies correlated with an increase of CD56bright NK cell levels in the peripheral blood and CSF [77, 78]. Despite some supporting publications on NK cells in MS, their functional role and relevance is currently incompletely understood. In EAE, depletion of NK cells led to an exacerbation of disease symptoms [79]. Observations from other treatment concepts show that therapy with IFN-β leads to an expansion of CD56bright and a decrease of cytotoxic CD56dim NK cells [80]. Intriguingly, the biological effects of daclizumab are most likely based on different modes of action, namely 1) a reduction of early T-cell activation by blocking IL-2 cross-presentation via dendritic cells; 2) an expansion and enhancement of immunregulatory CD56bright NK cells [81]; and 3) a reduction of proinflammatory lymphoid tissue-inducer cells [82]. It can be assumed that these different pathways, taken together, lead to a rebalancing of the immune regulatory network in patients with MS. The immunregulatory effects of daclizumab display novel modes of action than currently approved therapeutics, enhancing future treatment opportunities for patients with MS. Furthermore, owing to its effects on rebalancing autoimmune responses, a potential effect on NMOSD awaits further clarification. Studies in patients with NMOSD might be of special clinical interest in light of a lack of effect of other novel MS treatment options (natalizumab and probably also alemtuzumab).
The efficacy of daclizumab has been tested in clinical trials, including a randomized multicenter phase IIb trial (CHOICE), a phase II/III placebo-controlled, randomized trial (SELECT), and a phase III study comparing daclizumab and IFNβ-1a (DECIDE). In the CHOICE trial, daclizumab was tested at either 1 or 2 mg/kg as an add-on therapy to IFN-β, leading to a dose-dependent reduction of new or enlarged gadolinium-enhanced lesions [76]. In the SELECT trial, patients were randomized to 2 doses of daclizumab (150 and 300 mg) and placebo treatment every 4 weeks for 1 year. Daclizumab reduced the ARR by 54 % in the 150 mg group and 50 % in the 300 mg group compared with placebo, and the study initiators decided to focus on 150 mg daclizumab rather than IFN-β1a in the DECIDE trial. The ARR was significantly lower for daclizumab compared with IFN-β1a (0.216 vs 0.393, a reduction of 45 %). In addition, MRI revealed 54 % fewer new or enlarging CNS lesions. The effect on disability progression was less pronounced, with daclizumab being only slightly more effective than IFN-β at reducing the risk of increased disability lasting for 3 months [83]. In general, daclizumab has a favorable safety profile and has been in long-term use in oncology and transplantation medicine. However, the side effects of daclizumab in clinical trials include a higher rate of potentially serious infections, liver abnormalities, skin reactions, and a tendency to develop secondary autoimmune phenomena in a number of patients [84, 85]. The 2 most relevant safety problems seem to be various cutaneous events (e.g., rash, eczema, acne, erythema, pruritis) and liver function abnormalities. Skin reactions were reported in 37 % versus 19 % of patients in the daclizumab high-yield process and IFN-β1a groups, leading to treatment discontinuation in 5 % versus 1 % of patients. The most common skin reactions were rash and eczema, while serious cutaneous events were reported only in 2 % and 1 % of patients, respectively. Elevations of liver enzymes > 5 times the upper limit of normal occurred in 6 % and 3 % of patients, respectively. While these elevations were most commonly observed during the first year of IFN-β1a treatment, in the DECIDE study they were evenly distributed over time during daclizumab treatment. Of note, most side effects were self-limited or could be successfully managed by treatment discontinuation and/or treatment with cortisone. Approval has been applied for (2015) and approval is expected in 2016. Because of the associated adverse effects, this first self-administrable, subcutaneously injectable mAb should be accompagnied by a strict monitoring and safety program in clinical practice.
Failed Approaches or Therapeutic Concepts with Inconclusive Results
Even though multiple drugs have been approved since 1993 for the treatment of patients with RRMS (Table 1), a large number of therapeutic concepts failed to show benefit in clinical trials, despite a sound scientific rationale and promising data from preclinical animal models. Failure of these interventions has nonetheless provided important information for a better understanding of the pathophysiology of MS. Therefore, we will provide a short overview of important failed clinical trials aiming at (more or less direct) T-cell intervention in MS.
Anti-CD3 and Anti-CD4-directed Therapies (Muromonab and Priliximab)
One of the pivotal steps in initiating autoimmune inflammation is the activation of autoreactive T cells in the periphery via TCR-mediated recognition of autoantigens. After transmigration across the activated BBB, immune cells become reactivated by local CNS-resident antigen-presenting cells (APC). CD4+ and CD8+ cytotoxic T cells recruite further inflammatory cells types and trigger demyelination and axonal damage. Modulation of T-cell differentiation and rebalancing pathogenic Th1/Th17 cells towards a Treg/Th2 phenotype represents an attractive pathway for therapeutic interventions.
Initial T-cell-directed concepts aimed at a broad depletion of autoreactive T cells using monoclonal antibodies against pan-T-cell marker molecules [86]. Early phase I studies started nearly 30 years ago by initially demonstrating suppression of in vitro measures of human immune responses [87]. However, clinical trials targeting CD3 (muromonab) and CD4 (priliximab) unfortunately—and to the surprise of some within the community—yielded negative results in patients with MS. Muromonab (OKT3) was tested in an open-label trial in 16 patients with RRMS and progressive MS [88], and showed significant hematological and systemic toxicity without convincing beneficial clinical effects. In contrast, treatment with anti-CD4 monoclonal antibodies was better tolerated, without major toxic side effects [86, 89–91]. An open-label trial with the chimeric anti-CD4 antibody priliximab induced a sustained reduction of CD4+ T cells in the peripheral blood. However, in a phase II clinical trial in 71 patients, no significant effects were observed for gadolinium-enhancing lesions and expanded disability status scale progression over 9 months. Further analysis revealed that priliximab preferentially reduced circulating naïve T cells and nonactivated Th1 cells. The lessons from these first attemps was that targeting widely distributed and rather “unselective” T-cell surface markers failed because of side effects and a lack of clinical effect.
IL-12/23: p40 Neutralizing mAb (Ustekinumab)
Subsequently, research focus shifted towards cytokine- and subtype-directed therapeutic approaches. Initially, autoreactive Th1 cells were regarded as the main pathogenic T-cell subpopulation driving autoreactive immune processes. Th1 cells are induced by IL-12 and produce IFN-γ, while Th2 cells, in contrast, secrete IL-4, IL-5, and IL-13. However, more recent findings have implicated, in particular, Th17 cells as another major cell population involved in the pathogenesis of MS and EAE [92–95]. The Th1 cytokine IL-12 consists of the 2 heterodimeric subunits p40 and p35. Later, it was discovered that IL-23 shares the p40 subunit with IL-12. Intensive research demonstrated that IL-12(p35) knockout (KO) mice were still susceptible to EAE, while genetic deletion of IL-12(p40) resulted in resistance to EAE symptoms [92, 96]. Furthermore, the IL-12/23 subunit p40 is detected in MS lesions and administration of IL-12 induces relapses in the EAE model [86, 97]. In a direct approach, the use of IL-23 KO mice showed the crucial role of this cytokine for EAE induction. IL-23 drives the induction of IL-17-producing T cells and adoptive transfer of Th17 cells causes severe EAE symptoms, while IL-17 KO mice are protected from EAE symptoms [94].
Ustekinumab was developed as a mAb directed against the IL-12/IL-23 p40 subunit, showing efficacy in a preclinical marmoset model of EAE [98]. Subcutaneous administration was well tolerated in a phase I trial in patients with RRMS [99]. Based on this study, ustekinumab was tested over 19 weeks in a randomized, double-blind, placebo-controlled, dose-ranging phase II study in 249 patients with RRMS. No significant differences were observed for new gadolinium-enhancing T1-weighted lesions, which were defined as the primary end point, nor for clinical parameters [100]. The reason for the negative results found for ustekinumab are not yet fully understood. However, the antibody is currently approved for the treatment of psoriasis, an autoimmune disorder with many immunological similarities to MS. Basic immunological differences in MS have been proposed, for example wrong window of time for treatment and relative importance of Th17 cells [101]. Furthermore, specific properties of the IL-12 signaling pathway including independent levels of IL-12 p40 subunits and its respective heterodimers might present an alternative explanation [102].
Anti-CD40L Antibody (Toralizumab)
Blockade of co-stimulatory pathways or activation of co-inhibitory pathways have been proposed as promising strategies against autoimmune-mediated stimulation of T lymphocytes. The CD40–CD40L pathway plays an important role in interaction between T cells and B cells or dendritic cells. Activation of this pathway enhances antigen-specific T-cell proliferation and promotes B-cell differentiation. A pilot study with a humanized anti-CD40L antibody (toralizumab) showed favorable results and a phase II, double-blind, placebo-controlled study was initiated with 40 patients with RRMS [103]. However, the trial was halted prematurely because of safety concerns after 3 reports of thromboembolism in a concomitant study of toralizumab in patients with Crohn disease [104]. While it is still unclear whether this might have been due to anti-CD40L blockade itself, for example by crossreactivity with activated platelets, no further attempts in patients with MS have been conducted owing to unfavorable risk-to-benefit expectations.
Therapies Aiming at T-lymphocyte Migration [Anti-α4β Integrin (Firategrast) and Anti-LFA-1 (Rovelizumab)]
Apart from natalizumab, different therapies targeting T-cell migration pathways have been and still are being evaluated. Firategrast is an mAb therapy acting against anti α4β1/α4β7 integrins, preventing migration of inflammatory T lymphocytes into the CNS, similar to natalizumab therapy. In contrast to natalizumab, firategrast is taken orally and is short-acting, providing differences in bioavailability and pharmacodynamics. Results from a randomized, double-blind, placebo-controlled, phase II study in 343 patients with RRMS showed that the highest dose of firategrast resulted in a significantly lower cumulative number of new gadolinium-enhancing lesions (49 %) [105]. However, experience from the phase II natalizumab trial reported a 92 % reduction [106], pointing towards a lower efficacy of firategrast than expected. Furthermore, none of the doses of firategrast resulted in a significantly lower frequency of relapses compared with placebo. At present, it is unclear whether a phase III study will be initiated.
Leukocytes express lymphocyte function-associated antigen 1 (LFA-1) on their surface and LFA-1/intracellular adhesion molecule 1 interaction inhibits cell adhesion between leukocytes and vascular endothelial cells. An open phase I study in 24 patients and a subsequent phase II study were performed in 169 patients using a humanized monoclonal anti-LFA-1 antibody (rovelizumab). No significant benefits in clinical or MRI outcomes were observed [107, 108]. This demonstrates that some mechanisms regulating cell migration over the BBB seem to be redundant and that novel target molecules need to be chosen carefully. In this context, previous experiences using the LFA-1 neutralizing antibody efalizumab should be kept in mind [109]. This antibody was approved for the treatment of psoriasis until its withdrawal because of 2 cases of fatal PML. As evalizumab was not studied in patients with MS, general conclusions about a potential risk of LFA-1 blockade for PML development cannot be easily drawn. Interestingly, evalizumab had only small effects on CNS migration rather then acting on activation and proliferation of naïve T cells and antigen-specific restimulation of memory T cells.
Future T-cell-directed Therapeutic Approaches
Induction of Antigen-specific Immunotolerance
Potential autoreactive T lymphocytes are eliminated in thymic T-cell generation. However, this principle of thymic education (based on positive and negative selection) is incomplete and supported by several mechanisms of peripheral tolerance induction and maintenance. A limited number of self-reactive T cells escape the thymus and are present in the peripheral T-cell repertoire where they might become activated initiating autoimmune diseases. Inhibition of autoreactive T cells through induction of antigen-specific immune tolerance holds the promise of effective treatment of autoimmune pathology with few side effects and preservation of normal immune functions [110]. In MS, a number of different approaches have already been tested in clinical trials or in currently ongoing trials with the aim of inhibiting myelin-reactive immune responses.
Myelin Peptide-based Approaches
In the actively induced EAE model, antigen-specific inhibition of disease symptoms can be achieved by administration of tolerogenic myelin antigens. Different administration routes (intravenously, transdermally, intranasally, orally) have been assessed experimentally [111], and while all of them seem to achieve tolerance induction, different pathways seem to be involved, for example elimination of antigen-specific cells by induction of apoptosis or anergy, or induction of tolerogenic antigen-specific cells [110]. A number of attempts have so far been assessed in human trials. Nearly 20 years ago, the oral administration of myelin basic antigen protein or peptides was found to suppress EAE symptoms [112, 113]. Subsequently, a large phase III, placebo-controlled trial was performed and patients received bovine myelin-containing myelin basic protein (MBP) and protein lipoprotein (PLP) orally. Overall, the clinical outcome of this trial was negative [114], while some beneficial effects were observed for immunological parameters. Oral antigen uptake induced a shift towards tolerogenic immune cell populations: Th2, transforming growth factor-β-producing T cells, and suppressive CD4+CD25–LAP+ T cells [115].
Another attempt was made using intravenous administrations of soluble MBP peptide (MBP82-96). While this treatment was well tolerated and showed promising effects in phase I/II clinical trials [116, 117], a subsequent double-blind, placebo-controlled phase III trial in > 600 human leukocyte antigen (HLA)–DR2/4-positive patients with secondary progressive MS (SPMS) failed [118]. Negative results were also reported for intravenous injections of another approach using a soluble major histocompatibility complex (MHC)–peptide complex in patients with SPMS (MHC–MBP84-102 [119]). RTL1000 [myelin oligodendrocyte glycoprotein (MOG)35–55 peptide bound to single-chain domains of HLA-DR2) showed efficacy in the EAE model, and a phase I trial showed that this treatment was well tolerated [120]. At present, no information is available on whether a phase II trial will be initiated. Possible reasons for the failure of these trials include patient selection, as immune tolerance might be more effective in patients with early RRMS. Furthermore, the primary target antigen in MS is not known and it is likely that a mixture of different potential target proteins might yield better results. Based on that assumption, another phase I trial has tested a single infusion of autologous peripheral blood mononuclear cells chemically coupled with seven different myelin peptides (MOG1-20, MOG35–55, MBP13–32, MBP83–99, MBP111–129, MBP146–170, and PLP139–154) in 7 patients with RRMS and 2 with SPMS [121]. Patients receiving higher doses showed a decrease in antigen-specific T-cell responses, indicating a good feasibility and tolerability of this approach.
As an alternative application route, transdermal administration of a mixture of 3 myelin peptides (MBP85–99, MOG35–55, and PLP139–151) has been tested in a double-blind, phase II, placebo-controlled trial in 30 patients with RRMS [122]. After 1 year, patients treated with myelin peptide skin patches showed a significantly better outcome in clinical radiological parameters and treatment was well tolerated. So far, no official announcement concerning a phase III trial with this treatment has been made. ATX-MS-1467 is a mix of 4 synthetic myelin peptides derived from MBP protein. It has shown a favorable profile in 2 phase I trials in patients with SPMS and RRMS (NCT01097668; see [123]), and a phase II trial in patients with RRMS is currently ongoing (NCT01973491; Table 2). In summary, myelin peptide-based tolerization approaches are intruiging from a pathophysiological point of view, while currently 2 myelin peptide-based approaches using a mixture of different epitopes in patients with RRMS still have the potential to demonstrate clinical efficacy in the future.
Altered Peptide Ligands
TCR activation involves recognition of an immunogenic peptide bound to MHC receptors on APC by the TCRs, initiating a downstream signaling cascade. Altered peptide ligands (APL) are synthetic peptides derived from MHC-binding antigenic peptide and are modified in order to change MHC–TCR binding characteristics, leading to tolerogenic immune responses to the original peptide [110]. These can either be explained by partial activation of T cells, by induction of suppressive Th2 T cells, or by a direct antagonism of peptide and APL [124–126]. A phase II trial with an APL derived from the immunodominant MBP83–99 peptide was halted owing to increased relapses in 3/8 patients receiving the high-dose regime [127]. Immunological analysis revealed that APL treatment expanded proinflammatory encephalitogenic MBP83–99 T cells, leading to clinical relapses. At the same time, an ongoing larger clinical trial was stopped for safety reasons. Later analysis showed that, differently from the first trial, immunological and radiological results showed a tendency to beneficial effects [128, 129]. The reasons for the failure of the first trial remain unclear but might include dose-dependent immunological effects. In summary, data from first approaches using APL show inconclusive results and future clinical attempts need to be accompanied by a strict monitoring of unexpected disease activity.
Autologous T-cell Therapy
The concept of TCR vaccination is based on administration of either attenuated autologous antigen-specific T cells or peptides from the autoantigen-specific complementarity determining region (CDR) region of these T cells, aiming at the induction of an immune response directed against pathogenic T cells [130, 131]. Small open-label studies revealed that this treatment was well tolerated and showed a reduction in the frequency of myelin-reactive T cells. A randomized, double-blind trial of autologous attenuated myelin-specific T cells was conducted in 33 patients with RRMS [132]. T cells were isolated from the peripheral blood of patients, stimulated with 9 different peptides derived from MBP, MOG, and PLP protein, attenuated by irradiation, and administrated subcutaneously. This trial showed that this procedure was safe and provided indications for clinical efficacy.
A randomized, placebo-controlled, phase IIb trial was conducted using myelin-specific T cells restimulated with 6 myelin peptides from MBP, MOG, and PLP proteins [Tcelna and Imilecleucel-T (Opexa Therapeutics, The Woodlands, TX, USA)] in 150 patients with RRMS [133]. After 1 year, analysis revealed no difference in clinical and radiological parameters. However, based on an open-label study showing favorable effects of Tcelna [134], a phase II, double-blind, placebo-controlled trial is currently ongoing in patients with SPMS (NCT01684761; Table 2) and results are expected in 2016. Another currently ongoing phase I/II trial is SCLEROLYM (NCT02427776), which is assessing safety and radiological disease activity in 18 patients with RRMS receiving autologous CD4+ T cells stimulated and expanded ex vivo (Table 2). The antigen is derived from a MOG peptide modified by a thioreductase motif in the flanking residues of the cell epitopes. Inclusion criteria are, among others, a specific HLA allele (HLA DRB1*1501) and a positive in vitro test for patients CD4+ cell reactivity to the immunogenic peptide. These preselective criteria are expected to enhance the chances of a beneficial therapeutic effect. Results from this study are expected in 2016–17.
Therapeutic vaccination using peptides from the specific autoantigen-recognizing CDR region of myelin-reactive T cells is another concept that has been tested in a double-blind proof-of-concept trial in 23 patiens [135]. Further studies have resulted in the development of a vaccine containing 3 CDR2 peptides (Neurovax; Immune Response BioPharma, Atlantic City, NJ, USA) that has been in the focus of an open-label study in 27 patients with MS [136]. A high frequency of IL-10-secreting cells and an increased expression of FoxP3 in Tregs were observed in treated patients. Most patients remained clinically stable and further clinical studies in patients with RRMS [110], pediatric MS (NCT02200718), and SPMS (NCT02057159) have been announced for 2016 (Table 2).
Anti-IL17A-directed Therapies
Several mAbs are currently being developed to target IL-17 (and, indirectly, Th17 cells). Secukinumab is a mAb that neutralizes IL-17A aiming at suppression of pathogenic Th17 cells, a key population not only in MS, but also in T-cell-driven inflammatory pathways in different autoimmune disorders. So far, secukinumab has been tested in 3 different prototypic diseases. In psoriasis, it showed convincing results in a phase II trial in 404 patients and 2 phase III trials in > 2000 patients [137, 138], leading to its approval in 2015; however, it failed to show efficacy in a phase II trial in refractive rheumatoid arthritis (RA) [139]. In a placebo-controlled proof-of-concept study in 73 treatment-naïve patients with RRMS, secukinumab treatment resulted in a significant reduction (67 %) of new gadolinium-enhancing lesions in MRI measurements after 24 weeks [140]. Based on these results, the efficacy of secukinumab was assessed in a placebo-controlled, randomized, phase II trial in patients with RRMS (NCT01874340). However, this trial was terminated early after enrollment of only 28 patients, and effect on outcome measures cannot be determined. The sponsor indicated that this decision was based on the development of another anti-IL17 antibody, called CJM112, with superior potential to secukinumab. A phase II proof-of-concept study (MABINGO) in 360 patients with MS is currently being initiated [141].
The alternative anti-IL-17A antibody ixekizumab recently showed positive results in 2 phase III trial in 2500 patients with psoriasis and has subsequently been submitted to the US Food and Drug Administration for approval to be used as a treatment for moderate-to-severe plaque psoriasis [142]. So far, evaluation of ixekizumab for psoriatic arthritis has been announced, while a potential clinical program for MS or neuromyelitis optica remains unclear.
The third late-stage IL-17 pathway targeting antibody in psoriasis is brodalumab, which binds the IL-17 receptor A. Brodalumab has showed efficacy in phase III clinical trials for the treatment of psoriasis [143]. However, one of the responsible pharmaceutical companies decided to withdraw from the clinical development of brodalumab, based on results of clinical studies which showed an increase in suicidal thoughts and completed suicide risk. Further information on the future of brodalumab is pending. Moerover, it is as yet unclear whether a putative link between suicide ideation and IL-17 targeting might even be a class effect, and evaluation of preclinical and postmarketing data is currently awaited. It should be pointed out that brodalumab has a broader effect than secukinumab and ixekizumab as it also blocks the cytokines IL-17B, IL-17C, IL-17D, IL-17E, and IL-17 F. As targeting IL-17A has showed a remarkable clinical impact in psoriasis, clinical evaluation in MS is of great interest provided that the potential risks are acceptable.
Anti-CTLA-4-directed Therapy (Abatacept)
Co-stimulatory molecules deliver secondary signals enhancing or downregulating TCR responses. Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4; CD152) is an extensively studied coinhibitory molecule expressed on T lymphcoytes, dampening immune responses upon binding to CD80 or CD86 on APC [144]. Activation of the CTLA-4 pathway reduces immune activity, which has led to the clinical approval of CTLA4–Ig (fusion protein of CTLA-4 and antibodies; abatacept) for RA and a second-generation form, belatacept, for renal transplantation. In contrast, antagonistic antibodies for CTLA-4 increase immune activity. Ipilimumab has been approved for the treatment for melanoma and is currently under investigation for multiple types of cancer. Anti-CTLA-4 treatment increased clinical signs of EAE in animal models [145, 146]. Abatacept has showed safety and favorable immunological effects in a phase I clinical trial in patients with RRMS [147], while an initial placebo-controlled phase II trial in 219 patients was halted prematurely because of an increased relapse rate and MRI activity in an abatacept-treated group [148]. However, a post-hoc analysis revealed that patients in this treatment arm had a higher baseline activity at the time of study inclusion, providing the rationale for another attempt a few years later. A subsequent randomized, double-blind, placebo-controlled phase II clinical trial in 65 patients with RRMS (ACCLAIM study, NCT01116427; Table 2) has recently been completed, but the final results are still pending.
Cytokine-directed Therapeutic Approaches (IL-7, IL-2)
A few years ago, genome-wide association studies confimed the influence of genetic factors on the development of MS. Immune-related genes have repeatedly been confirmed, mainly in the HLA gene cluster, but also in specific signaling pathways like the IL-2 receptor, the IL-7 receptor, or CD58 [149]. IL-7 signaling plays an important role in T-cell development, homeostasis, and generation of memory T cells, and therapeutic targeting has been considered for various immune-mediated diseases [150]. RN168 is a mAb inhibiting the human IL-7 receptor and a dose-finding phase I study was started to evaluate the safety and tolerability of this approach (NCT02045732). The study was terminated early by the sponsor in April 2015 for reasons unrelated to safety. At present, further background information or clinical data from this trial have not been released.
The pleiotropic effects of IL-2 on T lymphocytes have been studied previously, in detail [151]. IL-2 induces proliferation and promotes expansion of TCR-stimulated T cells [152, 153]. Both activated effector T cells and CD4+CD25+Foxp3+ Tregs display high levels of the IL-2 receptor CD25. IL-2 deficiency in KO mice resulted in autoimmune-mediated lethal lymphoproliferation induction, a critical function of IL-2 in regulating peripheral T-cell tolerance [154–156]. Indeed, IL-2 receptor signaling is fundamental for the development and peripheral homeostasis of Tregs. Interestingly, the net biological effects of IL-2 are dose-dependent and, at a low dose, IL-2 preferentially expands Tregs. In patients with MS, Tregs show impaired functional properties (see [157] for a detailed review). A randomized, double-blind, placebo-controlled, phase II clinical trial with repeated administration of low-dose IL-2 has been announced (NCT02424396; Table 2). The primary end point is the increase in Tregs compared with baseline after 5 days, indicating a biological response to this treatment. Secondary end points include a longer-lasting effect after months and radiological disease parameters. The results of this trial are expected in 2017.
Regulatory T Lymphocytes as Therapeutic Targets
Strategies aiming to enhance the function of immunoregulatory CD4+CD25+ T cells have the potential to mediate beneficial effects in MS and T-cell-mediated autoimmune disorders. The anti-CD4 mAb tregalizumab (BT-061) selectively activates the suppressive properties of Tregs by binding to a unique conformational epitope on domain 2 of CD4 [158]. In contrast to other known CD4-binding antibodies, tregalizumab induces a suboptimal activation of TCR downstream pathways preferentially generating Tregs. An initial phase II study in RA did not meet primary end points and it is currently being re-evaluated whether pilot studies will be conducted in other autoinflammatory conditions such as psoriasis or MS.
Anti VLA-2 Therapy (Vatelizumab)
Based on successful clinical experience with natalizumab, the transmigration process of immune cells across the BBB into the inflamed CNS tissue has been brought to the fore of pharmacological research. Vatelizumab is a mAB directed against α2β1 integrin (VLA-2) on activated lymphocytes, preventing binding to collagen [159, 160]. It has shown efficacy in preclinical models and it is assumed that vatelizumab prevents lymphocytes from binding to collagen fibres that build up in inflammatory sites. Currently, a phase II, double-blind, randomized, placebo-controlled clinical trial evaluating multiple doses of vatelizumab in 168 patients with RRMS is ongoing (EMPIRE, NCT02222948; Table 2). The primary outcome measure is the cumulative number of new contrast-enhancing lesions. The treatment period is only 12 weeks, followed by a safety follow-up period of up to 92 weeks. The first results might be expected at the end of 2016, while study completion is due to be 2018.
Conclusion
Based on a growing understanding of the immunpathogenesis of MS, the field is rapidly evolving towards more effective therapies with biologicals [161]. More efficacious interventions in specific immune system pathways hold the potential towards a better control of disease efficacy and novel treatment concepts for a “freedom from disease activity” or even “continuous improvement of disability”. In MS, the targeting of T lymphocytes has evolved into a cornerstone of successful therapy. Novel recent approaches either target specific immune cell populations (alemtuzumab: T and B lymphocytes) or specific mechanistical pathways derived from pathophysiological studies (natalizumab). However, some of these therapeutic approaches also potentially involve novel unexpected adverse events such as potentially fatal infections, for example PML, or can induce secondary autoimmune phenomena. Furthermore, even the growng armamentarium of approved therapies for MS does not prevent disease progression in a substantial number of patients, underlining the necessity for both immunological and neuroprotective approaches. A number of clinical trials with novel T-cell-directed therapies is currently ongoing following different directions: targeted inhibition of CNS antigen-specific T cells, especially in the very early phase of the disease, has the potential to be a direct and highly specific way of inhibiting pathological CNS immune responses without major side effects. Furthermore, the modulation of specific T-cell-associated molecules and pathways provides the opportunity for individual tailored clinical therapies. In view of these current developments, targeting T cells will certainly remain a vital element in MS therapy. It remains to be speculated whether the goal of reinduction of CNS tolerance in MS (by very specific T-cell-tailored therapies) will ever be superior to the increasing armamentarium of biologicals generally modulating T-cell function via selective depletion, lymphocyte sequestration, or T cell/immune cell subset lineage deviation. One key goal with the latter approaches is certainly better individualization of treatments, with regard to principle choice in the individual patient and also to drug-specific prediction of response and risks.
References
Katrych O, Simone TM, Azad S, Mousa SA. Disease-modifying agents in the treatment of multiple sclerosis: a review of long-term outcomes. CNS Neurol Disord Drug Targets 2009;8:512-519.
Klotz L, Wiendl H. Monoclonal antibodies in neuroinflammatory diseases. Expert Opin Biol Ther 2013;13:831-846.
Hafler DA, Slavik JM, Anderson DE, O'Connor KC, De Jager P, Baecher-Allan C. Multiple sclerosis. Immunol Rev 2005;204:208-231.
O'Brien K, Gran B, Rostami A. T-cell based immunotherapy in experimental autoimmune encephalomyelitis and multiple sclerosis. Immunotherapy 2010;2:99-115.
Traugott U, Reinherz EL, Raine CS. Multiple sclerosis: distribution of T cell subsets within active chronic lesions. Science 1983;219:308-310.
Denic A, Wootla B, Rodriguez M. CD8(+) T cells in multiple sclerosis. Expert Opin Ther Targets 2013;17:1053-1066.
Babbe H, Roers A, Waisman A, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med 2000;192:393-404.
Jacobsen M, Cepok S, Quak E, et al. Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain 2002;125:538-550.
Brostoff SW, Mason DW. Experimental allergic encephalomyelitis: successful treatment in vivo with a monoclonal antibody that recognizes T helper cells. J Immunol 1984;133:1938-1942.
Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+ 2- T lymphocytes. J Immunol 1981;127:1420-1423.
Waldor MK, Sriram S, Hardy R, et al. Reversal of experimental allergic encephalomyelitis with monoclonal antibody to a T-cell subset marker. Science 1985;227:415-417.
Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214-219.
Compston A. The genetics of multiple sclerosis. J Neurovirol 2000;6(Suppl. 2):S5-S9.
Hafler DA, Compston A, Sawcer S, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007;357:851-862.
Disanto G, Kjetil Sandve G, et al. DNase hypersensitive sites and association with multiple sclerosis. Hum Mol Genet 2014;23:942-948.
Corvol JC, Pelletier D, Henry RG, et al. Abrogation of T cell quiescence characterizes patients at high risk for multiple sclerosis after the initial neurological event. Proc Natl Acad Sci U S A 2008;105:11839-11844.
Schulze-Topphoff U, Casazza S, Varrin-Doyer M, et al. Tob1 plays a critical role in the activation of encephalitogenic T cells in CNS autoimmunity. J Exp Med 2013;210:1301-1309.
Menge T, Weber MS, Hemmer B, et al. Disease-modifying agents for multiple sclerosis: recent advances and future prospects. Drugs 2008;68:2445-2468.
Meuth SG, Gobel K, Wiendl H. Immune therapy of multiple sclerosis—future strategies. Curr Pharm Des 2012;18:4489-4497.
Constantinescu CS, Gran B. The essential role of T cells in multiple sclerosis: a reappraisal. Biomed J 2014;37:34-40.
Kozovska ME, Hong J, Zang YC, et al. Interferon beta induces T-helper 2 immune deviation in MS. Neurology 1999;53:1692-1697.
Rudick RA, Ransohoff RM, Lee JC, et al. In vivo effects of interferon beta-1a on immunosuppressive cytokines in multiple sclerosis. Neurology 1998;50:1294-1300.
Mirandola SR, Hallal DE, Farias AS, et al. Interferon-beta modifies the peripheral blood cell cytokine secretion in patients with multiple sclerosis. Int Immunopharmacol 2009;9:824-830.
Aharoni R. The mechanism of action of glatiramer acetate in multiple sclerosis and beyond. Autoimmun Rev 2013;12:543-553.
Hartung HP, Gonsette R, Konig N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002;360:2018-2025.
Neuhaus O, Kieseier BC, Hartung HP. Mitoxantrone in multiple sclerosis. Adv Neurol 2006;98:293-302.
Rice GP, Hartung HP, Calabresi PA. Anti-alpha4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology 2005;64:1336-1342.
Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol 2010;33:91-101.
Cherwinski HM, Cohn RG, Cheung P, et al. The immunosuppressant leflunomide inhibits lymphocyte proliferation by inhibiting pyrimidine biosynthesis. J Pharmacol Exp Ther 1995;275:1043-1049.
Bomprezzi R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: an overview. Ther Adv Neurol Disord 2015;8:20-30.
Moharregh-Khiabani D, Linker RA, Gold R, Stangel M. Fumaric acid and its esters: an emerging treatment for multiple sclerosis. Curr Neuropharmacol 2009;7:60-64.
Hale G. The CD52 antigen and development of the CAMPATH antibodies. Cytotherapy 2001;3:137-143.
Wiendl H, Kieseier B. Multiple sclerosis: reprogramming the immune repertoire with alemtuzumab in MS. Nat Rev Neurol 2013;9:125-126.
Ransohoff RM, Hafler DA, Lucchinetti CF. Multiple sclerosis-a quiet revolution. Nat Rev Neurol 2015;11:134-142.
Engelhardt B, Kappos L. Natalizumab: targeting alpha4-integrins in multiple sclerosis. Neurodegener Dis 2008;5:16-22.
Rommer PS, Dudesek A, Stuve O, Zettl UK. Monoclonal antibodies in treatment of multiple sclerosis. Clin Exp Immunol 2014;175:373-384.
Archelos JJ, Previtali SC, Hartung HP. The role of integrins in immune-mediated diseases of the nervous system. Trends Neurosci 1999;22:30-38.
Coisne C, Mao W, Engelhardt B. Cutting edge: natalizumab blocks adhesion but not initial contact of human T cells to the blood–brain barrier in vivo in an animal model of multiple sclerosis. J Immunol 2009;182:5909-5913.
Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 1992;356:63-66.
McCormack PL. Natalizumab: a review of its use in the management of relapsing-remitting multiple sclerosis. Drugs 2013;73:1463-1481.
Schwab N, Schneider-Hohendorf T, Wiendl H. Therapeutic uses of anti-alpha4-integrin (anti-VLA-4) antibodies in multiple sclerosis. Int Immunol 2015;27:47-53.
Plavina T, Subramanyam M, Bloomgren G, et al. Anti-JC virus antibody levels in serum or plasma further define risk of natalizumab-associated progressive multifocal leukoencephalopathy. Ann Neurol 2014;76:802-812.
Schwab N, Schneider-Hohendorf T, Posevitz V, et al. L-selectin is a possible biomarker for individual PML risk in natalizumab-treated MS patients. Neurology 2013;81:865-871.
Schwab N, Schneider-Hohendorf T, Pignolet B, et al. PML risk stratification using anti-JCV antibody index and L-selectin. Mult Scler 2015 Oct 2.
Nieuwkamp DJ, Murk JL, van Oosten BW, et al. PML in a patient without severe lymphocytopenia receiving dimethyl fumarate. N Engl J Med 2015;372:1474-1476.
Van Schependom J, Gielen J, Laton J, Nagels G. Assessing PML risk under immunotherapy: if all you have is a hammer, everything looks like a nail. Mult Scler 2015 Jul 21
Schwab N, Schneider-Hohendorf T, Wiendl H. Trafficking of lymphocytes into the CNS. Oncotarget 2015;6:17863-17864.
Schneider-Hohendorf T, Rossaint J, Mohan H, et al. VLA-4 blockade promotes differential routes into human CNS involving PSGL-1 rolling of T cells and MCAM-adhesion of TH17 cells. J Exp Med 2014;211:1833-1846.
Havla J, Gerdes LA, Meinl I, et al. De-escalation from natalizumab in multiple sclerosis: recurrence of disease activity despite switching to glatiramer acetate. J Neurol 2011;258:1665-1669.
Killestein J, Vennegoor A, Strijbis EM, et al. Natalizumab drug holiday in multiple sclerosis: poorly tolerated. Ann Neurol 2010;68:392-395.
Rinaldi F, Seppi D, Calabrese M, Perini P, Gallo P. Switching therapy from natalizumab to fingolimod in relapsing-remitting multiple sclerosis: clinical and magnetic resonance imaging findings. Mult Scler 2012;18:1640-1643.
Barnett MH, Prineas JW, Buckland ME, Parratt JD, Pollard JD. Massive astrocyte destruction in neuromyelitis optica despite natalizumab therapy. Mult Scler 2012;18:108-112.
Kleiter I, Hellwig K, Berthele A, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol 2012;69:239-245.
Limmroth V, Barkhof F, Desem N, Diamond MP, Tachas G. CD49d antisense drug ATL1102 reduces disease activity in patients with relapsing-remitting MS. Neurology 2014;83:1780-1788.
Jovani M, Danese S. Vedolizumab for the treatment of IBD: a selective therapeutic approach targeting pathogenic a4b7 cells. Curr Drug Targets 2013;14:1433-1443.
Engelhardt B, Laschinger M, Schulz M, Samulowitz U, Vestweber D, Hoch G. The development of experimental autoimmune encephalomyelitis in the mouse requires alpha4-integrin but not alpha4beta7-integrin. J Clin Invest 1998;102:2096-2105.
Haanstra KG, Hofman SO, Lopes Estevao DM, et al. Antagonizing the alpha4beta1 integrin, but not alpha4beta7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J Immunol 2013;190:1961-1973.
Coles AJ, Cox A, Le Page E, et al. The window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapy. J Neurol 2006;253:98-108.
Rodig SJ, Abramson JS, Pinkus GS, et al. Heterogeneous CD52 expression among hematologic neoplasms: implications for the use of alemtuzumab (CAMPATH-1H). Clin Cancer Res 2006;12:7174-7179.
Ginaldi L, De Martinis M, Matutes E, et al. Levels of expression of CD52 in normal and leukemic B and T cells: correlation with in vivo therapeutic responses to Campath-1H. Leuk Res 1998;22:185-191.
Ruck T, Bittner S, Wiendl H, Meuth SG. Alemtuzumab in multiple sclerosis: mechanism of action and beyond. Int J Mol Sci 2015;16:16414-16439.
Thompson SA, Jones JL, Cox AL, Compston DA, Coles AJ. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J Clin Immunol 2010;30:99-105.
Jones JL, Thompson SA, Loh P, et al. Human autoimmunity after lymphocyte depletion is caused by homeostatic T-cell proliferation. Proc Natl Acad Sci U S A 2013;110:20200-20205.
Zhang X, Tao Y, Chopra M, et al. Differential reconstitution of T cell subsets following immunodepleting treatment with alemtuzumab (anti-CD52 monoclonal antibody) in patients with relapsing–remitting multiple sclerosis. J Immunol 2013;191:5867-5874.
Stinissen P, Hellings N. Activation of myelin reactive T cells in multiple sclerosis: a possible role for T cell degeneracy? Eur J Immunol 2008;38:1190-1193.
Coles AJ, Fox E, Vladic A, et al. Alemtuzumab more effective than interferon beta-1a at 5-year follow-up of CAMMS223 clinical trial. Neurology 2012;78:1069-1078.
Rau D, Lang M, Harth A, et al. Listeria meningitis complicating alemtuzumab treatment in multiple sclerosis—report of two cases. Int J Mol Sci 2015;16:14669-14676.
Daniels GH, Vladic A, Brinar V, et al. Alemtuzumab-related thyroid dysfunction in a phase 2 trial of patients with relapsing-remitting multiple sclerosis. J Clin Endocrinol Metab 2014;99:80-89.
Tuohy O, Costelloe L, Hill-Cawthorne G, et al. Alemtuzumab treatment of multiple sclerosis: long-term safety and efficacy. J Neurol Neurosurg Psychiatry 2015;86:208-215.
Havrdova E, Horakova D, Kovarova I. Alemtuzumab in the treatment of multiple sclerosis: key clinical trial results and considerations for use. Ther Adv Neurol Disord 2015;8:31-45.
Hartung HP, Aktas O, Boyko AN. Alemtuzumab: a new therapy for active relapsing-remitting multiple sclerosis. Mult Scler 2015;21:22-34.
Bandala-Sanchez E, Zhang Y, Reinwald S, et al. T cell regulation mediated by interaction of soluble CD52 with the inhibitory receptor Siglec-10. Nat Immunol 2013;14:741-748.
Watanabe T, Masuyama J, Sohma Y, et al. CD52 is a novel costimulatory molecule for induction of CD4+ regulatory T cells. Clin Immunol 2006;120:247-259.
Milo R. The efficacy and safety of daclizumab and its potential role in the treatment of multiple sclerosis. Ther Adv Neurol Disord 2014;7:7-21.
Bielekova B, Catalfamo M, Reichert-Scrivner S, et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A 2006;103:5941-5946.
Wynn D, Kaufman M, Montalban X, et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol 2010;9:381-390.
Bielekova B, Howard T, Packer AN, et al. Effect of anti-CD25 antibody daclizumab in the inhibition of inflammation and stabilization of disease progression in multiple sclerosis. Arch Neurol 2009;66:483-489.
Bielekova B, Richert N, Herman ML, et al. Intrathecal effects of daclizumab treatment of multiple sclerosis. Neurology 2011;77:1877-1886.
Zhang B, Yamamura T, Kondo T, Fujiwara M, Tabira T. Regulation of experimental autoimmune encephalomyelitis by natural killer (NK) cells. J Exp Med 1997;186:1677-1687.
Chanvillard C, Jacolik RF, Infante-Duarte C, Nayak RC. The role of natural killer cells in multiple sclerosis and their therapeutic implications. Front Immunol 2013;4:63.
Wuest SC, Edwan JH, Martin JF, et al. A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy. Nat Med 2011;17:604-609.
Wiendl H, Gross CC. Modulation of IL-2Ralpha with daclizumab for treatment of multiple sclerosis. Nat Rev Neurol 2013;9:394-404.
Kappos L, Wiendl H, Selmaj K, et al. Daclizumab HYP versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N Engl J Med 2015;373:1418-1428.
Bielekova B, Richert N, Howard T, et al. Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon beta. Proc Natl Acad Sci U S A 2004;101:8705-8708.
Rojas MA, Carlson NG, Miller TL, Rose JW. Long-term daclizumab therapy in relapsing-remitting multiple sclerosis. Ther Adv Neurol Disord 2009;2:291-297.
Wiendl H, Hohlfeld R. Therapeutic approaches in multiple sclerosis: lessons from failed and interrupted treatment trials. BioDrugs 2002;16:183-200.
Hafler DA, Ritz J, Schlossman SF, Weiner HL. Anti-CD4 and anti-CD2 monoclonal antibody infusions in subjects with multiple sclerosis. Immunosuppressive effects and human anti-mouse responses. J Immunol 1988;141:131-138.
Weinshenker BG, Bass B, Karlik S, Ebers GC, Rice GP. An open trial of OKT3 in patients with multiple sclerosis. Neurology 1991;41:1047-1052.
Lindsey JW, Hodgkinson S, Mehta R, Mitchell D, Enzmann D, Steinman L. Repeated treatment with chimeric anti-CD4 antibody in multiple sclerosis. Ann Neurol 1994;36:183-189.
Lindsey JW, Hodgkinson S, Mehta R, et al. Phase 1 clinical trial of chimeric monoclonal anti-CD4 antibody in multiple sclerosis. Neurology 1994;44:413-419.
van Oosten BW, Lai M, Barkhof F, et al. A phase II trial of anti-CD4 antibodies in the treatment of multiple sclerosis. Mult Scler 1996;1:339-342.
Becher B, Durell BG, Noelle RJ. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest 2002;110:493-497.
Chen Y, Langrish CL, McKenzie B, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest 2006;116:1317-1326.
Komiyama Y, Nakae S, Matsuki T, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 2006;177:566-573.
Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005;201:233-240.
Segal BM, Dwyer BK, Shevach EM. An interleukin (IL)-10/IL-12 immunoregulatory circuit controls susceptibility to autoimmune disease. J Exp Med 1998;187:537-546.
Hohlfeld R, Wekerle H. Autoimmune concepts of multiple sclerosis as a basis for selective immunotherapy: from pipe dreams to (therapeutic) pipelines. Proc Natl Acad Sci U S A 2004;101(Suppl. 2):14599-14606.
Brok HP, van Meurs M, Blezer E, et al. Prevention of experimental autoimmune encephalomyelitis in common marmosets using an anti-IL-12p40 monoclonal antibody. J Immunol 2002;169:6554-6563.
Kasper LH, Everitt D, Leist TP, et al. A phase I trial of an interleukin-12/23 monoclonal antibody in relapsing multiple sclerosis. Curr Med Res Opin 2006;22:1671-1678.
Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol 2008;7:796-804.
Aranami T, Yamamura T. Th17 Cells and autoimmune encephalomyelitis (EAE/MS). Allergol Int 2008;57:115-120.
Ulzheimer JC, Meuth SG, Bittner S, Kleinschnitz C, Kieseier BC, Wiendl H. Therapeutic approaches to multiple sclerosis: an update on failed, interrupted, or inconclusive trials of immunomodulatory treatment strategies. BioDrugs 2010;24:249-274.
Couzin J. Drug discovery. Magnificent obsession. Science 2005;307:1712-1715.
Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med 2000;6:114.
Miller DH, Weber T, Grove R, et al. Firategrast for relapsing remitting multiple sclerosis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2012;11:131-139.
O'Connor P, Miller D, Riester K, et al. Relapse rates and enhancing lesions in a phase II trial of natalizumab in multiple sclerosis. Mult Scler 2005;11:568-572.
Bowen JD, Petersdorf SH, Richards TL, et al. Phase I study of a humanized anti-CD11/CD18 monoclonal antibody in multiple sclerosis. Clin Pharmacol Ther 1998;64:339-346.
Kleinschnitz C, Meuth SG, Wiendl H. The trials and errors in MS therapy. Int MS J 2008;15:79-90.
Schwab N, Ulzheimer JC, Fox RJ, et al. Fatal PML associated with efalizumab therapy: insights into integrin alphaLbeta2 in JC virus control. Neurology 2012;78:458-467.
Lutterotti A, Martin R. Antigen-specific tolerization approaches in multiple sclerosis. Expert Opin Investig Drugs 2014;23:9-20.
Miller A, Hafler DA, Weiner HL. Tolerance and suppressor mechanisms in experimental autoimmune encephalomyelitis: implications for immunotherapy of human autoimmune diseases. FASEB J 1991;5:2560-2566.
Bitar DM, Whitacre CC. Suppression of experimental autoimmune encephalomyelitis by the oral administration of myelin basic protein. Cell Immunol 1988;112:364-370.
Higgins PJ, Weiner HL. Suppression of experimental autoimmune encephalomyelitis by oral administration of myelin basic protein and its fragments. J Immunol 1988;140:440-445.
Weiner HL, Mackin GA, Matsui M, et al. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science 1993;259:1321-1324.
Faria AM, Weiner HL. Oral tolerance. Immunol Rev 2005;206:232-259.
Warren KG, Catz I, Ferenczi LZ, Krantz MJ. Intravenous synthetic peptide MBP8298 delayed disease progression in an HLA Class II-defined cohort of patients with progressive multiple sclerosis: results of a 24-month double-blind placebo-controlled clinical trial and 5 years of follow-up treatment. Eur J Neurol 2006;13:887-895.
Warren KG, Catz I, Wucherpfennig KW. Tolerance induction to myelin basic protein by intravenous synthetic peptides containing epitope P85 VVHFFKNIVTP96 in chronic progressive multiple sclerosis. J Neurol Sci 1997;152:31-38.
Freedman MS, Bar-Or A, Oger J, et al. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology 2011;77:1551-1560.
Goodkin DE, Shulman M, Winkelhake J, et al. A phase I trial of solubilized DR2:MBP84-102 (AG284) in multiple sclerosis. Neurology 2000;54:1414-1420.
Yadav V, Bourdette DN, Bowen JD, et al. Recombinant T-cell receptor ligand (RTL) for treatment of multiple sclerosis: a double-blind, placebo-controlled, phase 1, dose-escalation study. Autoimmune Dis 2012;2012:954739.
Lutterotti A, Yousef S, Sputtek A, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med 2013;5:188ra75.
Walczak A, Siger M, Ciach A, Szczepanik M, Selmaj K. Transdermal application of myelin peptides in multiple sclerosis treatment. JAMA Neurol 2013;70:1105-1109.
Streeter HB, Rigden R, Martin KF, Scolding NJ, Wraith DC. Preclinical development and first-in-human study of ATX-MS-1467 for immunotherapy of MS. Neurol Neuroimmunol Neuroinflamm 2015;2:e93.
Evavold BD, Allen PM. Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand. Science 1991;252:1308-1310.
Racioppi L, Ronchese F, Matis LA, Germain RN. Peptide-major histocompatibility complex class II complexes with mixed agonist/antagonist properties provide evidence for ligand-related differences in T cell receptor-dependent intracellular signaling. J Exp Med 1993;177:1047-1060.
De Magistris MT, Alexander J, Coggeshall M, et al. Antigen analog-major histocompatibility complexes act as antagonists of the T cell receptor. Cell 1992;68:625-634.
Bielekova B, Goodwin B, Richert N, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med 2000;6:1167-1175.
Crowe PD, Qin Y, Conlon PJ, Antel JP. NBI-5788, an altered MBP83-99 peptide, induces a T-helper 2-like immune response in multiple sclerosis patients. Ann Neurol 2000;48:758-765.
Kappos L, Comi G, Panitch H, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat Med 2000;6:1176-1182.
Medaer R, Stinissen P, Truyen L, Raus J, Zhang J. Depletion of myelin-basic-protein autoreactive T cells by T-cell vaccination: pilot trial in multiple sclerosis. Lancet 1995;346:807-808.
Zhang J, Medaer R, Stinissen P, Hafler D, Raus J. MHC-restricted depletion of human myelin basic protein-reactive T cells by T cell vaccination. Science 1993;261:1451-1454.
Karussis D, Shor H, Yachnin J, et al. T cell vaccination benefits relapsing progressive multiple sclerosis patients: a randomized, double-blind clinical trial. PLoS One 2012;7:e50478.
Fox E, Wynn D, Cohan S, Rill D, McGuire D, Markowitz C. A randomized clinical trial of autologous T-cell therapy in multiple sclerosis: subset analysis and implications for trial design. Mult Scler 2012;18:843-852.
Loftus B, Newsom B, Montgomery M, et al. Autologous attenuated T-cell vaccine (Tovaxin) dose escalation in multiple sclerosis relapsing-remitting and secondary progressive patients nonresponsive to approved immunomodulatory therapies. Clin Immunol 2009;131:202-215.
Vandenbark AA, Chou YK, Whitham R, et al. Treatment of multiple sclerosis with T-cell receptor peptides: results of a double-blind pilot trial. Nat Med 1996;2:1109-1115.
Vandenbark AA, Culbertson NE, Bartholomew RM, et al. Therapeutic vaccination with a trivalent T-cell receptor (TCR) peptide vaccine restores deficient FoxP3 expression and TCR recognition in subjects with multiple sclerosis. Immunology 2008;123:66-78.
Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med 2014;371:326-338.
Sigurgeirsson B, Kircik L, Nemoto O, et al. Secukinumab improves the signs and symptoms of moderate-to-severe plaque psoriasis in subjects with involvement of hands and/or feet: subanalysis of a randomized, double-blind, placebo-controlled, phase 2 dose-ranging study. J Eur Acad Dermatol Venereol 2014;28:1127-1129.
Genovese MC, Durez P, Richards HB, et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann Rheum Dis 2013;72:863-869.
Fernandez O, Alvarez-Cermeno JC, Arnal-Garcia C, et al. Review of the novelties presented at the 29th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) (III). Rev Neurol 2014;59:371-379.
Wiendl H, Dahlke F, Bennett D, Rosenkranz G, Wolf C, Bar-Or A. IL-17 neutralisation by subcutaneous CJM112, a fully human anti-IL-17A monoclonal antibody for the treatment of relapsing-remitting multiple sclerosis: study desing of a phase 2 trial. ECTRIMS 2015. 2015;Poster Number P655.
Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet 2015;386:541-551.
Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med 2015;373:1318-1328.
Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459-465.
Hurwitz AA, Sullivan TJ, Krummel MF, Sobel RA, Allison JP. Specific blockade of CTLA-4/B7 interactions results in exacerbated clinical and histologic disease in an actively-induced model of experimental allergic encephalomyelitis. J Neuroimmunol 1997;73:57-62.
Karandikar NJ, Eagar TN, Vanderlugt CL, Bluestone JA, Miller SD. CTLA-4 downregulates epitope spreading and mediates remission in relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol 2000;109:173-180.
Viglietta V, Bourcier K, Buckle GJ, et al. CTLA4Ig treatment in patients with multiple sclerosis: an open-label, phase 1 clinical trial. Neurology 2008;71:917-924.
Dubey D, Kieseier BC, Hartung HP, Hemmer B, Miller-Little WA, Stuve O. Clinical management of multiple sclerosis and neuromyelitis optica with therapeutic monoclonal antibodies: approved therapies and emerging candidates. Expert Rev Clin Immunol 2015;11:93-108.
Hoffjan S, Akkad DA. The genetics of multiple sclerosis: an update 2010. Mol Cell Probes 2010;24:237-243.
Sasson SC, Zaunders JJ, Kelleher AD. The IL-7/IL-7 receptor axis: understanding its central role in T-cell homeostasis and the challenges facing its utilization as a novel therapy. Curr Drug Targets 2006;7:1571-1582.
Grigorian A, Mkhikian H, Demetriou M. Interleukin-2, Interleukin-7, T cell-mediated autoimmunity, and N-glycosylation. Ann N Y Acad Sci 2012;1253:49-57.
Kim HP, Imbert J, Leonard WJ. Both integrated and differential regulation of components of the IL-2/IL-2 receptor system. Cytokine Growth Factor Rev 2006;17:349-366.
Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 1976;193:1007-1008.
Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med 2005;201:723-735.
D'Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol 2005;6:1152-1159.
Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol 2005;6:1142-1151.
Kleinewietfeld M, Hafler DA. Regulatory T cells in autoimmune neuroinflammation. Immunol Rev 2014;259:231-244.
Helling B, Konig M, Dalken B, et al. A specific CD4 epitope bound by tregalizumab mediates activation of regulatory T cells by a unique signaling pathway. Immunol Cell Biol 2015;93:396-405.
Tsunoda I, Terry EJ, Marble BJ, Lazarides E, Woods C, Fujinami RS. Modulation of experimental autoimmune encephalomyelitis by VLA-2 blockade. Brain Pathol 2007;17:45-55.
Werr J, Johansson J, Eriksson EE, Hedqvist P, Ruoslahti E, Lindbom L. Integrin alpha(2)beta(1) (VLA-2) is a principal receptor used by neutrophils for locomotion in extravascular tissue. Blood 2000;95:1804-1809.
Warnke C, Kieseier BC, Hartung HP. Biotherapeutics for the treatment of multiple sclerosis: hopes and hazards. J Neural Transm 2013;120(Suppl. 1):S55-S60.
Acknowledgments
We report no conflict of interest. S.B. has received honoraria for lecturing, travel expenses for attending meetings, and financial research support from Bayer Schering AG, Biogen Idec, Novartis, and TEVA. H.W. has received honoraria for lecturing, advisory board activities, travel expenses for attending meetings, and financial research support from Bayer Vital GmbH, Bayer Schering AG, Biogen Idec, CSL Behring, Fresenius Medical Care, Genzyme, GlaxoSmithKline, GW Pharmaceuticals, Lundbeck, Merck Serono, Omniamed, Novartis, Roche, and Sanofi. This work was funded by the Deutsche Forschungsgesellschaft (DFG; grant CRC128 A9 and B1 to HW, and grant BI1822/1-1 to SB) and the Kompetenznetz Multiple Sklerose (Competence Network for Multiple Sclerosis) funded by the Federal Ministry of Education and Research (FKZ 01GI1308B 01GI0907) to HW.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1225 kb)
Rights and permissions
About this article

Cite this article
Bittner, S., Wiendl, H. Neuroimmunotherapies Targeting T Cells: From Pathophysiology to Therapeutic Applications. Neurotherapeutics 13, 4–19 (2016). https://doi.org/10.1007/s13311-015-0405-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-015-0405-3