
Abstract
Immunotherapy has been investigated in a small subset of peripheral neuropathies, including an acute one, Guillain-Barré syndrome, and 3 chronic forms: chronic inflammatory demyelinating polyradiculoneuropathy, multifocal motor neuropathy, and neuropathy associated with IgM anti-myelin-associated glycoprotein. Several experimental studies and clinical data are strongly suggestive of an immune-mediated pathogenesis. Either cell-mediated mechanisms or antibody responses to Schwann cell, compact myelin, or nodal antigens are considered to act together in an aberrant immune response to cause damage to peripheral nerves. Immunomodulatory treatments used in these neuropathies aim to act at various steps of this pathogenic process. However, there are many phenotypic variants and, consequently, there is a significant difference in the response to immunotherapy between these neuropathies, as well as a need to improve our knowledge and long-term management of chronic forms.
Similar content being viewed by others


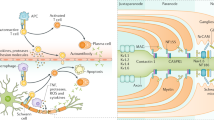
Background
Immunotherapy has investigated over the last few years with regard to a small subset of peripheral neuropathies classed as autoimmune diseases, that is, neuropathies in which an aberrant immune response is directed to components of the peripheral nervous system, leading to demyelination and sometimes axonal damage. The spectrum of these immune-mediated neuropathies mainly encompasses an acute one, Guillain-Barré syndrome (GBS), and 3 chronic forms, namely chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), multifocal motor neuropathy (MMN), and distal acquired demyelinating symmetric neuropathy associated with IgM anti-myelin-associated glycoprotein (MAG) (anti-MAG neuropathy). GBS was firstly reported almost 100 years ago [1], while CIDP, MMN, and anti-MAG neuropathy have been progressively identified since 1958, followed by characterization of clinical, electrophysiological, pathological, and immunological features [2]. In the same way, animal models have been elegantly produced by several researchers, and immune therapies have firstly been used in GBS [3], followed by CIDP [4], and more recently in MMN and anti-MAG neuropathy [5, 6]. Even though the exact mechanisms underlying the development of immunopathology remain unknown, immune-mediated neuropathies are considered treatable. The main issue for immunotherapy is the great variability in clinical presentation and course of these diseases, together with the different responses to treatment according to the clinical phenotypes. In the same vein, new subsets have been recently reported in the spectrum of CIDP, which may lead to different treatment options [7, 8].
In this review, we discuss different issues in the management of immune-mediated neuropathies, and give an update on current treatment options, in providing the best strategies in short- and long-term therapy.
GBS
Onset of GBS typically includes numbness, paresthesia, and weakness in the limbs. The main features of GBS are its rapid progression with bilateral and relatively symmetric weakness of the limbs with or without involvement of respiratory or cranial nerves. Maximal weakness is achieved within 4 weeks but most patients reach maximal involvement within 2 weeks. Patients then have a plateau phase of variable duration from days to several weeks or months. This phase is followed by a recovery phase, which extends over weeks to months. Despite immunotherapy, about 20 % of the “severely affected” patients remain unable to walk independently after 6 months. Moreover, many patients remain disabled, with motor or sensory persistent deficit, pain, or severe fatigue [9, 10]. GBS often remains a severe disease for which better treatment is required, at least in a subgroup of patients [11].
Plasma exchanges (PLEX) was first shown to be efficacious in GBS when given within the first 4 weeks from onset; however, it is more beneficial when applied within the first 2 weeks [12]. A second randomized controlled trial (RCT) found that 2 PLEX treatments are more effective than no PLEX in mildly affected patients (i.e., those that are able to walk) [13]. The first RCT with intravenous immunoglobulin (IVIG) was published in 1992 and demonstrated that IVIG given at 0.4 g/kg bodyweight/day for 5 consecutive days was as effective as PLEX [14]. A Cochrane review on the use of IVIG in GBS showed that there was no difference between IVIG and PLEX with respect to the improvement in disability grade after 4 weeks, the duration of mechanical ventilation, mortality, and residual disability [15]. In addition, in another RCT, the combination of PLEX followed by IVIG was not significantly better than PLEX or IVIG alone [16]. An interesting issue is the inefficacy of steroids in GBS, even when given as part of an IV regimen (IV methylprednisolone) in combination with IVIG [17].
Several issues remain unknown: 1) the appropriate treatment for patients presenting with various grades of disability, mainly mildly affected patients; 2) the appropriate treatment for patients who deteriorate after a first course of PLEX/IVIG; 3) the appropriate treatment in patients who deteriorate after initial improvement or stabilization following IVIG therapy, a condition known as “treatment-related clinical fluctuation”; 4) the distinction between patients with GBS with treatment-related clinical fluctuation and patients with acute-onset CIDP [18]. To try to answer to some of these points, the International GBS Outcome Study is a worldwide prospective outcome study that will recruit and follow 1000–1500 patients with GBS. It is likely that a wealth of new information on GBS will be available in the coming years. A key point is to identify patients with a poor prognosis; scales have been created to do this, including the Erasmus GBS Respiratory Insufficiency Scale, which evaluates the need for artificial ventilation at admission, and the Erasmus GBS Outcome Scale, which has been constructed and validated to determine outcome at 6 months [19]. In addition, it has been shown that a larger increase in serum IgG levels, 2 weeks after starting the first IVIG course, is associated with a better outcome [20]. Recently, some experts, under the umbrella of the Inflammatory Neuropathy Consortium, a special interest group of the Peripheral Nerve Society (PNS), aimed to explore the utility of a second IVIG infusion in patients who do not respond to a first IVIG infusion. An ongoing RCT (SID-GBS) is currently studying the effect of a second dose of IVIG given shortly after the first IVIG course only in patients with GBS with a poor prognosis, measured by the modified Erasmus GBS Outcome Scale. An international observational study on the effect of a second dose of IVIG in patients with GBS with a poor prognosis (I-SID-GBS) has just started.
CIDP
CIDP was first identified in 1958 [21]. During the 1970s increasingly large series of patients were published, notably by Dyck et al. [22] at the Mayo Clinic. It is the most common treatable neuropathy worldwide [4, 23–25], with a prevalence ranging from 2.0–3.0 to 8.9 per 100,000 people [26]. In a recently reported epidemiological study, mean age of onset was 57.7 years, prevalence increased with age and reached a maximum in the 70–79-year age group [27]. CIDP is believed to have an autoimmune etiology. However, the criteria for diagnosis are often vague, and there are no specific or reliable biomarkers to aid in establishing the diagnosis. In 1991, a first attempt to diagnose CIDP was based on clinical, electrophysiological, pathological, and cerebrospinal fluid features [28]. The more frequently used guideline for CIDP was revised in 2010, and proposed new criteria for diagnosis [29]. The validity of these criteria was established in a comparative study [30]. The usual clinical picture consists of symmetrical motor weakness in the proximal and distal muscles of the 4 extremities for at least 2 months, sensory involvement predominantly in large myelinated fibers, and hypo- or areflexia. Electrophysiological investigations show demyelinating features, that is, slowed motor nerve conduction velocities, prolongation of motor distal latencies, absence or prolongation of F-waves, and conduction blocks (CB) on motor nerves. In addition, supportive criteria obtained from cerebrospinal fluid examination and magnetic resonance imaging of the peripheral nerve roots may be helpful for diagnosis.
The presence on nerve biopsies of “onion bulb” formations, perivascular inflammatory infiltrates, and patterns of demyelination/remyelination are hallmarks of CIDP. The mechanisms that lead to demyelination of peripheral nerves are poorly understood but are believed to be mediated by both cellular and humoral immune factors. No clear correlation has yet been demonstrated between CIDP and specific serum autoantibodies, although antiganglioside antibodies and antimyelin antibodies are found in a proportion of patients. Recent challenges have focused on finding new serological biomarkers for CIDP. Antibodies binding to contactin-1 and contactin-associated protein 1 (CNTN1/CASPR) seem to correlate with older age and more aggressive onset, predominantly motor involvement with early secondary axonal degeneration and poor response to IVIG [31]. Similarly, patients with autoantibodies binding to neurofascin 155 of the IgG4 isotype could constitute a specific subgroup with severe deficit, poor response to IVIG, and disabling tremor [32].However, overall these antibodies have been found in a very small proportion in patients.
CIDP has an unpredictable course, although the majority of patients have a severe motor, sensory, or sensorimotor disabling status after several years. In a recent population-based study, the nadir overall neuropathy limitation scale score was 5, and 58 % of patients were unable to walk independently at some points of their illness [27]. In one of the largest reported studies between 3 % and 7 % of patients died during follow-up [33].
Based on consensus expert opinion, diagnosis of CIDP leads to typical CIDP being distinguished from other atypical presentations, including sensory CIDP [34, 35], multifocal acquired demyelinating sensory and motor neuropathy [36, 37], and distal acquired demyelinating symmetric neuropathy [38, 39]. Although there may be overlap between these presentations, this distinction seems appropriate as response to treatment may differ among each presentation, favoring the hypothesis that different pathogenic mechanisms may underlie the heterogeneity of CIDP [24, 25].
We will now consider first-line therapy of CIDP, then long-term treatment options.
First-line Treatment for CIDP
Corticosteroids, PLEX, and IVIg have proven efficacy for CIDP. In a recent study, up to 80 % of patients responded to at least 1 of 3 first-line therapies [27]. Patients with high relapse rates may become treatment dependent, which leads to a search for factors associated with treatment dependence [40].
Corticosteroids
Corticosteroids likely exert their benefits via several mechanisms. They act as inhibitors of T-cell proliferation and T-cell-dependent immunity. They have also been shown to interfere with the activity of cytokines, decreasing the expression of several proinflammatory cytokines, such as interleukin (IL)-1, IL-2, IL-6, and tumor necrosis factor, and increasing the expression of anti-inflammatory cytokines such as IL-4 and IL-10 [41].
In a historical RCT, corticosteroids were shown to be more effective than no treatment [42]. A recently reported trial compared pulsed high-dose dexamethasone (40 mg/day for 4 days followed by placebo for 24 days; 6 cycles) with standard prednisolone treatment (60 mg/day for 5 weeks and tapering to alternate day doses and then zero over the next 27 weeks) in 40 patients [43]. Twenty-four patients received dexamethasone and 16 received prednisolone. At 12 months, 16 patients were in remission (40 % of patients—10 in the dexamethasone group, 6 in the prednisolone group) without a significant difference between the 2 groups. Most adverse events were minor and did not differ substantially between groups. In long-term evaluation of these patients, median time to relapse ranged from 11 months for oral prednisolone to 17.5 months for pulsed dexamethasone [44].
In pure motor CIDP, corticosteroids have been associated with clinical increase of motor deficit. In these cases, corticosteroids should not be first treatment option and if they are used, close monitoring for deterioration is strongly recommended [29].
In summary, although corticosteroids are well documented and inexpensive treatment option for patients with CIDP, the serious complications that may arise during their prolonged use are a major limitation. In addition, some of these side effects (i.e., aseptic necrosis of the head, sepsis) may add substantially to the total cost of this therapy.
PLEX
It has been emphasized that humoral effector mechanisms may underlie or at least participate in the pathogenesis of CIDP. Removal of these pathogenic humoral factors may therefore be responsible for the recovery of nerve conduction velocity in patients with CIDP after PLEX, thereby providing a rationale for the use of PLEX in CIDP. PLEX is therefore a well-established and well-tolerated treatment of patients with CIDP [45]. However, as with corticosteroids, relapses occur in the majority of patients, and PLEX has severe limitations: it is invasive, time-consuming, expensive, and can only be performed in specialized centers, with specific equipment and well-trained teams. For these reasons, the European Federation of Neurological Societies (EFNS)/PNS guidelines concluded that either corticosteroids or IVIG should be considered first. However, the presence of relative contraindications to any of these treatments should favor the use of PLEX [29].
IVIG
The mechanism by which IVIG exerts its immunomodulatory effect in CIDP is poorly understood. Theories include neutralization of pathogenic autoantibodies by anti-idiotypes, attenuation of complement-mediated tissue damage, saturation and functional blockade of Fc receptors on macrophages, modulation of proinflammatory cytokines/T-cell mediators, and decrease in autoantibody production through the binding of anti-idiotypes to antigen receptors on B cells [46].
In the ICE study [47], 10 % IVIG [10 % caprylate-chromatography purified immune globulin (IGIV-C)] efficacy and safety has been studied in 117 patients with CIDP enrolled in a randomized, double-blind, placebo-controlled, response-conditional crossover trial. IGIV-C or placebo was given at the initial dose of 2 g/kg, then 1 g/kg every 3 weeks for up to 24 weeks. Absence of improvement in adjusted Inflammatory Neuropathy Cause and Treatment (INCAT) disability score of ≥ 1 point conducted to switch into the alternate treatment in a crossover period. Primary outcome was the percentage of patients who had maintained an improvement from baseline through to week 24 of follow-up in adjusted INCAT disability score of at least 1 point. Patients who showed an improvement and completed 24 weeks of treatment were eligible to be randomly re-assigned in a blinded 24-week extension phase. During the first period, 32/59 (54 %) patients treated with IGIV-C and 12/58 (21 %) patients who received placebo had an improvement in adjusted INCAT disability score that was maintained through to week 24 (p = 0.0002).. Similar results were observed during the crossover period. During the extension phase, participants who continued to receive IGIV-C had a longer time to relapse than placebo-treated patients (p = 0.011). Moreover long-term therapy with IGIV-C, showed a maintained improvement in health-related quality of life in patients with CIDP [48]. The PRIMA study [49] confirmed these results with another IVIG 10 % brand.
A Cochrane review recently summarized IVIG studies in CIDP, pointing to an improvement of disability, for at least 2–6 weeks, compared with placebo, and having a similar efficacy to PLEX, oral prednisolone, and intravenous methylprednisolone (see below) [50].
Despite efficacy, a high proportion of patients with CIDP patients remain dependent on IVIG treatment [40]. This fact justifies the search for new ways of administering IVIG, such as home infusions, in order to maximize the benefits of therapy and render side effects, and social and economic impacts, minimal. In that sense, some studies have reported the use of subcutaneous immunoglobulin (SCIG). SCIG has no established difference in half-life, and is usually given at lower doses and more frequent intervals than IVIG. The consequence is a higher and more stable IgG level, which may prevent end-of-dose reduction in efficacy and minimize side effects [51]. A double-blind, placebo-controlled RCT of SCIG, performed in 30 patients with CIDP who were previously responders to IVIG, showed that there was no significant difference in Medical Research Council (MRC) sum score or grip dynamometry, but there was a significant improvement in isokinetic muscle strength in SCIG-treated patients [52]. Moreover, 70 % of patients preferred SCIG administration. Finally, SCIG seems to be a valid option in extension studies for the majority of patients avoiding the need of repeated hospitalizations [53, 54]. New studies are currently ongoing to get approval from agencies for SCIG use in CIDP and MMN (see below).
In summary, the efficacy and safety of IVIG/SCIG have been documented in several RCTs for CIDP. However, this treatment remains expensive and time-consuming in the long-term.
Comparative Trials
A comparative study has been published [55], which assessed efficacy and tolerability of IVIG (0.5 g/kg/day for 4 consecutive days) and high-dose intravenous methylprednisolone (IVMP) (0.5 g/day for 4 consecutive days) given every month for 6 consecutive months. This double-blinded RCT was completed in 45 patients (24 IVIG, 21 IVMP). IVMP was more frequently stopped (p = 0.0085) because of lack of efficacy, adverse events, or voluntary withdrawal, compared with IVIG. However, after discontinuation of therapy, patients treated with IVIG had a higher frequency of worsening and more frequently required further treatment (p = 0.0317). In a follow-up study, the authors looked at the proportion of patients and the duration of relapse after therapy discontinuation with a median follow-up of 42 months (range 1–60 months) [56]. Twenty-four of 28 patients (85.7 %) treated with IVIG worsened after discontinuation of therapy, with a comparable number in the IVMP group (10/13 patients; 77 %). Interestingly, the median time to relapse in the IVIG group was 4.5 months compared with 14 months in the IVMP group (p = 0.013) suggesting that, when effective, IVMP may have a longer beneficial effect in CIDP than IVIG.
To summarize, recommendations for the induction of treatment are listed in Table 1, as good practice points of the EFNS/PNS guidelines [29].
Long-term Therapy for CIDP
The usual lack of long-term response to the first-line treatments has led to trials of immunosuppressive drugs. To date, no immunomodulatory treatment has proven efficacy for CIDP. However, in clinical practice, these drugs are often used for CIDP. A retrospective observational study including 110 patients refractory to conventional therapies intended to verify if immunosuppressors/immunomodulators (IA) could increase the number of patients with CIDP responding to treatments [57]. Thirty-seven percent of the 110 patients responded to one IA; the rate of response to each IA was 17–38 %, with the exception of interferon (IFN)-β1a, which was not effective in any treated patient. There was no significant difference in the clinical response to the different IAs. Ciclosporin was significantly associated with more adverse events (p = 0.01). Moreover, no relation of IA response was found with either the presence of axonal damage, age at CIDP onset, disease duration, or response to conventional therapies. We selected in the following the main reported results in some trials with IAs.
IFN-β1a
IFN-β1a has proven efficacy in the treatment of multiple sclerosis [58]. The immunomodulatory properties of IFN-β1a that are believed to mediate its beneficial effects in MS suggest that it could also be effective in CIDP, leading to a prospective, open-label clinical trial studying the safety, tolerability, and efficacy of once-weekly IFN-β1a (Avonex; Biogen, Research Triangle Park, NC, USA) in a cohort of 20 patients with IVIG-resistant CIDP [59]. It showed an improvement in clinical grading but not in grip strength scores. Then, a large RCT was conducted in 67 patients with IVIG-dependent CIDP, followed for 32 weeks, with either 30 or 60 mg of IFN-β1a once or twice weekly [60]. IFN-β1a was not more effective than placebo in reducing the mean dose of IVIG, with 47 % of patients in both groups not needing to restart IVIG after their suspension after 16 weeks of therapy with IFN-β1a. However, the subgroup of patients requiring higher doses of IVIG or with a greater baseline weakness (MRC < 51), could significantly reduce IVIG after IFN-β1a compared with placebo.
Methotrexate
Other possible immunomodulators to be tried in CIDP include methotrexate (MTX), which is a widely used disease-modifying treatment by rheumatologists, on account of its efficacy and safety profile. A multicentre, double-blind RCT aimed to compare MTX with placebo [61]. Response to treatment was considered if there was > 20 % dose reduction in IVIG or corticosteroid. There was no significant difference found between in patients receiving MTX given at 15 mg weekly and those in placebo groups.
Both this study and the study with IFN-β1a showed that a consistent proportion of patients with CIDP are probably overtreated with IVIG or corticosteroids, as therapy could be reduced or suspended without worsening in > 40 %. Another issue may be the lack of statistical correction for stable patients with poorly active disease randomized to placebo, or the choice of outcome measures that do not capture some changes in the disease symptoms and signs. Consequently, there is a need to improve outcome measures in future trials (see below).
Rituximab
Rituximab is a chimeric (mouse/human) monoclonal antibody against CD20+ B lymphocytes, that was mainly used as a disease-modifying therapy in hematological diseases, and more recently in anti-MAG neuropathy [62]. A retrospective study in 13 patients with CIDP (8 with a hematologically associated condition) showed that treatment with rituximab may clinically improve patients (at least 2 points in clinical scales) or reduce previous necessary therapies (9 patients; 7 with hematological diseases) [63]. This study also pointed to a significant correlation between response and shorter disease duration. It was then suggested that rituximab may be an option, especially in CIDP associated with hematological diseases.
Alemtuzumab
Alemtuzumab is a monoclonal antibody against CD52 antigen. Scarce information is reported in patients with CIDP. A study in 7 patients pointed to a prolonged remission or partial response in 4 patients [64]. However, 3/7 patients developed an autoimmune condition (high levels of antithyroperoxidase antibodies in 2 patients, 1 of whom was later diagnosed with Basedow disease, and 1 with autoimmune hemolytic anaemia).
In conclusion, the EFNS/PNS guidelines for maintenance treatment in CIDP are listed in Table 1. If the response is inadequate or the maintenance doses of the initial treatment (IVIG, steroids, or PLEX) result in adverse effects, the other first-line treatment alternatives should be tried before considering combination treatments, or adding an IA may be considered, but there is no sufficient evidence to recommend any particular drug (good pratice point 29).
An RCT with fingolimod as the IA to be used in CIDP is currently ongoing (FORCIDP study).
MMN
First reported in 1986 [65, 66], MMN is a very rare disease, with a prevalence of around 0.6 per 100,000 individuals. It is a purely motor neuropathy, characterized by progressive distal asymmetric limb weakness that usually starts and predominates in the upper limbs, with minimal or no sensory impairment. Nerve conduction studies have found multifocal persistent CB, which are the hallmark of the disease and allow MMN to be distinguished from motor neuron disease. The association of MMN with high serum levels of IgM antibodies against the ganglioside GM1 was then reported, together with the positive effects of immunomodulatory treatments [67].
The abovementioned clinical data and electrophysiologic abnormalities mainly support the diagnosis of MMN, with both of them having been classified as diagnostic criteria in the EFNS/PNS guideline on management of MMN, first revised in 2010 [68]. In addition, the diagnosis may be supported by laboratory and imaging data, as well as immunological features [5, 69].
In 1 study regrouping 88 patients with MMN [70], 18 % of patients reported minimal or no disability of the arms [Overall Disability Sum Score (ODSS) = 0 and 1), 61 % had moderate impairment of the arms (ODSS = 2), and 21 % had severe disability (ODSS = 3).
IVIG has been proven by RCT to improve weakness and disability in patients with MMN, and is therefore now considered to be the gold standard treatment of this disabling disease. Consequently, MMN is considered a treatable disease; however, MMN does not respond to some immunomodulatory treatments that are effective in CIDP, and the effect of IVIG on motor symptoms and signs may decline after several years.
IVIG
IVIG remains the treatment of choice for patients with MMN [5, 71–76]. IVIG is recommended as a first-line therapy, based on a meta-analysis of 4 randomized, double-blind, placebo-controlled, trials [73–76], involving a total of 34 patients: 78 % of study participants had a significant short-term improvement in strength, selected as primary outcome measure, following IVIG treatment compared with 4 % following placebo [77]. Another RCT was conducted in the USA and Canada to assess the efficacy, safety, and tolerability of 10 % liquid IVIG in 44 patients with MMN [78]. Patients were randomized 1:1 to either double-blind treatment of IVIG followed by placebo for 12 weeks each or vice versa. Open-label IVIG was administered for 12 weeks at the beginning and end of the study, for clinical stabilization, and between double-blind periods to prevent a carry-over effect. Mean maximal grip strength of the more affected hand declined 31.38 % during placebo treatment and increased 3.75 % during IVIG treatment (p = 0.005). In addition, in 37.5 % of participants, the disability scale (Guy’s Neurological Disability Score for upper limbs) worsened during placebo but not during IVIG, whereas the converse was observed in 11.9 % (p = 0.021); 69 % of participants switched prematurely from placebo to open-label treatment because of significant deterioration, and 2.4 % switched from blinded treatment to open-label IVIG (p < 0.001). The authors concluded that IVIG was effective in improving both muscle strength and disability in patients with MMN. In line with these results, good practice points for treatment recommended an initial dose of 2 g/kg given over 2–5 consecutive days [68], and, if effective, followed by maintenance therapy, ideally 1 g/kg every 2–4 weeks or 2 g/kg every 1–2 months (Table 2). Limitations in long-term IVIG therapy are the adverse side effects, mainly thromboembolic events; anaphylactic reactions, which may occur in patients with selective IgA deficiency and anti-IgA antibodies; renal tubular necrosis, which may be prevented by detection of pre-existent renal disease and correct hydration; and possible difficulties with venous access.
In the majority, MMN has a chronic slowly or stepwise progressive course. Therapies aim to reduce motor deficit along with CB, slow down ongoing axonal degeneration, or even promote reinnervation and remyelination. However, a minority of patients achieve prolonged remission upon treatment, and most patients need treatment for years. Several long-term retrospective studies have assessed this issue in the last few years. In the first [79], the authors performed a long-term follow-up of 11 patients with MMN, who were treated initially with 1 full course of IVIG (2 g/kg), then 0.4 g/kg every week, followed by maintenance therapy of 1 infusion every 1–7 weeks. During the 4–8-year follow-up period, patients were assessed by MRC sumscore of 20 muscle groups, and some disability scales. Muscle strength improved significantly within 3 weeks of the start of IVIG treatment and was significantly better at the last follow-up examination than before treatment, even though it decreased slightly and significantly during the follow-up period. CB disappeared in 6 nerve segments but new CB appeared in 8 nerve segments during the follow-up period. The authors concluded that IVIG maintenance therapy had a beneficial long-term effect on muscle strength and upper limb disability but may not prevent a slight decrease in muscle strength. Similar results were found by other authors who reported follow-up in 10 patients with MMN responding to an initial course of IVIg with periodic infusion for 5–12 years (mean 8.2 years) [80]. At the last follow-up, only 2 patients had maintained the maximal improvement achieved during therapy, while 8 worsened despite the immunoglobulin dosage. This decline started after 3–7 years (mean 4.8 years) of therapy and correlated with a reduction of distal compound muscle action potential amplitudes. However, another study reported significant and sustained improvement in muscle strength (assessed byMRC score in 8 muscle groups) [81], disability (assessed by Modified Rankin Score), CB, and signs of axonal degeneration on electrophysiological studies in 10 patients with MMN, with a follow-up of 3.5–12.0 years (mean 7.3 years). The authors concluded that long-term IVIG therapy improved muscle strength and functional disability, decreased the number of CBs and the extent of axonal degeneration, and promoted reinnervation. The difference from previous findings may be explained by the different regimens in giving IVIG, the patients in this last study being treated with significantly higher IVIG maintenance doses. In a retrospective study in 40 patients with MMN [82], we found that, within a mean follow-up of 2.2 ± 2.0 years, only 8 patients (22 %) had significant remission, defined as lasting stabilization of clinical improvement > 6 months, without further treatment after initial IVIG therapy during at least 6 months, whereas 25 patients (68 %) were dependent on maintenance IVIG infusions to stabilize their motor condition. Last, a recent retrospective study of 88 patients with MMN [70], within a mean follow-up of 6 years (range 0–17 years), found that 67 patients (76 %) received IVIG maintenance therapy at the time of the study, while 17 patients (19 %) did not use maintenance treatment for several reasons, including lack of beneficial effect (5 patients), and stable disease course without treatment (8 patients). Thirty-five (40 %) patients had unsuccessfully used other immunomodulators, mainly IFN-β and mycophenolate mofetil (MMF).
Subcutaneous Immunoglobulin
The limitations for IVIG led to investigation of immunoglobulin maintenance therapy by subcutaneous administration. A randomized, single-blinded, crossover study was conducted in 9 patients responsive to IVIG and assessed by the dynamometric strength of affected muscles and the SF-36 quality-of-life questionnaire [83]. IVIG and SCIG were equally effective, the mean change in muscle strength after SCIG being 3.6 % versus 4.3 % after IVIG. One patient presented sustained erythema and edema at the injection sites for a few weeks, but all other adverse events with SCIG were mild and transient. After the study, 5/9 patients preferred continuation with SCIG. Another open-label study conducted in 10 patients similarly found that SCIG therapy was feasible and safe and maintained strength as well as IVIG [84]. Finally, a 2-year follow-up study was reported by the authors of the first study in 6 patients with IVIG-responsive MMN [85]. The dosage of SCIG varied between 13 and 51 g per week, corresponding to a volume of 80–320 ml, infused twice or three times weekly. No major side events were reported, including local mild and transient skin reactions. The impairment and disability scores remained stable. Other trials are currently ongoing with SCIG for use in MMN.
Other Immunosuppressant and Immunomodulatory Agents
Corticosteroids and PLEX, which are effective in CIDP, have been reported not to be efficacious in most uncontrolled studies and therefore are not recommended for use in MMN, as they may lead to worsening of the motor condition [68].
However, some patients with MMN do not respond to IVIG, and others require progressively more frequent doses to maintain remission, or have involvement of new motor nerves, despite periodic IVIG infusions. Therefore, there is a need for searching for alternative or adjunctive immunosuppressive therapies [86]. The recommendations of the EFNS/PNS guideline are listed in Table 2 [68]. Among possible alternative/adjunctive therapies, cyclophosphamide and MMF have been the more frequently proposed. In addition, eculizumab has been tried as a specific immunomodulatory agent.
Cyclophosphamide
Several uncontrolled studies, including the historical study [67], have suggested the efficacy of cyclophosphamide as an alternative therapy. However, the EFNS/PNS guidelines concluded this immunosuppressor to be a less desirable therapeutic option, mainly because of its toxicity and lack of evidence of efficacy [68]. As an adjunctive treatment, only 1 uncontrolled study showed a reduction in the frequency of IVIG infusions in 6 patients [87]; however, 3 patients presented severe side effects.
MMF
A randomized, single-centre, placebo-controlled, “add-on” study of MMF 1 g twice a day for 1 year was conducted in 28 patients with MMN [88]. The results failed to show any significant dose reduction in IVIG, nor any difference in muscle strength, functional scores, or IgM anti-GM1 antibody titers between patients having received MMF or placebo.
Eculizumab
The rationale of the treatment with this original immunomodulator is linked to the fact that several experimental studies showed that the pathogenic effect of anti-GM1 antibodies is complement mediated; consequently, inhibition of complement factors may prevent nerve damage. The monoclonal antibody eculizumab binds and neutralizes human complement factor C5 preventing terminal complement activation and membrane lysis via membrane attack complex. Its safety and efficacy has been proven in complement-mediated diseases, mainly paroxysmal nocturnal hemoglobinuria. An open-label clinical trial of eculizumab treatment for 14 weeks was conducted in 13 patients with MMN (in association with IVIG in 10) [89]. The results disclosed a trend towards an improvement in patient-rated subjective scores and increased muscle strength as measured by myometry. In electrophysiological studies, there was a small yet significant net decrease in the median percentage CB across all nerves studied. The authors concluded there was a small treatment effect occurring in some patients that appeared supplementary to an independent of the IVIG treatment effect, and occurred more frequently in patients with higher baseline motor function. This novel trial raises new questions for further research concerning the use of complement inhibitors in the treatment of MMN.
Anti-MAG Neuropathy
Within the spectrum of chronic immune-mediated neuropathies, demyelinating neuropathy associated with IgM monoclonal gammopathy and antibodies against MAG is a distinct entity that typically presents with progressive sensory ataxia and painful paresthesias [90]. Patients present with a striking immunochemical profile, suggesting the possibility of an autoimmune mechanism: monoclonal IgM recognizes a carbohydrate MAG epitope, which is shared with a number of other glycoconjugates involved in cell adhesion, including the Po glycoprotein of myelin, peripheral myelin protein-22, sulfated sphingolipid, and other related glycolipids. The disease may progress slowly over many years in some patients, whereas others develop significant disability mostly due to dysesthesias and ataxia.
There is insufficient evidence from most pilot studies or RCTs on IgM anti-MAG demyelinating neuropathy to recommend any particular immunotherapy [91]. Efficacy of rituximab has been supported by uncontrolled studies [92–95]. In a first RCT conducted in 26 patients [96], intention-to-treat analysis showed improvement after 8 months in 4/13 patients treated with rituximab and none of the 13 patients on placebo (p = 0.096) Excluding 1 patient randomized to the rituximab group who had a normal INCAT score on inclusion, and could not therefore improve, the results were significant (p = 0.036). Another RCT, the rituximab versus placebo study in polyneuropathy associated with anti-MAG IgM monoclonal gammopathy (RIMAG study), involved 54 patients enrolled in 9 centers: 26 patients in the rituximab group (4 weekly infusions of rituximab 375 mg/m2) and 28 patients in the placebo group [62]. The results showed no significant changes in the INCAT sensory sum score, selected as the primary outcome, between rituximab and placebo groups at 1 year, in both intention-to-treat and per-protocol analyses. However, significant changes were observed in per-protocol analysis for the INCAT disability score, the self-evaluation score, and 2 subscores of the SF-36 questionnaire. Unless uncontrolled, some interesting results have been reported with either higher doses of rituximab or combined therapies [97–99].
Improvement in Assessment of Dysimmune Neuropathies for Clinical Trials: Outcome Measures
The selection of outcomes measures for use in clinical trials is a challenge for dysimmune neuropathies. The outcome measures should cover deficit, disability, impairment, and quality of life. They should be simple, valid, reliable, and responsive, and correlate with disease severity. A workshop organized by the European Neuromuscular Centre was held in 2013 and edited recommendations for GBS, CIDP, monoclonal gammopathy of undetermined significance-related polyneuropathy, and MMN, most of them being selected from the peripheral neuropathy outcome measurement standardization (PeriNomS) study [100]. It pointed out a better evaluation of data analysis with the Rasch method, which enables a transformation of ordinal data into interval metric data, increasing the level of precision in the assessment. The Inflammatory Rasch-built Overall Disability scale offers the promise of being a more sensitive measure and is highly recommended in future trials involving patients with GBS and CIDP [101].
Conclusion
This overview aimed to summarize past issues in improving the treatment options for GBS, CIDP, MMN, and anti-MAG neuropathy. They have been outlined in Table 3. The challenges for the future are mainly: 1) to look for potential new biomarkers for diagnosis, including autoantibodies, in line with defined subsets of the disease; 2) to improve the outcome measures for clinical trials, with specific attention to disability scales, mainly the Inflammatory Rasch-built Overall Disability scale; 3) to develop animal models as there is a lack of consistent data in this field; 4) to test a second IVIG dose in GBS and new immunomodulatory/immunosuppressive agents in the long-term treatment for CIDP, MMN, and anti-MAG neuropathy.
References
Guillain G, Barré JA, Strohl A. Sur un syndrome de radiculo-névrite avec hyperalbuminose du liquide céphalo-rachidien sans réaction cellulaire. Remarques sur les caractères cliniques et graphiques des réflexes tendineux. Bull Soc Med Hop Paris 1916; 40:1462-1470.
Hughes R, Léger JM. The discovery of the Guillain-Barré syndrome and related disorders. Presse Med 2013:42:e177-e179.
Hughes RA, Swan AV, Raphael JC, Annane D, van Koningsveld R, van Doorn PA. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain 2007;130:2245-2257.
Vallat JM, Sommer C, Magy L. Chronic inflammatory demyelinating polyradiculoneuropathy: diagnostic and therapeutic challenges for a treatable condition. Lancet Neurol 2010;9:402-412.
Léger JM, Guimaràes-Costa R, Iancu Ferfoglia R. The pathogenesis of multifocal motor neuropathy and an update on current management options. Ther Adv Neurol Disord 2015;8:109-122.
Dalakas MC. Pathogenesis and treatment of anti-MAG neuropathy. Curr Treat Options Neurol 2010;12:71-83.
Miura Y, Devaux JJ, Fukami Y, et al. Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain 2015;138:1484-1491.
Mathey EK, Park SB, Hughes RAC, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry 2015;86:720-728.
Ruts L, Drenthen J, Hop WC, et al. Pain in Guillain-Barré syndrome: a long-term follow-up study. Neurology 2010;75:1439-1447.
Merkies IS, Schmitz PI, Samjin JP, van der Méché FG, van Doorn PA. Fatigue in immune-mediated polyneuropathies. European INCAT Group. Neurology 1999;53:1648-1654.
Van Doorn PA. Diagnosis, treatment and prognosis of Guillain-Barré syndrome. Presse Med 2013;42:e193-e201.
The Guillain-Barré syndrome Study Group. Plasmapheresis and acute Guillain-Barré syndrome. Neurology 1985;35:1096-1104.
The French Cooperative Group on Plasma Exchanges in Guillain-Barré syndrome. Appropriate number of plasma exchanges in Guillain-Barré syndrome. Ann Neurol 1997;41:298-306.
Van der Méché FG, Schmitz PI and the Dutch Guillain-Barré Study Group A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. New Engl J Med 1992;326:1123-1129.
Hughes RA, Svan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev 2012;7:CD002063.
Plasma exchange/Sandoglobulin Guillain-Barré syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin and combined treatments in Guillain-Barré syndrome. Lancet 1997;349:225-230.
Van Koningsveld R, Schmitz PI, Van der Méché FG, Visser LH, Meulstee J, Van Doorn PA. Effects of methylprednisolone when added to standard treatment with intravenous immunoglobulin in Guillain-Barré syndrome: randomized trial. Lancet 2004;363:192-196.
Ruts L, Drenthen J, Jacobs BC, van Doorn PA. Distinguishing acute-onset CIDP from fluctuating Guillain-Barré syndrome: a prospective study. Neurology 2010;74:1680-1686.
Van Koningsveld R, Steyerberg EW, Hughes RA, Swan AV, Van Doorn PA, Jacobs BC. A clinical prognosis scoring system for Guillain-Barré syndrome. Lancet Neurol 2007;6:589-594.
Kuitwaard K, de Gelder J, Tio-Gillen AP, et al. Pharmacokinetics of intravenous immunoglobulin and outcome in Guillain-Barré syndrome. Ann Neurol 2009;66:599-603.
Austin JH. Recurrent polyneuropathies and their corticosteroid treatment. Brain 1958;81:157-192.
Dyck PJ, Lais AC, Ohta M, Bastron JA, Okazaki H, Groover RV. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc 1975;50:621-651.
Eftimov F, van Schaik I. Chronic inflammatory demyelinating polyradiculoneuropathy: update on clinical features, phenotypes and treatment options. Curr Opin Neurol 2013;5:496-502.
Nobile-Orazio E. Chronic inflammatory demyelinating polyradiculoneuropathy and variants: where we are and where we should go. J Peripher Nerv Syst 2014;19:2-13.
Guimaràes-Costa R, Iancu Ferfoglia R, Viala K, Léger JM. Challenges in the treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Rev Neurol 2014;170:595-601.
Laughlin RS, Dyck PJ, Melton LJ, Leibson C, Ransom J, Dyck PJB. Incidence and prevalence of CIDP and the association of diabetes mellitus. Neurology 2009;73:39-45.
Mahdi-Rogers M, Hughes RA. Epidemiology of chronic inflammatory neuropathies in southeast England. Eur J Neurol 2014;21:28-33.
Ad Hoc Subcommittee of the American Academy of Neurology Aids Task Force. Research criteria for the diagnosis of CIDP. Neurology 1991;41:617-618.
Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. J Peripher Nerv Syst 2010;15:1-9.
Rajabally YA, Nicolas G, Piéret F, Bouche P, Van den Bergh PY. Validity of diagnostic criteria for chronic inflammatory demyelinating polyneuropathy: a multicentre European Study. J Neurol Neurosurg Psychiatry 2009;80:1364-1368.
Querol L, Nogales-Gadea G, Rojas-Garcia R, et al. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol 2013;73:370-380.
Querol L, Nogales-Gadea G, Rojas-Garcia R, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology 2014;82:879-886.
Maisonobe T, Chassande B, Verin M, Jouni M, Léger JM, Bouche P. Chronic dysimmune demyelinating polyneuropathy: a clinical and electrophysiological study of 93 patients. J Neurol Neurosurg Psychiatry 1996;61:36-42.
Viala K, Maisonobe T, Stojkovic T, et al. A current view of the diagnosis, clinical variants, response to treatment and prognosis in chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst 2010;15:50-56.
Ayrignac X, Viala K, Morizot Koutlidis R, et al. Sensory chronic inflammatory demyelinating polyneuropathy: an under-recognized entity? Muscle Nerve 2013 48:727-732.
Viala K, Renié L, Maisonobe T, et al. Follow-up study and response to treatment in 23 patients with Lewis-Sumner syndrome. Brain 2004;127:2010-2017.
Attarian S, Verschueren A, Franques J, Salort-Campana E, Jouve E, Pouget J. Response to treatment in patients with Lewis-Sumner syndrome. Muscle Nerve 2011;44:179-184.
Katz JS, Saperstein DS, Gronseth G, Aamato AA, Barohn RJ. Distal acquired demyelinating symmetric neuropathy. Neurology 2000;54:615-620.
Larue S, Bombelli F, Viala K, et al. Non-anti-MAG DADS neuropathy as a variant of CIDP: clinical, electrophysiological, laboratory features and response to treatment in 10 cases. Eur J Neurol 2011;18:899-905.
Rabin M, Mutlu G, Stojkovic T, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: search for factors associated with treatment dependence or successful withdrawal. J Neurol Neurosurg Psychiatry 2014;85:901-906.
Mehendiratta MM, Hughes RA. Corticosteroids for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev 2012;8:CD002062.
Dyck PJ, O'Brien PC, Oviatt KD, et al. Prednisone improves chronic inflammatory demyelinating polyradiculoneuropathy more than no treament. Ann Neurol 1982;11:136-141.
Van Schaik IN, Eftimov F, van Doorn PA, et al. Pulsed high-dose dexamethasone versus standard prednisolone treatment for chronic inflammatory demyelinating polyradiculoneuropathy (PREDICT study): a double-blind, randomised, controlled trial. Lancet Neurol 2010;9:245-253.
Eftimov F, Vermeulen M, van Doorn PA, Brusse E, van Schaik IN. PREDICT. Long-term remission of CIDP after pulsed dexamethasone or short-term prednisolone treatment. Neurology 2012;78:1079-1084.
Mehndiratta MM, Hughes RA, Agarwal P. Plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev 2012;9:CD003906.
Kazatchkine M, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med 2001;345:747-755.
Hughes RA, Donofrio P, Bril V, et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol 2008;7:136-144.
Merkies IS, Bril V, Dalakas MC, et al. Health-related quality-of-life improvements in CIDP with immune globulin IV 10%: the ICE Study. Neurology 2009;72:1337-1344.
Léger JM, De Bleecker JL, Sommer C, et al. Efficacy and safety of PrIVIgen(®) in patients with chronic inflammatory demyelinating polyneuropathy: results of a prospective, single-arm, open-label Phase III study (the PRIMA study). J Peripher Nerv Syst 2013;18:130-140.
Eftimov F, Winer JB, Vermeulen M, de Haan R, van Schaik IN. Intravenous immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev 2013;12:CD0011797.
Rajabally YA. Subcutaneous immunoglobulin therapy for inflammatory neuropathy: current evidence base and future prospects. J Neurol Neurosurg Psychiatry 2013;85:631-637.
Markvardsen LH, Debost JC, Harbo T, et al. Subcutaneous immunoglobulin in responders to intravenous therapy with chronic inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol 2013;20:836-842.
Markvardsen LH, Harbo T, Sindrup SH, et al. Subcutaneous immunoglobulin preserves muscle strength in chronic inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol 2014;21:1465-1470.
Cocito D, Merola A, Peci E, et al. Subcutaneous immunoglobulin in CIDP and MMN : a short-term nationwide study. J Neurol 2014;261:2159-2164.
Nobile-Orazio E, Cocito D, et al. Intravenous immunoglobulin versus intravenous methylprednisolone for chronic inflammatory demyelinating polyradiculoneuropathy: a randomised controlled trial. Lancet Neurol 2012;11:493-502.
Nobile-Orazio E, Cocito D, Jann S, et al. Frequency and time to relapse after discontinuing 6-month therapy with IVIg or pulsed methylprednisolone in CIDP (IMC study follow-up). J Neurol Neurosurg Psychiatry 2015;86:729-734.
Cocito D, Grimaldi S, Paolasso I, et al. Immunosuppressive treatment in refractory chronic inflammatory demyelinating polyradiculoneuropathy. A nationwide retrospective analysis. Eur J Neurol 2011;18:1417-1421.
Goodin DS, Frohman EM, Garmany GP, et al. Disease modyfing therapies in multiple sclerosis. Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Neurology 2002;58:169-178.
Vallat JM, Hahn AF, Léger JM, et al. Interferon beta-1a as investigational treatment for CIDP. Neurology 2003;60(Suppl. 3):S23-S28.
Hughes RA, Gorson KC, Cros D, et al. Intramuscular interferon beta-1a in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology 2010;74:651-657.
RMC Trial Group. Randomised controlled trial of methotrexate for chronic inflammatory demyelinating polyradiculoneuropathy (RMC trial): a pilot, multicentre study. Lancet Neurol 2009;8:158-164.
Léger JM, Viala K, Nicolas G, et al. Placebo-controlled trial of rituximab in IgM anti-myelin-associated gycoprotein neuropathy. Neurology 2013;80:2217-2225.
Benedetti L, Briani C, Franciotta D, et al. Rituximab in patients with chronic inflammatory demyelinating polyradiculoneuropathy: a report of 13 cases and review of the literature. J Neurol Neurosurg Psychiatry 2011;82:306-308.
Marsh EA, Hirst CL, Llewelyn JG, et al. Alemtuzumab in the treatment of IVIG-dependent chronic inflammatory demyelinating polyneuropathy. J Neurol 2010;257:913-919.
Roth G, Roth J, Oschner F. Motor neuropathy with proximal multifocal persistent block, fasciculations and myokyma. Evolution to tetraplegia. Eur Neurol 1986;25:416-423.
Chad DA, Hammer K, Sargent J. Slow resolution of multifocal weakness and fasciculation: a reversible motor neuron syndrome. Neurology 1986;36:1260-1263.
Pestronk A, Cornblath DR., Ilyas A, et al. A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol 1988;24:73-78.
Van Schaik IN., Léger JM, Nobile-Orazio E, et al. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of multifocal motor neuropathy. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. Joint Task Force of the EFNS and the PNS. J Periph Nervous System 2010;15: 295-301.
Guimaràes-Costa R, Bombelli F, Léger JM. Multifocal motor neuropathy. Curr Opin Neurol 2010; 26:2503-2509.
Cats EA, Van der Pol WL, Piepers S, et al. Correlates of outcome and response to IVIg in 88 patients with multifocal motor neuropathy. Neurology 2010; 75:818-825.
Léger JM, Vargas S, Lievens I. Efficacy of intravenous immunoglobulin in multifocal motor neuropathy. Ann N Y Acad Sci 2007;1110:248-255.
Donofrio PD, Berger A, Brannagan TH, et al. Consensus statement: the use of intravenous immunoglobulin in the treatment of neuromuscular conditions: report of the AANEM ad hoc committee. Muscle Nerve 2009;40:890-900.
Azulay JP, Blin O, Pouget J, et al. Intravenous immunoglobulin treatment in patients with motor neuron syndromes associated with anti-GM1 antibodies: a double-blind, placebo-controlled study. Neurology 1994;44:429-432.
Van den Berg LH, Kerkhoff H, Oey PL, Franssen H, Mollee I, Vermeulen M. Treatment of multifocal motor neuropathy with high dose intravenous immunoglobulins: a double blind, placebo controlled study. J Neurol Neurosurg Psychiatry 1995;59:248-252.
Federico P, Zochodne DW, Hahn AF, Brown WF, Feasby TE. Multifocal motor neuropathy improved by IVIg: randomized, double-blind, placebo-controlled study. Neurology 2000;55:1256-1262.
Léger JM, Chassande B, Musset L, Meininger V, Bouche P, Baumann N. Intravenous immunoglobulin therapy in multifocal motor neuropathy: a double-blind, placebo-controlled study. Brain 2001;124:145-153.
Van Schaik IN, van den Berg LH, de Haan R, Vermeulen M. Intravenous immunoglobulin for multifocal motor neuropathy. Cochrane Database Syst Rev 2005;2:CD004429.
Hahn AF, Beydoun SR, Lawson V, et al. for the IVIg in MMN study. A controlled trial of intravenous immunoglobulin in multifocal motor neuropathy. J Peripher Nerv Syst 2013;18:321-330.
van den Berg-Vos RM, Franssen H, Wokke JH, Van den Berg LH. Multifocal motor neuropathy: long-term clinical and electrophysiological assessment of intravenous immunoglobulin maintenance treatment. Brain 2002;125:1875-1886.
Terenghi F, Cappellari A, Bersano A, Carpo M, Barbieri S, Nobile-Orazio E. How long is IVIg effective in multifocal motor neuropathy? Neurology 2004;62:666-668.
Vucic S, Black KR, Chong PS, Cros D. Multifocal motor neuropathy: decrease in conduction blocks and reinnervation with long-term IVIg. Neurology 2004; 63:1264-1269.
Léger JM, Viala K, Cancalon F, et al. Intravenous immunoglobulin as short- and long-term therapy of multifocal motor neuropathy: a retrospective study of response to IVIg and of its predictive criteria in 40 patients. J Neurol Neurosurg Psychiatry 2008;79:93-96.
Harbo T, Andersen H, Hess A, Hansen K, Sindrup SH, Jakobsen J. Subcutaneous versus intravenous immunoglobulin in multifocal motor neuropathy: a randomized, single-blinded cross-over trial. Eur J Neurol 2009;16:631-638.
Eftimov F, Vermeulen M, de Haan RJ, van den Berg H, van Schaik IN. Subcutaneous immunoglobulin therapy for multifocal motor neuropathy. J Peripher Nerv Syst 2009;14:93-100.
Harbo T, Andersen H, Jakobsen J. Long-term therapy with high doses of subcutaneous immunoglobulin in multifocal motor neuropathy. Neurology 2010;75:1377-1380.
Umapathi T, Hughes RAC, Nobile-Orazio E, Léger JM. Immunosuppressant and immunomodulatory treatments for multifocal motor neuropathy. Cochrane Database Syst Rev 2012;4:CD003217.
Meucci N, Cappellari A, Barbieri S, Scarlato G, Nobile-Orazio E. Long term effect of intravenous immunoglobulins and oral cyclophosphamide in multifocal motor neuropathy. J Neurol, Neurosurg Psychiatry 1997;63:765-769.
Piepers S, van den Berg-Vos R, van der Pol WL, Franssen H, Wokke J, van den Berg L. Mycophenolate mofetil as adjunctive therapy for MMN patients: a randomized, controlled trial. Brain 2007;130:2004-2010.
Fitzpatrick AM, Mann CA, Barry S, Brennan K, Overell JR, Willison HJ. An open label clinical trial of complement inhibition in multifocal motor neuropathy. J Peripher Nerv Syst 2011;16:84-91.
Chassande B, Léger JM, Younes-Chennoufi A, et al. Peripheral neuropathy associated with IgM monoclonal gammopathy: correlations between M-protein antibody activity and clinical/electrophysiological features in 40 cases. Muscle Nerve 1998;21:55-62.
Lunn MTP, Nobile-Orazio E. Immunotherapy for IgM anti-myelin-associated glycoprotein paraprotein-associated peripheral neuropathies. Cochrane Database Syst Rev 2012;5:CD002827.
Pestronk A, Florence J, Miller T, Choksi R, Al-Lozi MT, Levine TD. Treatment of IgM antibody associated polyneuropathies using rituximab. J Neurol Neurosurg Psychiatry 2003;74:485-489.
Renaud S, Gregor M, Fuhr P, et al. Rituximab in the treatment of polyneuropathy associated with anti-MAG antibodies. Muscle Nerve 2003;27:611-615.
Benedetti L, Briani C, Franciotta D, et al. Long-term effect of rituximab in anti-MAG polyneuropathy. Neurology 2008;71:1742-1744.
Niermeijer JM, Eurelings M, Lokhors HL, et al. Rituximab for polyneuropathy with IgM monoclonal gammopathy. J Neurol, Neurosurg Psychiatry 2009;80:1036-1039.
Dalakas MC, Rakocevic G, Salajegheh, et al. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein antibody demyelinating neuropathy. Ann Neurol 2009;65:286-293.
Renaud S, Fuhr P, Gregor M, et al. High-dose rituximab and anti-MAG-associated polyneuropathy. Neurolog 2006;66:742-744.
Gruson B, Kamel G, Beaumont M, et al. Long-term response to rituximab and fludarabine combination in IgM anti-myelin-associated glycoprotein neuropathy. J Periph Nerv Syst 2011;16:180-185.
Hospital M-A, Viala K, Dragomir S, et al. Immunotherapy-based regimen in anti-MAG neuropathy: results in 45 patients. Hematologica 2013;98:155-157
Vanhoutte EK, Faber CG, Merkies IS. PeriNomS study group. 196th ENMC International Workshop: outcome measures in inflammatory peripheral neuropathies 8–10 February, Naarden, The Netherlands. Neuromuscul Disord 2013;23:924-933.
Draak THP, Vanhoutte EK, van Nes SI, et al. Changing outcome in inflammatory neuropathies. Rasch-comparative responsiveness? Neurology 2014;83:1-9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Disclosures
Dr Léger received departmental research grants or honoraria from Biogen-Idec, Baxter, CSL-Behring, Kedrion, LFB, Novartis, and Octapharma. Drs. Guimaraes-Costa and Muntean have nothing to disclose.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article. (PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Léger, JM., Guimarães-Costa, R. & Muntean, C. Immunotherapy in Peripheral Neuropathies. Neurotherapeutics 13, 96–107 (2016). https://doi.org/10.1007/s13311-015-0401-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-015-0401-7