
Abstract
Introduction
Semaglutide is a glucagon-like peptide-1 (GLP-1) analog approved for the once-weekly treatment of type 2 diabetes. The objective of this 16-week, double-blind, single-center thorough QT study was to confirm that semaglutide treatment does not prolong cardiac repolarization versus placebo. Prolongation of the QT interval is a biomarker for ventricular tachyarrhythmia.
Methods
In a parallel design, 168 healthy subjects were randomized to the treatment or placebo arms, of whom 166 were treated with subcutaneous semaglutide (N = 83; escalated to a supratherapeutic dose of 1.5 mg) or placebo (N = 83). The subjects (60% males) had a mean age of 38.2 years and body mass index of 25.1 kg/m2. To assess QT assay sensitivity, subjects in the placebo group received a single 400 mg moxifloxacin dose as positive control, and placebo in a crossover fashion. The primary endpoint was the time-matched change from baseline in QT interval corrected individually for heart rate (ΔQTcI), calculated from 11 electrocardiogram recordings from 0 to 48 h after the last 1.5 mg dose. Similar assessments were made for the therapeutic 0.5 and 1.0 mg semaglutide dose levels.
Results
No QTcI prolongation occurred with any semaglutide dose; the upper limits of two-sided 90% confidence intervals of the placebo-subtracted ΔQTcI were < 10 ms at all doses and time points. Exposure–response analysis showed no dependence of QTcI on semaglutide concentration. QT assay sensitivity was confirmed. The semaglutide safety profile was similar to that of other GLP-1 receptor agonists.
Conclusion
Based on investigations of QT/QTc, no concern with regard to ventricular arrhythmias was raised as semaglutide did not prolong the cardiac repolarization duration in healthy subjects.
Trial Registration
ClinicalTrials.gov identifier: NCT 02064348.
Funding
Novo Nordisk.
Similar content being viewed by others

Introduction
Semaglutide is a glucagon-like peptide-1 (GLP-1) analog approved for the once-weekly treatment of type 2 diabetes. Semaglutide has 94% amino acid homology with native GLP-1, with structural modifications which include amino acid substitutions at position 8 (alanine to alpha-aminoisobutyric acid) and 34 (lysine to arginine), as well as acylation of the lysine at position 26 with a spacer and a C-18 fatty diacid chain [1,2,3]. The substitution at position 8 makes semaglutide less susceptible to degradation by dipeptidyl peptidase-4 (DPP-4), whereas improved albumin binding is facilitated through the lysine acylation and fatty diacid side chain [3]. Overall, these modifications extend the half-life of semaglutide to approximately 1 week, making it suitable for once-weekly subcutaneous administration. The therapeutic maintenance doses are 0.5 and 1.0 mg [1, 2].
The QT interval, measured in milliseconds from the start of the QRS complex to the end of the T wave on a body surface electrocardiogram (ECG), represents the duration of electrical depolarization and repolarization of the ventricular myocardium [4]. Prolongation of the QT interval is a biomarker for ventricular tachyarrhythmia, such as Torsade de Pointes, and is regarded as a risk factor for sudden cardiac death [5]. Some drugs are known to influence ion channels in cardiac cells, causing a delay in cardiac repolarization [6, 7]. For this reason, it is a regulatory requirement to test for prolongation of the QT interval for most new drugs with systemic exposure [8, 9]. No prolongation of the QT interval has been observed previously for GLP-1 receptor agonists (GLP-1RAs) [10,11,12].
The trial reported here was conducted during the clinical development program to investigate the potential of semaglutide, at steady-state therapeutic and supratherapeutic doses, to prolong the QT or corrected QT (QTc) interval in healthy subjects in a thorough QT/QTc study setting.
Methods
Trial Conduct and Population
This clinical pharmacology trial was conducted at a single clinical research site in Germany (PAREXEL International GmbH, Berlin, Germany) and was registered at ClinicalTrials.gov (identifier: NCT 02064348).
Healthy male and female subjects aged between 18 and 55 years with a body mass index (BMI) of 20–30 kg/m2 and body weight of 60–110 kg were eligible to participate in the trial. Subjects were to be in good health on the basis of medical history, physical examination and routine clinical laboratory data, including ECG. Key exclusion criteria included: (1) known or suspected hypersensitivity to trial products or ECG electrodes; (2) participation in another clinical trial 90 days before screening; (3) a QTcF interval (the Fridericia heart rate [HR] corrected interval) [13] of > 450 ms; (4) hypotension (systolic blood pressure [SBP] of < 90 mmHg or diastolic BP [DBP] of < 50 mmHg) or hypertension (SBP > 140 mmHg or DBP > 90 mmHg) or HR of < 50 beats per minute (bpm) or > 90 bpm; (5) a history of seizures, epilepsy, syncope, cardiac arrest, cardiac arrhythmia or Torsade de Pointes, atrioventricular block or structural heart disease; (6) a family history of long QT syndrome; (7) a family history of sudden cardiac death.
Trial Design and Treatment
This was a randomized, double-blind and placebo-controlled parallel trial. A parallel design was chosen in favor of a crossover design because of the inherent properties of semaglutide (long half-life and the need for gradual dose escalation to mitigate gastrointestinal adverse events). According to the regulatory guidelines [8, 9, 14], a supratherapeutic dose level should be tested. Therefore, to obtain the intended supratherapeutic dose of 1.5 mg for semaglutide, dose escalation was performed every 4 weeks (0.25, 0.5, 1.0 and 1.5 mg). As a consequence, a crossover design would have prolonged the trial duration considerably, increasing the risk of greater variability of the QT measurements.
The trial design is shown in Fig. 1. Subjects were randomized to treatment with either semaglutide (Arm 1) or placebo (Arm 2). In addition, a single oral dose of 400 mg moxifloxacin (Avelox®) was given as a positive control to assess the sensitivity of the QT assay. This was done in a nested crossover design for only the placebo-treated subjects in order to reduce the number of subjects receiving the positive control [15] and to avoid interaction with semaglutide. Thus, the placebo treatment arm was further divided into two subgroups (Arms 2A and 2B). The single dose of moxifloxacin or moxifloxacin–placebo was administered before and after the treatment with placebo. To keep the investigators and the study subjects blinded, the semaglutide-treated subjects in Arm 1 received moxifloxacin–placebo both before and after the semaglutide treatment.

Trial design. ECG Electrocardiogram, PK pharmacokinetics
Treatment allocation took place at the trial site according to a randomization list in a 2:1:1 manner: Arm 1 subjects received semaglutide, and moxifloxacin–placebo both before and after the semaglutide treatment; Arm 2A subjects received placebo, and moxifloxacin before and moxifloxacin–placebo after the placebo treatment; Arm 2B subjects received placebo, and moxifloxacin–placebo before and moxifloxacin after the placebo treatment.
Semaglutide (Novo Nordisk A/S, Bagsværd, Denmark) or placebo doses were injected subcutaneously once weekly. The initial dose was 0.25 mg for 4 weeks, which was ultimately escalated to a supratherapeutic 1.5 mg dose for 4 weeks via steps of 0.5 mg and 1.0 mg doses for 4 weeks, respectively. In total, the treatment duration was 16 weeks, followed by a 5- to 7-week follow-up period. To ensure compliance, the first and the last dose of each dose level and all injections of the highest 1.5 mg dose were administered at the trial site. The remaining injections were self-administered at home.
Due to slightly differing appearances, the dose of moxifloxacin or moxifloxacin–placebo was administered orally by an independent person who was not blinded to treatment and not otherwise involved in the trial. The investigator, subjects, ECG core laboratory and sponsor personnel remained blinded throughout the trial.
Endpoints Related to QT, Pharmacokinetics and Safety
The primary objective of the trial was to confirm that treatment with a once-weekly supratherapeutic dose of semaglutide 1.5 mg at steady state does not result in an unacceptable prolongation in cardiac repolarization compared with placebo. The primary endpoint was the time-matched change from baseline in the individual-specific QT interval corrected individually for HR (ΔQTcI). This was calculated from 11 ECG recordings after the last 1.5 mg dose at all measured time points: before dosing and at 12, 18, 24, 25, 26, 27, 30, 36, 42 and 48 h after dosing. These time points were selected due to the long half-life of semaglutide of approximately 1 week [1, 2]. If the upper limit of the two-sided 90% confidence interval (CI) (equivalent to the upper limit of a one-sided 95% CI) of the mean placebo-subtracted ΔQTcI (ΔΔQTcI) for semaglutide 1.5 mg was < 10 ms at all time points studied, it was confirmed that there was no unacceptable prolongation of the QT interval. Similar assessments were made at steady state for the 0.5 and 1.0 mg dose levels of semaglutide as secondary endpoints.
For evaluation of the sensitivity of the QT assay with a single dose of moxifloxacin as positive control, ECG recordings were evaluated at eight time points: before dosing and at 1, 2, 3, 6, 12, 18 and 24 h after dosing. As confirmatory endpoints for assay sensitivity, the QTcI based on the ECG recordings obtained at 3 and 6 h after dosing were used, based on the well-known pharmacokinetic profile of moxifloxacin [16]. Supportive secondary endpoints were the QTcI based on ECG recordings obtained at the remaining five post-dose time points. Assay sensitivity was confirmed if the lower limit of the one-sided 95% CI of the mean placebo-subtracted QTcI for moxifloxacin was above 5 ms for at least one of the two confirmatory time points [8, 9, 14].
To further explore the possible influences of semaglutide on the cardiac conduction system, we included the following supportive secondary endpoints: the uncorrected QT interval, QTcL (the linear regression HR-corrected QT interval), QTcF [13] and QTcB (Bazett’s corrected QT interval) [17]. Other ECG parameters included HR, the PR interval and QRS complex duration. All parameters were based on ECG recordings obtained at 11 time points from before dosing to 48 h after the last doses of semaglutide and placebo at the 0.5, 1.0 and 1.5 mg dose levels.
In addition to assessing the central tendency analyses of ECG parameters, we also examined the occurrence of outliers. QTc outliers were defined as a QTc interval longer than 450, 480 or 500 ms and as increases from baseline of more than 30 or 60 ms. HR outliers were defined as values of < 50 bpm or > 100 bpm. PR interval outlier thresholds were defined as values of > 220 ms and/or changes from baseline of > 25%. QRS complex outliers were defined as occurrences of absolute QRS duration of > 110 ms and/or changes from baseline of > 25%. Endpoints for the evaluation of T and U wave abnormalities, identified by the cardiologist, were also included in the assessment.
The pharmacokinetics of semaglutide were assessed at steady state of each dose level using the following endpoints: the area under the semaglutide concentration–time curve from time zero to 48 and 168 h (AUC0–48h and AUC0−168h, respectively), maximum plasma concentration (Cmax), time to Cmax (tmax) and trough plasma concentrations (Ctrough). Dose proportionality of semaglutide was assessed by the endpoints AUC0–48h, AUC0–168h and Cmax.
An exposure–response analysis was done in which the association between baseline-adjusted and placebo-corrected QTcI of semaglutide dose levels of 0.5, 1.0 and 1.5 mg at steady state and the corresponding semaglutide plasma concentrations was investigated.
The following endpoints completed the assessment of the safety and tolerability of semaglutide: the incidence of adverse events (AEs) and hypoglycemic events; the occurrence of positive anti-semaglutide antibodies at follow-up; changes from baseline to follow-up in the following: hematology, biochemistry and urinalysis parameters, calcitonin concentrations, vital signs, ECG findings, physical examination, body weight and fasting plasma glucose. AEs were recorded from randomization to the end of the follow-up period.
QTc Corrections for HR
The QT interval is influenced by many factors, including HR, and several methodologies have been developed to adequately correct for HR and its changes [4, 7]. While Bazett’s method is still the most widely used method in clinical practice [17, 18], there is evidence to support that, due to inter-individual variability, HR correction is best estimated for each individual [7]. As GLP-1RAs are known to increase the HR [19], in the trial reported here, four correction methods for HR were applied: the individual exponential, the linear regression, Fridericia’s [13], and Bazett’s, as outlined below [17]. The uncorrected QT interval was also evaluated.
-
(1)
The individual exponential correction was used for the primary endpoint. The correction was calculated using QTcI = QT/RRβ, where RR denotes the RR interval. The coefficient β for each subject was derived by fitting all baseline recordings in a model of the form logQTij = αi +βilogRRij + eij, where QTij denotes the QT value for subject i recording j.
-
(2)
The linear regression correction was calculated using QTcL = QT − γ(1-RR), where the coefficient γ was determined using linear regression on all baseline recordings in a model of the form QTk = α + γ (1-RRk) + ek. QTk denotes the QT value for recording k (across all subjects).
-
(3)
Fridericia’s correction (QTcF) was calculated using QT/RR1/3.
-
(4)
Bazett’s correction (QTcB) was calculated using QT/RR1/2.
ECG Recording and Evaluation
Electrocardiograms were recorded using continuous digital 12-lead Holter devices (model CM 3000-12; GETEMED Medizin und Informationstechnik AG, Teltow, Berlin, Germany) and a modified Einthoven-Goldberger lead system (the Mason-Likar lead system) [20]. ECG recording periods had a duration of 48 h at each visit (Fig. 1) and occurred at week 0 (baseline), week 7 (last week of the 0.5 mg dose period), week 11 (last week of the 1.0 mg dose period) and week 15 (last week of the 1.5 mg dose period). In addition, at weeks 0 and 15, the 48-h ECG period was extended by 24 h after the administration of moxifloxacin or moxifloxacin–placebo in each arm. At each of the predefined time points before and after administration of the treatment, each subject had to lie down in a supine position to achieve that individual’s resting HR. At each time point, standard 10-s triplicate ECGs were extracted from the continuous recordings within a 2-min period. These extractions were done shortly prior to the blood draws used for the pharmacokinetic evaluation and were assessed at a central core laboratory where also the measurement and cardiologist evaluation took place. The primary cardiac intervals (QT, PR, QRS and RR) were measured semi-automatically for each ECG in four, if possible, consecutive complexes, in the same lead (primary lead II) within each subject. A board-certified cardiologist evaluated one randomly selected ECG at each time point and assessed the appearance of abnormal findings, including the occurrence of T and U wave abnormalities. For the statistical evaluation of the intervals, the means over all evaluable single-complex values for each triplicate ECG measurement were calculated.
Bioanalytical Methods
Plasma semaglutide concentrations were analyzed as previously reported using liquid chromatography with tandem mass spectrometry (Celerion Inc., Fehraltorf, Switzerland), with a lower limit of quantification for semaglutide of 0.729 nmol/L [1].
Statistical Analyses
The sample size was calculated to achieve 80% overall power for establishing that the upper limits of the standard one-sided 95% CIs for the 11 comparisons between semaglutide 1.5 mg and placebo in the first 48 h after dosing were < 10 ms. A sample size based on 80% overall power, a standard deviation (SD) of 11 ms and a true unobserved treatment difference between semaglutide and placebo of 3 ms, based on a previous trial [12], was considered to be sufficient to evaluate the primary objective. A total sample size of 168 subjects was selected to ensure 140 subjects completing the trial and assuming a drop-out rate of approximately 16%.
The primary endpoint was analyzed using a mixed-effects model for repeated measurements with the 11 time-matched ΔQTcI measurements as the dependent variables. The two placebo arms (Arms 2A and 2B) were pooled. Treatment (semaglutide 1.5 mg or placebo) was included as a fixed factor in the model and the baseline QTcI measurements were included as covariates, assuming an unstructured covariance matrix. Furthermore, treatment and baseline QTcI measurements were nested within the time point. Using the model, the mean ΔΔQTcI was estimated for each of the 11 time points and presented with two-sided 90% CIs. Eleven one-sided tests of the null hypothesis that the mean treatment difference is > 10 ms were performed at the 5% level. The primary objective was fulfilled if the null hypothesis was rejected in all 11 tests or, equivalently, if the upper limits of all 11 two-sided 90% CIs were < 10 ms.
To establish QT assay sensitivity, we analyzed eight time-matched QTcI measurements taken during the 24 h after administration of moxifloxacin/placebo (at baseline and week 15). The two time points of 3 and 6 h after dosing were considered to be confirmatory. The mean placebo-subtracted QTcI for moxifloxacin was estimated separately for each time point using an analysis of variance with treatment (moxifloxacin/placebo), period (baseline or week 15) and subject as fixed effects, and pre-dose QTcI as a covariate. QT assay sensitivity was established if the lower limit of the two-sided 95% CI was > 5 ms for at least one of the two confirmatory time points, corresponding to a Bonferroni correction for multiplicity. The remaining five post-dose time points were considered to be supportive and were analyzed in a similar way together with 90% CIs.
The uncorrected QT as well as the QTcL, QTcF and QTcB endpoints were analyzed using the same model as for the primary endpoint. The correlation between the various QT correction methods and RR was evaluated by plotting the corrected QT values against the RR values. The plot was used to assess the appropriateness of the different correction methods.
HR and PR intervals and QRS duration were analyzed separately using the same model as for the primary endpoint.
An exploratory analysis of the changes in QTcI interval and the relationship of these changes to semaglutide plasma concentrations was made. The 11 ΔQTcI measurements in the 48 h after dosing at steady state of each semaglutide dose level were placebo-corrected by subtracting the time-matched estimated means of ΔQTcI for the placebo-treated subjects. The exposure–response relationship was analyzed using a linear mixed-effects model with the placebo-subtracted ΔQTcI as the dependent variable, the log-transformed semaglutide plasma concentration and baseline QTcI as covariates and the subject as a random effect.
To test for dose proportionality, the AUC and Cmax were log-transformed and analyzed using a linear mixed model with log-dose as a fixed effect and subject as a random effect. The estimated doubling constant (2β, where β was the log-dose slope in the above analysis) was presented with the corresponding 95% CI. A sensitivity analysis of each dose–proportionality pharmacokinetic endpoint was performed, which excluded seven subjects who were suspected of being noncompliant with the dosing-schedule. This analysis was conducted to explore the robustness of the prespecified dose-proportionality analysis.
All other pharmacokinetic endpoints and safety endpoints were summarized descriptively.
Statistical analyses were performed using SAS software, version 9.3 (SAS Institute, Cary, NC, USA).
Compliance with Ethics Guidelines
The trial was conducted in accordance with the Declaration of Helsinki [21] and Good Clinical Practice guidelines [22]. The trial protocol was approved by the health authority of The Federal Institute for Drugs and Medical Devices (Germany) and the independent ethics committee of Berlin, Germany. Prior to commencement of any trial-related activities, participants were informed of the risks and benefits of the trial, and all participants provided written informed consent.
Results
Subjects and Data
Of 372 screened subjects, 168 subjects were enrolled, and 166 were exposed to treatment: 83 received semaglutide and 83 received placebo and the positive control. Eight subjects in each group withdrew after randomization. In the semaglutide group, withdrawals were due to AEs (2 subjects), protocol violations (2) and withdrawal of consent (2); in addition, one subject was lost to follow-up and one withdrew due to ‘other’ reasons. In the placebo group, withdrawals were due to AEs (4 subjects) and withdrawal of consent (4). In total, 152 subjects completed the trial, which was conducted between 26 February 2014 and 23 April 2015. Analyses were based on all 166 randomized subjects who received at least one treatment dose.
Subject demographics are presented in Table 1. In summary, the subjects had a mean age of 38.2 years and a mean BMI of 25.1 kg/m2. Men made up approximately 60% of the trial population.
Time-Matched QTc Treatment Differences
An evaluation of the different correction methods was made by assessing the correlation between the individual uncorrected and corrected QT intervals and the corresponding RR intervals. The plots are shown in Electronic Supplementary Material (ESM) Fig. S1. As expected, a clear positive correlation between the uncorrected QT interval and the RR interval was apparent (the QT interval decreased with increasing HR). For the corrected intervals, a negative correlation was present when using QTcB; this negative correlation was also present but less steep for the QTcF. For the QTcI and QTcL intervals, no correlation was apparent.
The estimated mean baseline-adjusted placebo-subtracted QTcI (ΔΔQTcI) profiles in the 48 h after dosing at steady state of each semaglutide dose (0.5, 1.0 and 1.5 mg) are presented with 90% CIs in Fig. 2. It was confirmed that there was no unacceptable prolongation of QTcI with semaglutide 1.5 mg; the upper limits of the 11 two-sided 90% CIs were all < 10 ms (Table 2, Fig. 2). The maximum upper limit of the two-sided 90% CI for the time-matched ΔΔQTcI was 0.3 ms. Similarly, for semaglutide at the 0.5 and 1.0 mg dose levels, the upper limits of all 11 two-sided 90% CIs for the mean ΔΔQTcI were < 10 ms (Fig. 2), indicating no QTcI prolongation.

Time-matched mean placebo-subtracted ΔQTcI (ΔΔQTcI) for each semaglutide dose (0.5, 1.0 and 1.5 mg) over the 48-h post-dose period. Data are mean baseline-adjusted and placebo-subtracted QTcI with error bars representing the corresponding two-sided 90% confidence intervals. The dotted line corresponds to the ΔΔQTcI interval threshold of regulatory concern (< 10 ms). QTcl QT interval corrected individually for heart rate, ΔQTcI time-matched change from baseline in the QTcI
The observed mean changes from baseline in QTcI were shorter with semaglutide than with placebo. The estimated mean ΔΔQTcI ranged from − 9.6 (90% CI − 13.0; − 6.2) ms to − 4.5 (− 8.0; − 1.0) ms with semaglutide 0.5 mg, from − 9.0 (− 12.6; − 5.4) ms to − 2.3 (− 6.0; 1.3) ms with semaglutide 1.0 mg and from − 6.6 (− 10.1; − 3.0) ms to − 3.2 (− 6.6; 0.3) ms with semaglutide 1.5 mg.
No prolongation was observed for the uncorrected QT interval, QTcL or QTcF with doses of semaglutide of 0.5, 1.0 and 1.5 mg at steady state. Data for the highest 1.5 mg semaglutide dose are shown in ESM Tables S1–S3. For QTcB, the upper limits of the 90% CIs were > 10 ms for at least one time point at all dose levels (ESM Table S4).
QT Assay Sensitivity
Moxifloxacin prolonged the mean QTcI interval at the 3- and 6-h confirmatory endpoints by 12.3 and 8.9 ms, respectively, compared to placebo (Table 3), declining towards baseline levels after 24 h. This was in concordance with the observed changes in other thorough QT trials [16]. Thus, QT assay sensitivity was achieved as the lower limits of the 95% CIs for the mean placebo-subtracted QTcI were > 5 ms at both time points, 3 and 6 h after dosing.
Pharmacokinetics
The pharmacokinetic endpoints for each semaglutide dose level at steady state are presented in ESM Table S5. Dose-dependent increases were observed for AUC0−168h, AUC0−48h, Cmax and Ctrough. The median tmax was 26 h for semaglutide 0.25 mg and 27 h for semaglutide doses 0.5, 1.0 and 1.5 mg.
The increase in AUC0−48h and Cmax with increasing dose was consistent with dose proportionality (ESM Table S6). For AUC0−168h, a statistically significant deviation from dose proportionality was observed (P = 0.047). However, with an estimated doubling constant of 2.02 and a very narrow 95% CI (2.00; 2.04), this deviation was not considered to be clinically relevant. The sensitivity analysis showed that the increase in each of the three dose proportionality endpoints was consistent with dose proportionality (P = 0.073) (ESM Table S6).
Exposure–Response Relationship
There was no indication of a dependency of the placebo-subtracted ΔQTcI interval on semaglutide concentration (Fig. 3). Statistical analysis showed an estimated slope close to zero (0.43 ms/log[nmol/L]) (95% CI −0.03; 0.89; P = 0.065).

Placebo-corrected ΔQTcI interval versus semaglutide plasma concentration assessed at steady state at each dose. The QTcI intervals are individually placebo-corrected by subtracting the estimated placebo mean at each time point
Other ECG Assessments
Semaglutide treatment was associated with an increase in HR at all dose levels compared to placebo (ESM Fig. S2). There appeared to be a dose-dependent ordering of the estimated mean baseline-adjusted treatment differences in HR, ranging between 5.3 and 8.5 bpm across the 11 time points for the 0.5 mg dose, between 6.7 and 9.7 bpm for the 1.0 mg dose, and between 7.7 and 11.1 bpm for the 1.5 mg dose.
A small prolongation of the PR interval was observed at all dose levels of semaglutide compared to placebo, with an estimated mean ΔΔPR ranging from 6.1 to 10.7 ms with semaglutide 0.5 mg, from 3.5 to 9.2 ms with semaglutide 1.0 mg and from 4.6 to 10.0 ms with semaglutide 1.5 mg. No indication of a dose- or time-dependency was observed.
No significant change in QRS duration was observed with semaglutide treatment. The estimated mean ΔΔQRS ranged from − 1.2 to − 0.5 ms with semaglutide 0.5 mg, from − 1.5 to − 0.9 ms with semaglutide 1.0 mg and from − 1.9 to − 1.1 ms with semaglutide 1.5 mg.
A few T wave abnormalities were observed for three female subjects treated with semaglutide doses 0.5 or 1.0 mg; all were evaluated to be of no clinical relevance. No subjects in the placebo group had T wave abnormalities, and no U wave abnormalities were observed in either group.
Only a few QTcI outliers (i.e. QTcI intervals of > 450, > 480 or > 500 ms, or increases from baseline of > 30 or > 60 ms) were observed, and these were more frequent in the placebo group than in the semaglutide group (10 vs. 3 events across doses and outlier types). Results for the other correction methods are presented in the ESM Appendix.
At baseline, a similar number of subjects in the semaglutide and the placebo groups (20 vs. 21) had a HR of < 50 bpm. As expected, due to the HR-increasing effect of semaglutide, few subjects had a HR of < 50 bpm after treatment with all semaglutide doses (8 subjects across doses vs. 63 subjects with placebo). Two subjects treated with semaglutide 1.5 mg each showed a HR of > 100 bpm at one of the 11 time points during the 48-h ECG recording. No subjects with a HR of > 100 bpm were observed in the placebo group or in the other semaglutide dose groups.
With regard to the PR interval, more subjects in the semaglutide group than in the placebo group had PR interval values of > 220 ms after baseline (29 events vs. 12 events across doses). With semaglutide, five events of increased PR interval of > 25% above baseline were observed in the 1.0 and 1.5 mg treatment groups (none in the 0.5 mg group), whereas no subjects treated with placebo had any such increases. There were no observations of a second-degree atrioventricular block or higher-grade atrioventricular blocks for any subject for all ECGs evaluated by the cardiologist.
ECGs with a longer QRS duration of > 110 ms were observed more frequently for subjects treated with placebo (169 events after baseline) than for those treated with semaglutide (40 events) at all dose levels. No changes in QRS complex duration of > 25% above baseline were observed for any of the treatment arms.
Safety and Tolerability
In this population of healthy subjects, no unexpected safety findings related to treatment with subcutaneous semaglutide up to a 1.5 mg dose were identified. All but one reported AEs were of mild or moderate severity (ESM Table S7); one severe event of ear pain was reported by a subject in the placebo group. Gastrointestinal disorders, predominantly nausea, vomiting and diarrhea, were more frequent in the semaglutide group (56.6% of subjects) than in the placebo group (33.7%). Metabolism and nutrition disorders and predominantly decreased appetite were also more frequent in those receiving semaglutide (51.8% of subjects) than in subjects in the placebo group (12.0%). One serious AE (clavicle fracture) was reported by one subject treated with semaglutide (onset was 23 days after the last 1.5 mg dose); there were no serious AEs in the placebo group. Two subjects treated with semaglutide (2.4%) and four subjects treated with placebo (4.8%) withdrew from the trial due to non-serious AEs. In the semaglutide group, these events comprised decreased appetite, abdominal discomfort, nausea, fatigue, redness at ECG electrode sites and eczema, and in the placebo group, the events were urinary tract infection, headache, limb pain, ankle sprain, nausea, amenorrhea and dizziness; all subjects recovered.
No severe hypoglycemic episodes were reported. One blood glucose-confirmed symptomatic hypoglycemic episode (blood glucose < 3.1 mmol/L [56 mg/dL]) was reported in the semaglutide group. Symptomatic episodes (blood glucose < 3.9 mmol/L [70 mg/dL]) according to the American Diabetes Association definition [23] were reported by 10.8% of subjects in the semaglutide group compared with 2.4% of those in the placebo group. The observed mean (± SD) levels of fasting plasma glucose decreased with semaglutide (− 0.33 ± 0.39 mmol/L [− 6.0 ± 7.0 mg/dL]), whereas levels appeared to be relatively unchanged with placebo (− 0.07 ± 0.42 mmol/L [− 1.2 ± 7.5 mg/dL]).
In terms of vital signs, observed mean SBP and DBP were almost unchanged during the 16-week treatment period for both treatment groups, with mean (± SD) SBP changes from baseline to last dose of − 2 ± 11 mmHg (semaglutide) versus 4 ± 11 (placebo) and diastolic BP changes of 5 ± 8 mmHg (semaglutide) versus 6 ± 7 mmHg (placebo). In concordance with the ECG results, the HR increased from baseline to last dose for subjects treated with semaglutide (mean ± SD change of 10 ± 9 bpm) but showed almost no change for subjects treated with placebo [2 ± 9 bpm).
A decrease in body weight was observed for subjects treated with semaglutide, with a mean (± SD) change from baseline of − 7.6 ± 3.4 kg compared with 0.0 ± 2.4 kg for the placebo group.
No safety findings were identified based on clinical laboratory parameters or physical examination, and no subjects developed anti-semaglutide antibodies.
Discussion
The aim of this single-center clinical pharmacology trial in healthy individuals was to investigate possible effects of the glucose-lowering drug semaglutide on the ECG and other safety parameters in a thorough QT study setting. Therapeutic and supratherapeutic doses of 0.5, 1.0 and 1.5 mg at steady state were evaluated in a parallel, placebo- and actively-controlled trial for their ability to prolong cardiac repolarization duration, as assessed by HR-corrected QT intervals. Healthy males and females were both included in the evaluation of effect, safety and tolerability. The parallel design was chosen as dose escalation every 4 weeks to the supratherapeutic dose was necessary to limit gastrointestinal tolerability issues. The parallel design included three arms, of which two consisted of a nested placebo and positive control (moxifloxacin) crossover treatment.
As GLP-1RAs are known to be associated with increased HR [19] and based on previous experience [11, 12], we chose the mean change in individually corrected QT interval (QTcI) as the primary endpoint. Other correction formulae (QTcB, QTcF and QTcL) were also applied in this trial. The QTcI method proved to be least biased by the HR changes and, thus, the choice of QTcI as the primary correction method in the present trial was appropriate [8, 9].
Clinically relevant drug-induced delays in cardiac repolarization can lead to cardiac arrhythmias, notably Torsade de Pointes and ventricular fibrillation, which could be fatal [5]. The current trial did not show a prolongation of the mean ΔΔQTcI interval at any of the time points studied, thus meeting the criteria for a negative QT study [8, 9]. All upper limits of the two-sided 90% CIs of the placebo-subtracted mean QTcI changes from baseline before, during and after maximum semaglutide plasma concentration were below or only slightly above zero, and were thus below the threshold of regulatory concern (10 ms) [8, 9]. Moxifloxacin was used as a positive control and, as expected, elicited statistically significant increases in the QTcI interval. Thus, assay sensitivity was established [24].
A separate exposure–response analysis did not reveal a dependency of the placebo-subtracted ΔQTcI interval on semaglutide concentration when all doses of semaglutide up to the 1.5 mg supratherapeutic dose were pooled.
In addition to applying the individual QT correction method, the analysis was also performed on the other selected HR correction methods. Similar to the results obtained for the QTcI, no prolongation using QTcL and QTcF was observed at any of the time points or semaglutide dose levels. The prolongation observed for at least one time point at all dose levels with the QTcB correction method can be explained by the effect of semaglutide on increasing the HR, as the QTcB correction method is known to overestimate when the HR is increased [7,8,9].
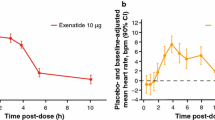
The lack of an effect of semaglutide on cardiac repolarization is consistent with results for other GLP-1RAs. The receptor agonist dulaglutide administered once weekly is not associated with QTc prolongation [10]. Similarly, neither albiglutide administered once weekly nor liraglutide administered once daily have been observed to prolong the QTcI interval, or indeed the QTc interval using other correction methods, in healthy subjects [11, 12]. While a single dose of exenatide was associated with a positive correlation between exposure and QTc interval in one study [25], another study with supratherapeutic concentrations of exenatide administered intravenously demonstrated no prolongation of a population-based correction interval (QTcP) or of QTcI in healthy subjects [26]. In the current trial, semaglutide plasma concentration increased in a manner that was consistent with dose proportionality. The lack of a prolongation of the QTcI interval with increasing semaglutide plasma concentration as assessed by the exposure–response analysis was likewise observed with liraglutide and exenatide [12, 26].
The median tmax was similar for semaglutide doses between 0.5 and 1.5 mg at 27 h post-dosing in these healthy subjects, and was overall comparable with results from other trials investigating the 1.0 mg dose of semaglutide in subjects with type 2 diabetes or obesity, with observed tmax values of 1–3 days [1, 27, 28]. In the current trial, samples for pharmacokinetic assessment were taken regularly between 0 and 48 h, but only once afterwards, at 168 h, which could have affected the results. Nevertheless, the results of this and other trials suggest that semaglutide is suitable for once-weekly administration [1, 27].
In this short-term trial in healthy subjects, there appeared to be a dose-dependent increase in HR with semaglutide as compared with placebo, with maximum treatment differences at each dose level ranging from 8.5 bpm with semaglutide 0.5 mg to 11.1 bpm with the 1.5 mg dose across the 11 time points. HR differences were lower in phase 3 trials, with mean increases of up to 3 bpm with semaglutide versus placebo [29, 30]. While GLP-1RAs are known to increase the HR in both healthy subjects and those with type 2 diabetes, the mechanism underlying the increased HR associated with GLP-1RAs is not known [31]. GLP-1 receptors can be found on the sinoatrial node [32], and GLP-1RAs may increase the HR not only through persistent, relative sympathetic enhancements but also by direct sinoatrial node stimulation [33]. The increased HR with semaglutide does not appear to present an increased cardiovascular risk. In a pre-registration cardiovascular-outcomes trial in individuals with type 2 diabetes, subjects treated with semaglutide had a lower rate of cardiovascular death, nonfatal myocardial infarction or nonfatal stroke than those treated with placebo (hazard ratio 0.74, 95% CI 0.58; 0.95) [30].
Other ECG assessments evaluated in this trial included PR interval, QRS duration and general assessment of ECG findings by a board-certified cardiologist. Semaglutide treatment was associated with a small prolongation of the PR interval versus placebo (range 3.5–10.0 ms across doses) which, however, was neither dose nor time dependent. There were also no observations of second-degree or higher-grade atrioventricular block, both of which may be associated with a lengthened PR interval. A PR interval prolongation has also been observed with other GLP-1RAs, such as liraglutide, dulaglutide and albiglutide [10, 11, 34], but not with exenatide [26]. The QRS duration was unaffected by semaglutide, which is in accordance with the results of other trials on GLP-1RAs [11, 26].
In general, semaglutide was well tolerated, and no new safety or tolerability issues were observed in comparison with previous trials of semaglutide [29, 35, 36], or other GLP-1RAs [34, 37,38,39]. Most side effects were due to gastrointestinal disorders, predominantly nausea, vomiting and diarrhea, or decreased appetite.
A limitation of the trial is the long half-life of semaglutide, making a crossover design unfeasible. The time span from baseline assessment to the QT evaluation during treatment may adversely affect the assay stability, but in the current trial, this was mitigated by use of a positive control at the start and end of semaglutide treatment, which exhibited no sign of assay drift over time. Also, the maximum semaglutide dose tested was not a ‘substantial multiple of the expected therapeutic dose,’ but exposures in the current trial were well above those associated with the therapeutic doses used in phase 3 trials [27]. Both males and females were enrolled in the trial according to the guidelines, as women appear to be more susceptible to medication-induced QTc prolongation and have a longer baseline QT interval after puberty [7, 18]. However, no sex-specific subanalyses were performed as such studies are not required unless a positive signal for QTc prolongation is demonstrated.
Conclusions
Semaglutide treatment did not prolong the cardiac repolarization duration in healthy subjects at therapeutic and supratherapeutic doses of up to 1.5 mg once weekly. Based on the trial results, no concern was raised with regard to ventricular arrhythmias. The results on cardiac repolarization were supported by an exposure–response analysis that indicated no dependency of the placebo-subtracted ΔQTcI interval on the semaglutide concentration. The assay sensitivity was confirmed as the positive control, moxifloxacin, showed the expected QTc prolongation. The impact of semaglutide on other ECG parameters was in accordance with observations in other GLP-1RAs. Semaglutide was generally well tolerated and exhibited a safety profile consistent with the GLP-1RA drug class.
References
Kapitza C, Nosek L, Jensen L, Hartvig H, Jensen CB, Flint A. Semaglutide, a once-weekly human GLP-1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J Clin Pharmacol. 2015;55:497–504.
Nauck MA, Petrie JR, Sesti G, et al. A phase 2, randomized, dose-finding study of the novel once-weekly human GLP-1 analog, semaglutide, compared with placebo and open-label liraglutide in patients with type 2 diabetes. Diabetes Care. 2016;39:231–41.
Lau J, Bloch P, Schaffer L, et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem. 2015;58:7370–80.
van Noord C, Eijgelsheim M, Ch. Stricker BH. Drug- and non-drug-associated QT interval prolongation. Br J Clin Pharmacol. 2010;70:16–23.
Straus SMJM, Kors JA, De Bruin ML, et al. Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. J Am Coll Cardiol. 2006;47:362–7.
Darpo B. Spectrum of drugs prolonging QT interval and the incidence of torsades de pointes. Eur Heart J Suppl. 2001;3:K70–80.
Nachimuthu S, Assar MD, Schussler JM. Drug-induced QT interval prolongation: mechanisms and clinical management. Ther Adv Drug Saf. 2012;3:241–53.
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). The clinical evaluation of QT/QTc interval. Prolongation and proarrhythmic potential for non-antiarrhytmic drugs E14. 2005. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf.
US Food and Drug Administration (FDA). Guidance for industry. E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. 2005. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073153.pdf.
Smith LL, Mosley IJF, Parke C, Brown J, Barris LS, Phan LD. Dulaglutide (trulicity): the third once-weekly GLP-1 agonist. Pharm Ther. 2016;41:357–60.
Darpo B, Zhou M, Matthews J, et al. Albiglutide does not prolong QTc interval in healthy subjects: a thorough ECG study. Diabetes Ther. 2014;5:141–53.
Chatterjee DJ, Khutoryansky N, Zdravkovic M, Sprenger CR, Litwin JS. Absence of QTc prolongation in a thorough QT study with subcutaneous liraglutide, a once-daily human GLP-1 analog for treatment of type 2 diabetes. J Clin Pharmacol. 2009;49:1353–62.
Fridericia LS. Die Systolendauer im Elektrokardiogramm bei normalen Menchan und bei Herzdranken. Acta Med Scand. 2002;53:469–86.
Committee for Medicinal Products for Human Use. Note for guidance on the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for nonantiarrhythmic drugs (CHMP/ICH/2/04). 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002879.pdf.
Zhang J. More lessons learned for TQT studies—FDA perspectives. Presentation at the DIA meeting ′QT and arrhythmia issues in drug development’. 2008. Washington DC.
Yan LK, Zhang J, Nq MJ, Dang Q. Statistical characteristics of moxifloxacin-induced QTc effect. J Biopharm Stat. 2010;20:497–507.
Bazett HC. An analysis of the time-relations of electrocardiograms. Heart. 1920;7:353–70.
Desai M, Li L, Desta Z, Malik M, Flockhart D. Variability of heart rate correction methods for the QT interval. Br J Clin Pharmacol. 2003;55:511–7.
Lorenz M, Lawson F, Owens D, et al. Differential effects of glucagon-like peptide-1 receptor agonists on heart rate. Cardiovasc Diabetol. 2017. https://doi.org/10.1186/s12933-016-0490-6.
Mason RE, Likar I. A new system of multiple-lead exercise electrocardiography. Am Heart J. 1966;71:196–205.
World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. JAMA. 2000;284:3043–5.
International Conference on Harmonisation. ICH Harmonised tripartite guideline. Good Clinical Practice, 1996. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf.
Seaquist ER, Anderson J, Childs B, et al. Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society. Diabetes Care. 2013;36:1384–95.
Bloomfield DM, Kost JT, Ghosh K, et al. The effect of moxifloxacin on QTc and implications for the design of thorough QT studies. Clin Pharmacol Ther. 2008;84:475–80.
Linnebjerg H, Seger M, Kothare P, Hunt T, Wolka AM, Mitchell MI. A thorough QT study to evaluate the effects of single dose exenatide 10 µg on cardiac repolarization in healthy subjects. Int J Clin Pharmacol. 2011;49:594–604.
Darpo B, Sager P, MacConell L, et al. Exenatide at therapeutic and supratherapeutic concentrations does not prolong the QTc interval in healthy subjects. Br J Clin Pharmacol. 2013;75:979–89.
Kapitza C, Dahl K, Jacobsen JB, Axelsen MB, Flint A. Effects of semaglutide on beta cell function and glycaemic control in participants with type 2 diabetes: a randomised, double-blind, placebo-controlled trial. Diabetologia. 2017;60:1390–9.
Blundell J, Finlayson G, Axelsen M, et al. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diab Obesity Metabol. 2017;19:1242–51.
Sorli C, Harashima S, Tsoukas GM, et al. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017;5:251–60.
Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834–44.
Robinson LE, Holt TA, Rees K, Randeva HS, O’Hare JP. Effects of exenatide and liraglutide on heart rate, blood pressure and body weight: systematic review and meta-analysis. BMJ Open. 2013;3:e001986.
Pyke C, Heller RS, Kirk RK, et al. GLP-1 receptor localization in monkey and human tissue; Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology. 2014;155:1280–90.
Nakatani Y, Kawabe A, Matsumura M, Aso Y, Yasu T, Banba N, Nakamoto T. Effects of GLP-1 receptor agonists on heart rate and the autonomic nervous system using Holter electrocardiography and power spectrum analysis of heart rate variability. Diabetes Care. 2015;39:e22–3.
Miyagawa J, Odawara M, Takamura T, Iwamoto N, Takita Y, Imaoka T. Once-weekly glucagon-like peptide-1 receptor agonist dulaglutide is non-inferior to once-daily liraglutide and superior to placebo in Japanese patients with type 2 diabetes: a 26-week randomized phase III study. Diabetes Obes Metab. 2015;17:974–83.
Ahren B, Masmiquel L, Kumar H, et al. Efficacy and safety of once weekly semaglutide versus sitagliptin as add-on to metformin and/or thiazolidinediones in subjects with type 2 diabetes (SUSTAIN 2): a 56-week randomised, controlled clinical trial. Lancet Diabetes Endocrinol. 2017;5:341–54.
Aroda VR, Bain SC, Cariou B, et al. Efficacy and safety of once-weekly semaglutide versus once-daily insulin glargine in insulin-naïve subjects with type 2 diabetes (SUSTAIN 4): a randomised open-label clinical trial. Lancet Diabetes Endocrinol. 2017;5:355–66.
Aroda VR, Ratner R. The safety and tolerability of GLP-1 receptor agonists in the treatment of type 2 diabetes: a review. Diabetes Metab Res Rev. 2017;27:528–42.
Prasad-Reddy L, Isaacs D. A clinical review of GLP-1 receptor agonists: efficacy and safety in diabetes and beyond. Drugs in Context. 2015;4:212238. https://doi.org/10.7573/dic.212283.
Trujillo JM, Nuffer W, Ellis SL. GLP-1 receptor agonists: a review of head-to-head clinical studies. Ther Adv Endocrinol Metab. 2015;6:19–28.
Acknowledgements
We thank all the participants, investigators and trial-site staff who were involved in the conduct of the trial.
Funding
This clinical trial was supported by Novo Nordisk A/S, who provided the trial products and funded the article processing charges. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Medical Writing and Editorial Assistance
We thank Desirée Thielke, MD (Novo Nordisk), for her review and input to the manuscript, and Angela Stocks, PhD (Larix A/S, Copenhagen, Denmark), for medical writing and editorial services, which were funded by Novo Nordisk.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole and have given their approval for this version to be published.
Disclosures
Valentin Demmel is an employee of Nabios GmbH, the ECG core laboratory that analyzed the ECGs for this trial. Anne Sandberg-Schaal is employed by and holds stocks in Novo Nordisk. Jacob B. Jacobsen is employed by and holds stocks in Novo Nordisk. Jonas Pettersson is employed by and holds stocks in Novo Nordisk. Anne Flint is employed by and holds stocks in Novo Nordisk. Georg Golor has no conflicts of interest to disclose.
Compliance with Ethics Guidelines
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.6236495.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Demmel, V., Sandberg-Schaal, A., Jacobsen, J.B. et al. No QTc Prolongation with Semaglutide: A Thorough QT Study in Healthy Subjects. Diabetes Ther 9, 1441–1456 (2018). https://doi.org/10.1007/s13300-018-0442-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-018-0442-0