
Abstract
Introduction
In a previous pooled analysis of 19 double-blind clinical studies conducted by Merck, which included data available as of July 2009 on 10,246 patients with type 2 diabetes (T2DM), treatment with sitagliptin was shown to be generally well tolerated compared with treatment with control agents. As the sitagliptin clinical development program continues, additional studies with sitagliptin have been completed. The present analysis updates the safety and tolerability assessment of sitagliptin by examining pooled data from 25 double-blind clinical studies.
Methods
The present analysis included data from 14,611 patients in 25 studies with T2DM who received either sitagliptin 100 mg/day (n = 7,726; sitagliptin group) or a comparator agent (n = 6,885; non-exposed group). These studies represent all randomized, double-blind trials conducted by Merck that included patients treated with the usual clinical dose of sitagliptin (100 mg/day) for between 12 weeks and 2 years, and for which results were available as of December 2011. These studies assessed sitagliptin, versus comparator agents, taken as monotherapy, initial combination therapy with metformin or pioglitazone, or as add-on combination therapy with other antihyperglycemic agents (metformin, pioglitazone, a sulfonylurea ± metformin, insulin ± metformin, or metformin + pioglitazone or rosiglitazone). Patient-level data from each study were used to evaluate between-group differences in the exposure-adjusted incidence rates of adverse events (AEs).
Results
Overall incidence rates of AEs and drug-related AEs were higher in the non-exposed group compared with the sitagliptin group. Incidence rates of specific AEs were generally similar between the two groups, except for higher incidence rates of hypoglycemia related to the greater use of a sulfonylurea and diarrhea related to the greater use of metformin in the non-exposed group, and of constipation in the sitagliptin group. Treatment with sitagliptin was not associated with an increased risk of major adverse cardiovascular events, malignancy, or pancreatitis.
Conclusion
In this updated pooled safety analysis of data from 14,611 patients with T2DM, sitagliptin 100 mg/day was generally well tolerated in clinical trials of up to 2 years in duration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the introduction of sitagliptin into the diabetes therapeutic armamentarium in 2006, the use of dipeptidyl peptidase-4 (DPP-4) inhibitors for the management of hyperglycemia in patients with type 2 diabetes has increased worldwide. The role of DPP-4 inhibitors in diabetes treatment guidelines has similarly evolved, with the most recent American Diabetes Association (ADA)/European Association for the Study of Diabetes (EASD) consensus guidelines considering DPP-4 inhibitors to be an appropriate second-line therapy after the initiation of metformin, and in the same category as other available antihyperglycemic therapies (including sulfonylureas, thiazolidinediones, glucagon-like peptide-1 (GLP-1) receptor agonists, and insulin) [1]. This represented a distinct departure from prior ADA/EASD guidelines, which considered only sulfonylureas and insulin to be “well-validated” second-line agents [2]. The emergence of the DPP-4 inhibitors has been driven in large part by the safety and tolerability profile of this class of agents compared with other antihyperglycemic agents. In particular, the low risk of hypoglycemia, the weight-neutrality, and the generally excellent tolerability when compared with other classes of drugs appear to have distinguished this class of incretin-based therapies.
In that context, it is important to continue to evaluate the safety and tolerability of this newer class of antihyperglycemic therapy in well-designed, randomized, controlled clinical trials. Recently, Monami et al. [3] performed an updated meta-analysis of 53 trials of at least 24 weeks in duration, which included over 33,000 patients with type 2 diabetes. In this analysis, which comprised 20,312 patients treated with a DPP-4 inhibitor and 13,569 patients treated with either placebo or an active comparator, outcomes of interest included the incidences of cancer, pancreatitis, all-cause and cardiovascular mortality, and major adverse cardiovascular events (MACE). There was no evidence of an increase in the incidence of cancer [Mantel–Haenszel odds ratio (MH-OR) 1.020, 95% CI 0.742, 1.402] or pancreatitis (MH-OR 0.786, 95% CI 0.357, 1.734) with DPP-4 inhibitor therapy. The overall MH-OR for all-cause and cardiovascular death in patients treated with DPP-4 inhibitor was 0.668 (95% CI 0.396, 1.124) and 0.505 (95% CI 0.252, 1.011), respectively. Additionally, a significantly lower risk of MACE (MH-OR 0.689, 95% CI 0.528, 0.899) was observed. While meta-analyses of published studies can provide an assessment of large numbers of patients across the class of DPP-4 inhibitors, the absence of patient-level data for specific adverse events and the focus, in most publications, on serious adverse experiences limit the ability of such analyses to provide a comprehensive assessment of the overall safety and tolerability profile of an individual DPP-4 inhibitor.
As part of the assessment of the safety and tolerability profile of sitagliptin, pooled analyses of patient-level clinical trial data have been previously reported [4–6]. This current pooled analysis includes data from 25 double-blind, randomized studies of sitagliptin 100 mg/day, and incorporates approximately 40% more patients and approximately 36% more patient-years of exposure than the prior pooled analysis. The availability of patient-level data coupled with a larger patient exposure allow for an enhanced ability to assess the incidence of less common adverse experiences, and also allow for more precise estimates of the incidence rates of reported adverse experiences.
Methods
This post hoc analysis used a pooled population (n = 14,611) drawn from all 25 multicenter, US or multinational, double-blind, parallel-group studies conducted by Merck & Co., Inc., in which patients were randomized to receive sitagliptin 100 mg/day (n = 7,726) or a comparator (n = 6,885) for at least 12 weeks and up to 2 years (the duration of the longest studies) and for which results were available as of December 1, 2011 (complete study listing in Table 6 in Appendix). Each protocol was reviewed and approved by appropriate ethical review committees and authorities for each clinical site. All patients were to have provided written informed consent. The studies evaluated sitagliptin as monotherapy, initial combination therapy with either metformin or pioglitazone, or add-on combination therapy with other antihyperglycemic agents, including metformin, pioglitazone, a sulfonylurea (with and without metformin), insulin (with and without metformin), or metformin with rosiglitazone or pioglitazone. Patients not receiving sitagliptin (i.e., the non-exposed group) received placebo, metformin, pioglitazone, a sulfonylurea (with and without metformin), insulin (with and without metformin), or metformin with rosiglitazone or pioglitazone. From each contributing study, the pooling was conducted by including those portions of each study that had parallel treatment groups with concurrent exposures to sitagliptin 100 mg/day (primarily administered as 100 mg once daily) or other treatments (either placebo or active comparator). Studies conducted only in Japan were excluded from all analyses; a lower starting dose of sitagliptin has been separately developed in Japan. The pooling excluded studies conducted in patients with moderate-to-severe renal insufficiency, because these patients received sitagliptin at doses less than 100 mg/day. Studies describing the safety and tolerability of sitagliptin in patients with moderate and severe renal insufficiency have been previously published [7–9].
In each study, investigators were to report adverse events (serious and non-serious) that occurred during the conduct of the study, as well as serious adverse events occurring within 14 days following the last dose of blinded study drug. These events were encoded in a uniform manner using the Medical Dictionary for Regulatory Activities® (MedDRA version 14.1; MedDRA MSSO, Chantilly, VA, USA), in which terms for specific adverse events that are alike or pertain to the same organ system are categorized by System Organ Class (SOC). To account for potential differences between groups in duration of exposure to treatment, reports of adverse events are expressed as exposure-adjusted incidence rates (numbers of patients with events per 100 patient-years). These analyses were based on the time to the first (incident) event, calculated as follows: incident event rate = 100 × (total number of patients with ≥1 event during eligible exposure period per total patient-years of exposure). The incident event rate per 100 patient-years is referred to as the “incidence rate” throughout the manuscript. For those patients for whom an event was reported, the patient-years of exposure were calculated as the time from the first dose of sitagliptin (or comparator) at randomization to the time that the first post-randomization event occurred. For patients without an event, the patient-years of exposure were calculated as the time from the first dose to 14 days after the last dose of study medication (i.e., sitagliptin or comparator). Differences between treatment groups and the associated 95% CI were calculated using the Miettinen and Nurminen method, stratified by study [10]. For endpoints occurring in fewer than four patients in both groups, 95% CIs were not computed because they did not have the potential of excluding zero. No statistical adjustments were performed for multiple comparisons. All analyses were performed using SAS® version 9.1; SAS Institute, Inc., Cary, NC, USA.
The present analysis used patient-level data from each study to assess the incidence rates of adverse events that occurred following initiation of double-blind study drug. Many studies in this analysis included open-label glycemic rescue therapy, which was to have been initiated based on protocol-specified hyperglycemia criteria that were progressively stricter over the course of the study. When initiated, glycemic rescue therapy was added to the ongoing, blinded study medication to which patients had been randomized. Except where mentioned otherwise, the analyses presented below include all post-randomization events reported to have occurred during a given study, including those events with onset after the initiation of glycemic rescue therapy.
The analysis in this article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by any of the authors.
Adverse Events of Interest
Hypoglycemia
For most studies, hypoglycemia was prespecified as an adverse event of interest. For all of the trials that were pooled for this analysis, hypoglycemia was based on investigator interpretation of clinical symptoms, without the requirement for a concurrent glucose determination. In contrast to the general analysis of adverse events, analyses of hypoglycemia adverse events excluded data following initiation of glycemic rescue therapy to avoid the confounding influence of medications that could cause hypoglycemia. In addition, a separate pooled analysis was performed including only those studies and portions of studies that did not include a sulfonylurea or insulin, to characterize the rate of hypoglycemia with sitagliptin relative to comparators not generally associated with an increased risk for hypoglycemia (i.e., metformin and pioglitazone, as well as placebo).
Gastrointestinal
The incidence of a composite endpoint of gastrointestinal (GI) adverse events (including diarrhea, nausea, vomiting, constipation, and a composite abdominal pain term, which included abdominal pain, upper and lower abdominal pain, abdominal and epigastric discomfort, and GI pain) was calculated. An additional analysis of these GI endpoints was conducted, excluding studies and portions of studies in which patients initiated metformin, to characterize the rate of these GI events with sitagliptin relative to comparators generally not associated with an increased risk for GI events. This separate analysis excluded data following initiation of glycemic rescue therapy.
MACE
An analysis of adverse cardiovascular events comprised of cardiovascular death in addition to ischemic events considered to be MACE was performed. For the MACE-related analysis, an exact method for Poisson processes [11], stratified by study, was used to calculate the exposure-adjusted incidence rate ratios (sitagliptin relative to comparator) and the associated 95% CI.
Neoplasms
All adverse event terms for neoplasms were reviewed in a blinded fashion and classified as corresponding to malignant or non-malignant neoplasms. All terms for malignant neoplasms were contained within the “Neoplasms benign, malignant, and unspecified” SOC, whereas terms for non-malignant neoplasms were contained both within and outside of the “Neoplasms benign, malignant, and unspecified” SOC. Incidence rates and between-group differences were computed for individual neoplasms as well as for the composite endpoints of all malignant neoplasms, all non-malignant neoplasms in the “Neoplasms benign, malignant, and unspecified” SOC, and all non-malignant neoplasms regardless of SOC.
Angioedema
Angioedema events and angioedema-related events, based on an expanded version of the Standard MedDRA Query (SMQ) that included anaphylactic reactions and hypersensitivity, were summarized by treatment group for the periods with and without exposure to an angiotensin-converting enzyme (ACE) inhibitor. Exposure to an ACE inhibitor was defined as the total days of use of an ACE inhibitor during the double-blind treatment period, with patients contributing to patient-years of exposure to an ACE inhibitor for the actual period of time that they were reported to have been taking an ACE inhibitor and to patient-years of non-exposure for the actual period of time that they were reported not to have been taking an ACE inhibitor.
Composite Endpoints of Interest
Incidence rates and between-group differences were calculated for a variety of composite endpoints, consisting of a collection of MedDRA adverse event terms related to the safety issue of interest. These composite endpoints included pancreatitis, pancreatic cancer, acute renal failure, proteinuria, bronchitis, pneumonia, upper respiratory infection, urinary tract infection, atrial fibrillation/flutter, and rash.
Laboratory Abnormalities
Percentages of patients meeting predefined laboratory abnormality criteria for liver enzyme abnormalities [alanine aminotransferase (ALT) and aspartate aminotransferase (AST)] and for serum creatinine were compared between groups.
Results
Patient Characteristics and Exposure
In the entire 25-study cohort, patients (55% male) had an mean age of 54 years (range 19–91 years; 17% ≥65 years), a mean duration of diabetes of 5.1 years, and a mean glycosylated hemoglobin (HbA1c) of 8.4% at baseline (with 29% of patients having a baseline HbA1c ≥9.0%) (Table 1). The majority of patients were White (61%), with 18% Asian and 6% Black. At baseline, 10% of patients had a history of cardiovascular disease, and 81% had additional cardiovascular risk factors besides type 2 diabetes mellitus and cardiovascular disease, including hypertension (53%), history of dyslipidemia/hypercholesterolemia (49%), and history of smoking (39%). There were no meaningful differences between groups in these baseline characteristics.
The mean exposure to study drug was slightly greater in the sitagliptin group relative to the non-exposed group: 284 dosing days (range 1–791) and 264 dosing days (range 1–801), respectively. In the sitagliptin group, 2,457 (32%) patients were treated for at least 1 year, with 584 (8%) of these patients treated for 2 years; the corresponding numbers of patients in the non-exposed group were 1,775 (26%) and 470 (7%). The proportions of patients discontinuing treatment were 27.2% in the sitagliptin group and 28.8% in the non-exposed group.
Summary Measures of Adverse Events
The incidence rate of patients reporting one or more adverse events was higher in the non-exposed group compared with the sitagliptin group (Table 2). The incidence rate of drug-related adverse events was also higher in the non-exposed group, as was the incidence of patient discontinuations due to a drug-related adverse event; this was primarily due to the greater incidence rate of adverse events of drug-related hypoglycemia reported for the non-exposed group (data not shown). The incidence of serious adverse events was similar for the two groups, both overall (Table 2) and by SOC category (data not shown). The incidence of adverse events resulting in death, overall, was similar in the two treatment groups; for the Neoplasms SOC, however, the incidence of adverse events resulting in death was lower in the sitagliptin group compared with the non-exposed group (one event in 6,388 patient-years of follow-up compared with six events in 5,378 patient-years of follow-up, respectively, with a difference in rates of −0.1 events per 100-patient-years (95% CI −0.2, −0.0).
Incidence rates for adverse events in each SOC are in Table 3. There were three SOCs (Metabolism and nutrition disorders; Neoplasms benign, malignant, and unspecified; and Skin and subcutaneous tissue disorders) for which the 95% CI for the between-group difference in incidence rates excluded 0. The between-group difference in the incidence rates of adverse events in the Metabolism and nutrition disorders SOC was primarily due to a higher incidence rate of hypoglycemia in the non-exposed group. The between-group difference in the Neoplasms benign, malignant, and unspecified SOC was related to a higher incidence rate in the sitagliptin group for non-malignant adverse events within the Neoplasms benign, malignant, and unspecified SOC, and was not the result of an imbalance in any single adverse event or any group of biologically related adverse events. The incidence rates of malignancy were similar for the two groups: 0.90 per 100 patient-years in the sitagliptin group and 0.93 per 100 patient-years in the non-exposed group [between-group difference of −0.05 (95% CI −0.41, 0.30)]. For the Skin and subcutaneous disorders SOC, the three most common adverse events were rash, pruritus, and urticaria; the 95% CI included zero for all three of these adverse events.
Adverse Events of Interest
Hypoglycemia
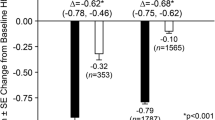
The incidence rates of hypoglycemia were based on symptomatic reports of hypoglycemia, regardless of a concurrent glucose measurement. The predefined analysis for hypoglycemia (i.e., excluding data after initiation of glycemic rescue therapy) showed a between-group difference of −6.2 events per 100 patient-years (95% CI −7.6, −5.0), favoring the sitagliptin group. The difference observed for hypoglycemia was mainly due to the use of a sulfonylurea as a comparator agent in three studies of up to 2 years in duration, as well as a study in which patients were switched from placebo to a sulfonylurea during a double-blind continuation period (P020 in Table 6 in Appendix). Results from some individual studies included in this pooled analysis (in which sitagliptin was added to either a sulfonylurea with or without metformin or to insulin with or without metformin) demonstrated an increased risk for hypoglycemia with sitagliptin used in combination with these agents relative to placebo. In a separate pooled analysis of hypoglycemia in which confounding effects of a sulfonylurea or insulin as either background or comparator therapies were removed, the incidence rates of hypoglycemia were 5.6 and 5.1 per 100 patient-years in the sitagliptin (n = 5,956) and non-exposed (n = 5,122) groups, respectively, with a between-group difference of 0.5 events per 100 patient-years (95% CI −0.7, 1.6).
GI Symptoms
The primary analysis of select GI adverse events demonstrated similar incidence rates for the pooled select GI terms, the composite of abdominal pain terms, nausea, and vomiting (Table 4). The incidence rate of the adverse event of constipation was higher in the sitagliptin group (2.3) than in the non-exposed group (1.8). For the specific adverse event of diarrhea, a lower incidence was observed in the sitagliptin group. The differences observed for diarrhea mainly reflected the use of metformin as a comparator; when the confounding effects of initiation of metformin were removed, the incidence rates were 4.3 and 4.9 per 100 patient-years in the sitagliptin (n = 5,940) and non-exposed (n = 5,122) groups, respectively.
MACE
Detailed description of the analyses of MACE has been previously published [12]. The exposure-adjusted incidence of MACE was 0.65 per 100 patient-years in the sitagliptin group, and 0.74 per 100 patient-years in the non-exposed group, with an adjusted incidence rate ratio of 0.83 (95% CI 0.53, 1.30).
Neoplasms
As noted above, the analysis of all events of malignancies revealed similar incidences in the two treatment groups: 0.90 per 100 patient years in the sitagliptin group and 0.93 per 100 patient-years in the non-exposed group [between-group difference of −0.05 (95% CI −0.41, 0.30)]. Low incidence rates of a wide range of specific malignancies were reported, with similar rates in both treatment groups; the 95% CI did not exclude zero for any of the specific malignancies that were reported. The most common malignancies observed were basal cell carcinoma, prostate cancer, and breast cancer (Table 8 in Appendix). Analyses were performed for the pool of terms representing the category of pancreatic cancer (adenocarcinoma of pancreas, pancreatic carcinoma, pancreatic carcinoma metastatic). The exposure-adjusted incidence rates for the pooled terms related to the category of pancreatic cancer were similar in the two treatment groups (0.05 and 0.06 events per 100 patient-years in the sitagliptin and non-exposed groups, respectively). The number of adverse events (three in each group) was below the pre-defined threshold for calculating a 95% CI.
The incidence rate of adverse events in the Neoplasms benign, malignant, and unspecified SOC overall was 2.03 per 100 patient-years in the sitagliptin group and 1.52 per 100 patient-years in the non-exposed group [between-group difference of 0.52 (95% CI 0.03, 1.01)]. The higher rate in the sitagliptin group was related to a higher rate of non-malignant neoplasms in the Neoplasms benign, malignant, and unspecified SOC [incidence rates of 1.18 and 0.60 per 100 patient-years in the sitagliptin and non-exposed groups, respectively; between-group difference of 0.60 (95% CI 0.25, 0.96)]. This difference was not the result of an imbalance in any single adverse event or any group of biologically related adverse events. The most common non-malignant neoplasm adverse event terms observed were uterine leiomyoma/leiomyoma, lipoma, and skin papilloma. The only term for which the 95% CI around the between-group difference excluded zero was lipoma [between-group difference 0.15 (95% CI 0.02, 0.29)]. A sensitivity analysis, performed to assess the incidence of non-malignant neoplasms across any SOC, revealed a similar pattern, with incidences of 1.58 and 1.12 per 100 patient-years in the sitagliptin and non-exposed groups, respectively [between-group difference of 0.45 (95% CI 0.02, 0.89)]; in this sensitivity analysis, the adverse event term “colonic polyp” was the most common, with similar incidences in the two treatment groups (0.25 and 0.26 per 100-patient years, respectively).
Angioedema
At baseline, 29.4% and 28.1% of sitagliptin-treated and non-exposed patients, respectively, were treated with ACE inhibitors. In the subgroup defined by ACE inhibitor use, the exposure-adjusted incidence of events was 0.99 per 100-patient-years in the sitagliptin group and 1.35 per 100-patient-years in the non-exposed group; for those patients not treated with ACE inhibitors, the incidence rates were 1.14 and 1.16, respectively.
Other Composite Endpoints
The following composite endpoints, primarily of interest due to theoretical mechanistic concerns and/or post-marketing case reports, were analyzed.
For the composite endpoint of pancreatitis (which included the MedDRA terms “pancreatitis” and “pancreatitis acute”), the incidence rates were similar for both groups (Table 4), with a difference in rate of −0.0 (95% CI −0.2, 0.1). A similar pattern was observed with an expanded composite that included the MedDRA term “pancreatitis chronic”.
The incidence of acute renal failure was assessed using both the narrow SMQ and the broad SMQ (Table 4); low and similar rates were observed in both treatment groups for both composite endpoints, as well as for the composite endpoint of proteinuria, which comprised the MedDRA terms “albumin urine present” or “protein urine present”.
Separate analyses were done on the composite endpoints of bronchitis, pneumonia, and upper respiratory infection (Table 4). Similar incidences were seen in both treatment groups for all three of these infection composites. Similar incidence rates were also observed for the composite endpoint of urinary tract infections (with or without cystitis).
The incidence of the composite endpoint of rash was higher in the sitagliptin group compared with the non-exposed group (Table 4). The difference in the composite endpoint was primarily related to a higher incidence of the individual terms “rash” and “rash macular”.
The incidence of the individual adverse event term “atrial fibrillation” was higher in the sitagliptin group (Table 4). For the predefined composite endpoint of atrial fibrillation/atrial flutter, the between-group difference was 0.2 event per 100 patient-years, and the 95% CI did not exclude zero (95% CI −0.0, 0.4).
Specific Adverse Events for which CI Excluded Zero
The incidences of adverse events for which the 95% CI excluded zero are depicted in Table 5. There were 17 specific adverse events in which the incidence was higher in the sitagliptin group, and 23 specific adverse events in which the incidence was higher in the non-exposed group. For those adverse events in which the between-group difference was ≥0.5 events per 100 patient-years, there were two (constipation and dyspepsia) and seven (diarrhea, fatigue, edema peripheral, blood glucose decreased, hypoglycemia, blood glucose increased, and weight increased) in which the incidences were higher in the sitagliptin and non-exposed groups, respectively. Apart from the adverse event of hypoglycemia, the between-group differences in adverse events for which the 95% CI excluded zero were all less than 1.5 events per 100 patient-years.
Predefined Laboratory Abnormality Criteria
Liver Enzymes
The proportions of patients in the sitagliptin and non-exposed groups with their last measurement (obtained either at the time of discontinuation or at the final scheduled study visit) of AST ≥3 times the upper limit of normal (ULN) were both 0.3% [between-group difference 0.0 (95% CI −0.2, 0.2)]; the proportion of patients whose last ALT measurement was ≥3 times the ULN were 0.8% and 0.6%, respectively [between-group difference 0.0 (95% CI −0.0, 0.5)]. One patient in each group had a last value of ALT or AST ≥3 times the ULN with a simultaneous elevation of the total serum bilirubin ≥2 times the ULN.
Serum Creatinine
Similar proportions of patients had a last measurement of serum creatinine with an increase of ≥0.3 mg/dL (1.8% and 1.7% in the sitagliptin and the non-exposed groups, respectively). The proportions of patients who met the predefined criterion of two or more consecutive serum creatinine measurements with an increase from baseline of ≥0.3 mg/dL, or an increase from baseline of ≥50% were also similar in the two groups (0.8% and 0.6%, respectively).
Discussion
An increase in the number of classes of antihyperglycemic therapy options available for the treatment of patients with type 2 diabetes offers patients more choices of effective and well-tolerated therapies that are needed for management of this chronic disease. Assessment of the risk/benefit profile of each class, and specific agents within each class, determines their value for patient management, and this has been acknowledged by the continued evolution of treatment guidelines [1]. Selective DPP-4 inhibitors, which provide physiologic increases in the incretins GLP-1 and gastric inhibitory polypeptide (GIP), offer the potential to be a preferred option for the management of hyperglycemia, since they lack many of the adverse effects observed with other diabetes medications (e.g., hypoglycemia, weight gain) [13]. Nevertheless, continued assessment of the safety and tolerability profile of newer agents, including DPP-4 inhibitors, is necessary as more patients are exposed to such treatments, both through expanded analyses of controlled clinical trial data as well as ongoing pharmacovigilance activities. While pharmacovigilance activities, which include assessment of post-marketing adverse event reports, are of value in identifying potential safety signals, it is well-recognized that these voluntary, spontaneous adverse event reports are derived from a population of uncertain size; thus, it is generally not possible to reliably establish the incidence of such events or to establish a causal relationship between a medication and a specific adverse event. Assessment of the incidence of adverse events from randomized, controlled, clinical trials remains the gold standard for rigorous evaluation of potential safety issues.
Prior pooled analyses of randomized, controlled, clinical trials with sitagliptin, the first-marketed DPP-4 inhibitor, indicated that this agent was generally well tolerated in studies up to 2 years in duration. These data were generally consistent with subsequent pooled analyses of other DPP-4 inhibitors using patient-level data [14–16] as well as with meta-analyses of the DPP-4 class using study-level data [3, 17, 18].
In this current report, the safety and tolerability of sitagliptin was assessed in an expanded pool of studies that comprised over 14,000 patients, representing the largest patient-level data set published to date for a DPP-4 inhibitor. This updated analysis, which expanded on the prior analysis by the addition of six clinical trials, 4,365 patients and 3,114 patient-years of exposure, revealed that treatment with sitagliptin was generally well tolerated, with exposure-adjusted incidence rates of adverse events generally similar to those observed with control therapy that did not include sitagliptin or other DPP-4 inhibitor.
The attainment of currently recommended glycemic goals is limited, in large part, by the increased incidence of hypoglycemia seen with intensive therapies, and particularly with glucose-independent regimens, which include sulfonylureas and insulin. Incretin-based therapies, which provide a glucose-dependent mechanism for enhanced insulin secretion and reduced glucagon secretion, should theoretically be devoid of this risk. Consistent with this mechanistic consideration, the analysis of symptomatic hypoglycemia in studies in which sitagliptin was used as monotherapy or combination therapy (where there was no use of sulfonylureas or insulin) revealed similar rates of symptomatic hypoglycemia for sitagliptin-treated patients compared with non-exposed patients (who received either placebo, metformin, or pioglitazone as comparator agents). The incidence of symptomatic hypoglycemia was lower in the pooled sitagliptin-treated population, mainly related to the use of sulfonylureas as a comparator in several studies. As reported in several clinical trials, the addition of sitagliptin to regimens containing sulfonylurea or insulin resulted in an expected increase in the incidence of symptomatic hypoglycemia related to improvements in glycemic control and a general lowering of ambient glucose concentrations [19, 20]. These findings are consistent with those seen with other classes of antihyperglycemic agents that do not cause hypoglycemia when used as monotherapy, but do so when added onto sulfonylureas or insulin [21, 22]. Thus, in the context of combinations of antihyperglycemic therapies, the risk of hypoglycemia should be carefully considered in choosing appropriate treatment combinations.
An increase in the incidence of GI symptoms is characteristic of treatment with GLP-1 receptor agonists and with metformin. In the current pooled analysis, similar exposure-adjusted incidences were seen in both treatment groups for nausea, vomiting, a composite endpoint of terms related to abdominal pain, and a composite of diverse GI adverse events. Consistent with earlier pooled analyses [5, 6], there was a lower incidence of diarrhea and a higher incidence of constipation observed in the sitagliptin treatment group. These findings were, in part, related to the known effects of metformin on increasing the incidence of diarrhea. However, in a sensitivity analysis in which the confounding effects of metformin as a comparator was removed, a modest increase in the incidence of constipation was still observed. The mechanism underlying this observation is not understood; while DPP-4 inhibitors have not been observed to slow gastric emptying, it remains possible that the physiologic elevations in GLP-1 may have an impact on intestinal motility.
Interest in the relationship between antihyperglycemic agents and pancreatitis was triggered originally by post-marketing reports of acute pancreatitis in patients with type 2 diabetes treated with exenatide [23, 24]. Post-marketing reports of acute pancreatitis in patients treated with all currently marketed GLP-1 mimetics and DPP-4 inhibitors have been observed, and are noted in the labeled information for these products, including sitagliptin [25]. Post-marketing reports represent voluntary, spontaneous adverse event reports regardless of etiology or probability that the medication caused the adverse event. Additionally, post-marketing events are reported from a population of uncertain size; thus, it is generally not possible to reliably establish the frequency of such events or to establish a causal relationship between a medication and a specific adverse event. As noted by the US Food and Drugs Administration (FDA), spontaneous reports such as those contained in the FDA’s Adverse Event Reporting System (AERS) database cannot be used to calculate the incidence of an adverse event [26]. Thus, an analysis of the AERS database that revealed an increase in the reporting rates for pancreatitis with sitagliptin and with exenatide is difficult to interpret, in part due to these intrinsic methodological limitations [27]. In a recently published analysis using a case–control study design, Singh et al. [28] reported a higher rate of hospitalizations for acute pancreatitis in patients with type 2 diabetes associated with the use of incretin-based therapies (sitagliptin or exenatide). This analysis has a number of methodological limitations, including the absence of data on pre-disposing baseline characteristics to allow for robust adjustment for confounding factors, a lack of confirmation of the diagnostic codes used, and lack of adjustment for potential channeling bias [29], which could result in preferential prescribing of incretin-based therapies to patients who were at greater risk for pancreatitis prior to treatment due to age, obesity or other risk factors. Randomized, controlled clinical trial data provide a more robust assessment of the incidence of adverse events. In the current pooled analysis, the incidence of acute pancreatitis was similar in the sitagliptin-treated and the non-exposed group, with exposure-adjusted incidence rates of 0.1 and 0.1 events per 100 patient-years, respectively. Similar findings were observed in the analysis of the composite endpoint of acute and chronic pancreatitis. These data are consistent with those reported previously in a smaller pooled analysis [4], and are also consistent with the systematic pharmacoepidemiologic retrospective cohort assessments performed in two large insurance claims databases [30, 31]. Events of pancreatitis will also be assessed in the sitagliptin cardiovascular outcome study TECOS [32], in which over 14,000 patients are currently enrolled; all cases of pancreatitis will be investigated by an adjudication committee (blinded to treatment assignment) using standard criteria for confirmation of the diagnosis of pancreatitis.
The relationship between antihyperglycemic therapies and malignancy has recently been a focus of increased attention. This is of particular importance in view of the reported association between both obesity and diabetes with an increased risk of malignancy [33], and recent associations of pioglitazone with bladder cancer [34], and dapagliflozin with bladder and breast cancer [35]. In the current pooled analysis, the exposure-adjusted incidence of malignancy was similar for sitagliptin-treated patients and non-exposed patients. The most common malignancies observed (basal cell carcinoma, prostate cancer and breast cancer) were reflective of the demographics of the population, and the incidence rates for these malignancies were similar in patients treated with sitagliptin and those not treated with sitagliptin. Of additional note was the similar incidence of pancreatic cancer in the two treatment groups. The relatively short-term duration of exposure (≤2 years) precludes definitive conclusions regarding any potential association with malignancy, but the lack of any signal in this randomized, controlled, clinical trial database is reassuring. Additionally, the incidence of cancer will be assessed in the long-term cardiovascular outcome study TECOS [32], in which a median duration of follow-up of 4 years is anticipated.
As had been observed in a previous pooled analysis [6], there was a slightly higher incidence of non-malignant neoplasms in the sitagliptin treatment group compared with the non-exposed group (1.18 versus 0.60 events per 100 patient-years). The between-group difference in incidence rates did not exclude zero for any non-malignant neoplasm other than lipoma. The most commonly observed non-malignant neoplasms (i.e., colonic polyp, uterine leiomyoma, and lipoma) were reflective of the expected pattern in the general adult population [36–38], and included a collection of disparate and diverse types of lesions of varying histology and biology. The large number of unrelated adverse event terms assessed in these pooled analyses and the varying and diverse histologies that underlie the reported non-malignant neoplasms suggest that the small increase in the incidence rate of non-malignant neoplasms in the sitagliptin group relative to the non-exposed group may be a stochastic finding and not related to the use of sitagliptin.
The incidence rate ratio of MACE in this pooled analysis was 0.83 (95% CI 0.53, 1.30). It is of interest that both preclinical and clinical mechanistic studies have demonstrated benefits of incretin-based therapies on cardiovascular function and outcomes [39, 40]. These data from the pooled analysis are consistent with a potential beneficial effect of sitagliptin on cardiovascular outcomes, but definitive evaluation of the cardiovascular effects of sitagliptin awaits the completion of the TECOS trial.
Over 17% of patients with diabetes are reported to have chronic kidney disease, and diabetes is associated with progressive renal insufficiency [41]. Clinical trials of sitagliptin in patients with moderate and severe renal insufficiency have indicated that sitagliptin is generally well tolerated in this population [7–9]. In this current pooled analysis of patients with normal or mildly impaired renal function, the evaluation of the impact of sitagliptin on renal function included an assessment of predefined changes in serum creatinine, and the incidence of adverse events related to progressive renal dysfunction (proteinuria and acute renal failure). For all of these measures, no difference between the two treatment groups was observed for the proportion of patients reaching the predefined laboratory abnormality thresholds or in the incidence of adverse events of proteinuria or acute renal failure.
The following are limitations of the present pooled analysis: the results are from patients included in randomized, controlled clinical studies of up to years in duration and, thus, may not be fully reflective of the use in the general population, nor of more prolonged use; the analysis focused on sitagliptin 100 mg/day, the usual clinical dose; and there were multiple comparisons made without an adjustment for multiplicity, which increased the chances for spurious findings. The strengths of these analyses include the ability to account for all reported adverse events using patient-level data, and the large number of clinical trials and patients analyzed.
Conclusion
In this updated pooled safety analysis based on data available as of December 2011 from over 14,000 patients with type 2 diabetes, treatment with sitagliptin 100 mg/day was generally well tolerated as monotherapy, as initial combination therapy, and as add-on therapy in double-blind, randomized clinical studies of up to 2 years in duration. Continued assessment of adverse events reported from clinical trials and from the post-marketing environment is ongoing.
References
Inzucchi SE, Bergenstal RM, Buse JB, American Diabetes Association; European Association for the Study of Diabetes, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364–79.
Nathan DM, Buse JB, Davidson MB, American Diabetes Association; European Association for the Study of Diabetes, et al. Medical management of hyperglycaemia in type 2 diabetes mellitus: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia. 2009;52:17–30.
Monami M, Dicembrini I, Martelli D, Mannucci E. Safety of dipeptidyl peptidase-4 inhibitors: a meta-analysis of randomized clinical trials. Curr Med Res Opin. 2011;27(Suppl 3):57–64.
Engel SS, Williams-Herman DE, Golm GT, et al. Sitagliptin: review of preclinical and clinical data regarding incidence of pancreatitis. Int J Clin Pract. 2010;64:984–90.
Williams-Herman D, Round E, Swern AS, et al. Safety and tolerability of sitagliptin in patients with type 2 diabetes: a pooled analysis. BMC Endocr Disord. 2008;8:14.
Williams-Herman D, Engel SS, Round E, et al. Safety and tolerability of sitagliptin in clinical studies: a pooled analysis of data from 10,246 patients with type 2 diabetes. BMC Endocr Disord. 2010;10:7.
Arjona Ferreira JC, Marre M, Barzilai N, et al. Efficacy and safety of sitagliptin versus glipizide in patients with type 2 diabetes and moderate-to-severe chronic renal insufficiency. Diabetes Care. 2013;36:1067–73.
Arjona Ferreira JC, Corry D, Mogensen CE, et al. Efficacy and safety of sitagliptin in patients with type 2 diabetes and ESRD receiving dialysis: a 54-week randomized trial. Am J Kidney Dis. 2013;61:579–87.
Chan JC, Scott R, Arjona Ferreira JC, et al. Safety and efficacy of sitagliptin in patients with type 2 diabetes and chronic renal insufficiency. Diabetes Obes Metab. 2008;10:545–55.
Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med. 1985;4:213–26.
Breslow NE, Day NE. Statistical methods in cancer research. The design and analysis of cohort studies, vol. II. Lyon: IARC Sci Pub; 1987.
Engel SS, Golm GT, Shapiro D, Davies MJ, Kaufman KD, Goldstein BJ. Cardiovascular safety of sitagliptin in patients with type 2 diabetes mellitus: a pooled analysis. Cardiovasc Diabetol. 2013;12:3.
Deacon CF. Dipeptidyl peptidase 4 inhibition with sitagliptin: a new therapy for type 2 diabetes. Expert Opin Investig Drugs. 2007;16:533–45.
Cobble ME, Frederich R. Saxagliptin for the treatment of type 2 diabetes mellitus: assessing cardiovascular data. Cardiovasc Diabetol. 2012;11:6.
Schernthaner G, Barnett AH, Emser A, et al. Safety and tolerability of linagliptin: a pooled analysis of data from randomized controlled trials in 3572 patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2012;14:470–8.
Schweizer A, Dejager S, Foley JE, Kothny W. Assessing the general safety and tolerability of vildagliptin: value of pooled analyses from a large safety database versus evaluation of individual studies. Vasc Health Risk Manag. 2011;7:49–57.
Goossen K, Graber S. Longer term safety of dipeptidyl peptidase-4 inhibitors in patients with type 2 diabetes mellitus: systematic review and meta-analysis. Diabetes Obes Metab. 2012 (Epub ahead of print).
Karagiannis T, Paschos P, Paletas K, Matthews DR, Tsapas A. Dipeptidyl peptidase-4 inhibitors for treatment of type 2 diabetes mellitus in the clinical setting: systematic review and meta-analysis. BMJ. 2012;344:e1369.
Vilsboll T, Rosenstock J, Yki-Jarvinen H, et al. Efficacy and safety of sitagliptin when added to insulin therapy in patients with type 2 diabetes. Diabetes Obes Metab. 2010;12:167–77.
Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab. 2007;9:733–45.
Buse JB, Henry RR, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care. 2004;27:2628–35.
Rosenstock J, Einhorn D, Hershon K, Glazer NB, Yu S. Efficacy and safety of pioglitazone in type 2 diabetes: a randomised, placebo-controlled study in patients receiving stable insulin therapy. Int J Clin Pract. 2002;56:251–7.
Bain SC, Stephens JW. Exenatide and pancreatitis: an update. Expert Opin Drug Saf. 2008;7:643–4.
Denker PS, Dimarco PE. Exenatide (exendin-4)-induced pancreatitis: a case report. Diabetes Care. 2006;29:471.
Januvia prescribing information. 2013. Whitehouse Station, New Jersey, USA, Merck Sharp & Dohme Corp, a subsidiary of Merck & Co., Inc.
US Food and Drug Administration. Adverse event reporting system; 2013. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/default.htm (Accessed 28 Feb 2013).
Elashoff M, Matveyenko AV, Gier B, Elashoff R, Butler PC. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology. 2011;141:150–6.
Singh S, Chang HY, Richards TM, Weiner JP, Clark JM, Segal JB. Glucagonlike peptide 1-based therapies and risk of hospitalization for acute pancreatitis in type 2 diabetes mellitus: a population-based matched case–control study. JAMA Intern Med. 2013;173:534–9.
Cai B, Katz L, Alexander CM, Williams-Herman D, Girman CJ. Characteristics of patients prescribed sitagliptin and other oral antihyperglycaemic agents in a large US claims database. Int J Clin Pract. 2010;64:1601–8.
Dore DD, Seeger JD, Arnold CK. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin. 2009;25:1019–27.
Garg R, Chen W, Pendergrass M. Acute pancreatitis in type 2 diabetes treated with exenatide or sitagliptin: a retrospective observational pharmacy claims analysis. Diabetes Care. 2010;33:2349–54.
Bethel MA, Green J, Califf M, Holman RR. Rationale and design of the Trial Evaluating Cardiovascular Outcomes with Sitagliptin (TECOS) [abstract]. Diabetologia. 2009;52(Suppl 1):S480.
Giovannucci E, Michaud D. The role of obesity and related metabolic disturbances in cancers of the colon, prostate, and pancreas. Gastroenterology. 2007;132:2208–25.
Neumann A, Weill A, Ricordeau P, Fagot JP, Alla F, Allemand H. Pioglitazone and risk of bladder cancer among diabetic patients in France: a population-based cohort study. Diabetologia. 2012;55:1953–62.
Burki TK. FDA rejects novel diabetes drug over safety fears. Lancet. 2012;379:507.
Hofstad B. Colon polyps: prevalence rates, incidence rates, and growth rates. In: Waye JD, Rex DK, Williams CB, editors. Colonoscopy: principles and practice. 2nd ed. New York: Blackwell Publishing Ltd; 2009. p. 358–78.
Okolo S. Incidence, aetiology and epidemiology of uterine fibroids. Best Pract Res Clin Obstet Gynaecol. 2008;22:571–88.
Rydholm A, Berg NO. Size, site and clinical incidence of lipoma. Factors in the differential diagnosis of lipoma and sarcoma. Acta Orthop Scand. 1983;54:929–34.
Chrysant SG, Chrysant GS. Clinical implications of cardiovascular preventing pleiotropic effects of dipeptidyl peptidase-4 inhibitors. Am J Cardiol. 2012;109:1681–5.
Deacon CF, Marx N. Potential cardiovascular effects of incretin-based therapies. Expert Rev Cardiovasc Ther. 2012;10:337–51.
United States Renal Data System. Annual Data Report (Volume 1): Atlas of Chronic Kidney Disease in the United States. Bethesda: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2010. http://www.usrds.org/2010/view/default.asp.
Scott R, Wu M, Sanchez M, Stein P. Efficacy and tolerability of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy over 12 weeks in patients with type 2 diabetes. Int J Clin Pract. 2007;61:171–80.
Hanefeld M, Herman GA, Wu M, Mickel C, Sanchez M, Stein PP. Once-daily sitagliptin, a dipeptidyl peptidase-4 inhibitor, for the treatment of patients with type 2 diabetes. Curr Med Res Opin. 2007;23:1329–39.
Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther. 2006;28:1556–68.
Charbonnel B, Karasik A, Liu J, Wu M, Meininger G. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care. 2006;29:2638–43.
Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care. 2006;29:2632–7.
Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia. 2006;49:2564–71.
Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab. 2007;9:194–205.
Seck T, Nauck M, Sheng D, Sitagliptin Study 024 Group, et al. Safety and efficacy of treatment with sitagliptin or glipizide in patients with type 2 diabetes inadequately controlled on metformin: a 2-year study. Int J Clin Pract. 2010;64:562–76.
Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care. 2007;30:1979–87.
Williams-Herman D, Johnson J, Teng R, et al. Efficacy and safety of initial combination therapy with sitagliptin and metformin in patients with type 2 diabetes: a 54-week study. Curr Med Res Opin. 2009;25:569–83.
Williams-Herman D, Johnson J, Teng R, et al. Efficacy and safety of sitagliptin and metformin as initial combination therapy and as monotherapy over 2 years in patients with type 2 diabetes. Diabetes Obes Metab. 2010;12:442–51.
Mohan V, Yang W, Son HY, et al. Efficacy and safety of sitagliptin in the treatment of patients with type 2 diabetes in China, India, and Korea. Diabetes Res Clin Pract. 2009;83:106–16.
Barzilai N, Guo H, Mahoney EM, et al. Efficacy and tolerability of sitagliptin monotherapy in elderly patients with type 2 diabetes: a randomized, double-blind, placebo-controlled trial. Curr Med Res Opin. 2011;27:1049–58.
Aschner P, Katzeff HL, Guo H, Sitagliptin Study 049 Group, et al. Efficacy and safety of monotherapy of sitagliptin compared with metformin in patients with type 2 diabetes. Diabetes Obes Metab. 2010;12:252–61.
Dobs AS, Goldstein BJ, Aschner P, et al. Efficacy and safety of sitagliptin added to ongoing metformin and rosiglitazone combination therapy in a randomized placebo-controlled 54-week trial in patients with type 2 diabetes (542). J Diabetes. 2013;5:68–79.
Raz I, Chen Y, Wu M, et al. Efficacy and safety of sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes. Curr Med Res Opin. 2008;24:537–50.
Yoon KH, Shockey GR, Teng R, et al. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and pioglitazone on glycemic control and measures of beta-cell function in patients with type 2 diabetes. Int J Clin Pract. 2011;65:154–64.
Yoon KH, Steinberg H, Teng R, et al. Efficacy and safety of initial combination therapy with sitagliptin and pioglitazone in patients with type 2 diabetes: a 54-week study. Diabetes Obes Metab. 2012;14:745–52.
Wainstein J, Katz L, Engel SS, et al. Initial therapy with the fixed-dose combination of sitagliptin and metformin results in greater improvement in glycaemic control compared with pioglitazone monotherapy in patients with type 2 diabetes. Diabetes Obes Metab. 2012;14:409–18.
Perez-Monteverde A, Seck T, Xu L, et al. Efficacy and safety of sitagliptin and the fixed-dose combination of sitagliptin and metformin vs. pioglitazone in drug-naive patients with type 2 diabetes. Int J Clin Pract. 2011;65:930–8.
Yang W, Guan Y, Shentu Y, et al. The addition of sitagliptin to ongoing metformin therapy significantly improves glycemic control in Chinese patients with type 2 diabetes. J Diabetes. 2012;4:227–37.
Olansky L, Reasner C, Seck TL, et al. A treatment strategy implementing combination therapy with sitagliptin and metformin results in superior glycaemic control versus metformin monotherapy due to a low rate of addition of antihyperglycaemic agents. Diabetes Obes Metab. 2011;13:841–9.
Reasner C, Olansky L, Seck TL, et al. The effect of initial therapy with the fixed-dose combination of sitagliptin and metformin compared with metformin monotherapy in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2011;13:644–52.
Henry RR, Staels B, Fonseca VB, et al. Efficacy and safety of initial combination treatment with sitagliptin and pioglitazone -a factorial study [abstract]. 2011 International Diabetes Federation, 21st World Diabetes Congress; 2011.
Fonseca V, Staels B, Morgan JD, et al. Efficacy and safety of sitagliptin added to ongoing metformin and pioglitazone combination therapy in a randomized, placebo-controlled, 26-week trial in patients with type 2 diabetes. J Diabetes Complications. 2012;27:177–83.
Scott R, Loeys T, Davies MJ, Engel SS. Efficacy and safety of sitagliptin when added to ongoing metformin therapy in patients with type 2 diabetes. Diabetes Obes Metab. 2008;10:959–69.
Arechavaleta R, Seck T, Chen Y, et al. Efficacy and safety of treatment with sitagliptin or glimepiride in patients with type 2 diabetes inadequately controlled on metformin monotherapy: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab. 2011;13:160–8.
Acknowledgments
Funding for the studies and article publication charges was provided by Merck Sharp & Dohme Corp. a subsidiary of Merck & Co., Inc, Whitehouse Station, New Jersey. Dr. S Engel is the guarantor for this article, and takes responsibility for the integrity of the work as a whole. Alan G. Meehan, Michael Davies, and Kathleen Newcomb, Merck Sharp & Dohme Corp. a subsidiary of Merck & Co., Inc, Whitehouse Station, NJ provided editorial support for this manuscript. The authors acknowledge Helen Wang (Merck Sharp & Dohme Corp.) for her statistical programming efforts. Funding for the studies was provided by Merck Sharp & Dohme Corp. a subsidiary of Merck & Co., Inc, Whitehouse Station, NJ.
Conflict of interest
Samuel S. Engel, Elizabeth Round, Gregory T. Golm, Keith D. Kaufman, Barry J. Goldstein are employees of Merck Sharp & Dohme Corp. a subsidiary of Merck & Co., Inc, Whitehouse Station, New Jersey, and may own stock or stock options in the company.
Compliance with ethics guidelines
The analysis in this article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Engel, S.S., Round, E., Golm, G.T. et al. Safety and Tolerability of Sitagliptin in Type 2 Diabetes: Pooled Analysis of 25 Clinical Studies. Diabetes Ther 4, 119–145 (2013). https://doi.org/10.1007/s13300-013-0024-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-013-0024-0