
Abstract
Aims/hypothesis
The aim of this study was to assess the efficacy and safety of oral semaglutide vs sitagliptin in a predominantly Chinese population with type 2 diabetes inadequately controlled with metformin treatment.
Methods
The Peptide Innovation for Early Diabetes Treatment (PIONEER) 12 trial was a randomised, double-dummy, active-controlled, parallel-group, Phase IIIa trial conducted over 26 weeks at 90 sites across the China region (including mainland China, Taiwan and Hong Kong) and five other countries. Adults aged ≥18 years (≥20 years in Taiwan) with a diagnosis of type 2 diabetes, HbA1c between 53 and 91 mmol/mol (inclusive) and treated with a stable daily dose of metformin were eligible for inclusion. Participants were randomised (1:1:1:1) using a web-based randomisation system to either once-daily oral semaglutide (3 mg, 7 mg or 14 mg) or once-daily oral sitagliptin 100 mg. Treatment allocation was masked to both participants and investigators. Randomisation was stratified according to whether participants were from the China region or elsewhere. The primary endpoint was change in HbA1c from baseline to week 26. The confirmatory secondary endpoint was change in body weight (kg) from baseline to week 26. All randomised participants were included in the full analysis set (FAS). All participants exposed to at least one dose of trial product were included in the safety analysis (SAS).
Results
Of 1839 participants screened, 1441 were randomly assigned to oral semaglutide 3 mg (n=361), 7 mg (n=360), 14 mg (n=361) or sitagliptin 100 mg (n=359) and included in the FAS. A total of 1438 participants were included in the SAS. In total, 75.2% of participants were from the China region. A total of 1372 (95.2%) participants completed the trial and 130 participants prematurely discontinued treatment (8.3%, 8.6% and 15.0% for oral semaglutide 3 mg, 7 mg and 14 mg, respectively; 4.2% for sitagliptin 100 mg). Significantly greater reductions in HbA1c from baseline to week 26 were reported for all doses of oral semaglutide vs sitagliptin 100 mg. For oral semaglutide 3 mg, 7 mg and 14 mg vs sitagliptin 100 mg, the estimated treatment differences (ETDs [95% CI]) were –2 (–4, –1) mmol/mol, –8 (–9, –6) mmol/mol and –11 (–12, –9) mmol/mol, respectively. The corresponding ETDs (95% CI) in percentage points vs sitagliptin 100 mg were –0.2 (–0.3, –0.1), –0.7 (–0.8, –0.6) and –1.0 (–1.1, –0.8), respectively. Reductions in body weight were significantly greater for all doses of oral semaglutide vs sitagliptin 100 mg (ETD [95% CI] –0.9 [–1.4, –0.4] kg, –2.3 [–2.8, –1.8] kg and –3.3 [–3.8, –2.8] kg for 3 mg, 7 mg and 14 mg, respectively). In the subpopulation of participants from the China region (75.2% of trial participants), reductions in HbA1c and body weight from baseline to week 26 were similar to those seen in the overall population. The most frequent adverse events in the semaglutide treatment arms were gastrointestinal, although these were mostly transient and mild/moderate in severity.
Conclusions/interpretation
Significantly greater reductions in both HbA1c and body weight over 26 weeks were seen with oral semaglutide 3 mg, 7 mg and 14 mg than with sitagliptin 100 mg in a predominantly Chinese population with type 2 diabetes inadequately controlled with metformin treatment. Oral semaglutide was generally well tolerated, with a safety profile consistent with that seen in the global PIONEER trials.
Trial registration
ClinicalTrials.gov NCT04017832.
Funding
This trial was funded by Novo Nordisk A/S, Søborg, Denmark.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.

Introduction
As one of the most common chronic diseases, diabetes is a major health problem both in China and worldwide. As of 2021, a total of 140.9 million people in China had been diagnosed with diabetes, with this number predicted to rise to 174.4 million by 2045 [1]. China now accounts for a quarter of all adults living with diabetes worldwide [1]; hence, there is a need for effective treatment approaches for Chinese populations.
To prevent long-term microvascular and macrovascular complications associated with hyperglycaemia, achieving and maintaining glycaemic control remains the primary goal of type 2 diabetes treatment. Chinese Diabetes Society (CDS), American Diabetes Association (ADA) and ADA/European Association for the Study of Diabetes (EASD) guidelines all recommend an HbA1c target of <53 mmol/mol (<7.0%) [2,3,4]. However, despite lifestyle modification and the availability of numerous pharmacotherapies, in 2018, only 50.1% of Chinese people with diabetes on glucose-lowering therapies had HbA1c levels within the target range [5].
Overweight and obesity are well-known risk factors for type 2 diabetes. In China, 34.3% and 16.4% of adults had overweight or obesity, respectively, between 2015 and 2019 [6]. CDS guidelines recommend a target of 5–10% body weight loss in people with overweight or obesity [2]. Glucose-lowering therapies that reduce body weight can therefore provide additional clinical benefits in people with type 2 diabetes, with weight loss shown to improve glycaemic control [7]. Glucagon-like peptide-1 receptor agonists (GLP-1RAs) are a drug class capable of reducing both HbA1c and body weight in people with type 2 diabetes [8]. The most recent CDS guidelines recommend the use of GLP-1RAs in combination with metformin, regardless of glycaemic targets, in people with type 2 diabetes with established atherosclerotic CVD or who are at high risk of CVD [2]. It is also recommended that people with type 2 diabetes and overweight or obesity consider treatment with GLP-1RAs or other glucose-lowering drugs with weight loss effects [2].
Globally, there are delays in treatment intensification in people with type 2 diabetes, with one systematic review of therapeutic inertia reporting a median time to treatment intensification ranging from 0.3 to 2.7 years after at least one HbA1c measurement above target [9]. Studies of GLP-1RAs have shown that the mode of administration associated with this drug class (injection) is a barrier to initiation [10], highlighting a need for a variety of effective therapies administered by different routes in this population.
Oral semaglutide, the first GLP-1RA developed for oral administration [11], may be a new option for people experiencing clinical inertia with injectable GLP-1RAs and other currently available therapies [10]. The global Phase IIIa Peptide Innovation for Early Diabetes Treatment (PIONEER) 1–8 trials assessed the efficacy and safety of oral semaglutide in people with type 2 diabetes across a broad range of background medications and comorbidities [12,13,14,15,16,17,18,19]. Oral semaglutide was shown to be effective at improving glycaemic control, with a safety profile consistent with that of the GLP-1RA class [12,13,14,15,16,17,18,19]. Although Asian racial and ethnic groups were included in the PIONEER trials [12,13,14,15,16,17,18,19], and the efficacy and safety of oral semaglutide in a predominantly Japanese population were demonstrated during PIONEER 9 and 10 [20, 21], there is limited evidence available for oral semaglutide from a predominantly Chinese population.
The multiregional PIONEER 12 trial aimed to assess the efficacy and safety of oral semaglutide vs the dipeptidyl peptidase-4 inhibitor (DPP-4i) sitagliptin in a predominantly Chinese population with type 2 diabetes inadequately controlled with metformin.
Methods
Trial design
PIONEER 12 (ClinicalTrials.gov NCT04017832) was a 26 week, randomised, double-blind, double-dummy, active-controlled, parallel-group Phase IIIa trial conducted at 90 sites across the China region (including mainland China, Taiwan and Hong Kong) and Brazil, the Czech Republic, Romania, Serbia and South Africa. The trial protocol was approved by the appropriate health authorities according to local guidelines and by an institutional review board/independent ethics committee. The trial was conducted in accordance with the Declaration of Helsinki 2013 and International Council for Harmonisation Good Clinical Practice guidelines. A list of investigators is provided in electronic supplementary material (ESM) Appendix 1. Written informed consent was obtained from all participants prior to any trial-related activities.
Participants
Participants were aged ≥18 years (≥20 years in Taiwan), had been diagnosed with type 2 diabetes ≥60 days prior to screening, had HbA1c 53–91 mmol/mol inclusive (7.0–10.5% inclusive) and had been on a stable dose of metformin (≥1500 mg or the maximum tolerated dose) for ≥60 days prior to screening. Exclusion criteria included treatment with any other medication for diabetes or obesity in the 60 days prior to screening (except metformin or short-term [14 days] insulin usage), history of pancreatitis, renal impairment and unstable diabetic retinopathy or maculopathy. Full eligibility criteria are provided in ESM Table 1. Data regarding sex, race and ethnicity were self-reported by participants.
Procedures, randomisation and masking
Following a 2 week screening period, eligible participants were randomised 1:1:1:1 using a web-based randomisation system to once-daily oral semaglutide (3 mg, 7 mg or 14 mg) or once-daily oral sitagliptin (100 mg) for 26 weeks, with the follow-up visit taking place at 31 weeks (ESM Fig. 1). Randomisation was stratified according to whether participants were from the China region or elsewhere. The population of participants from the China region is referred to hereafter as the Chinese subpopulation. The study was double-blinded, meaning that both participants and investigators were masked to the treatment allocation. As oral semaglutide and sitagliptin are not visually identical, a double-dummy trial design was used to mask treatment allocation. This meant that participants allocated to oral semaglutide also received a sitagliptin placebo, and participants allocated to sitagliptin also received an oral semaglutide placebo.
All participants underwent an 8 week escalation regimen to support the double-blinded trial design. Oral semaglutide was initiated at 3 mg and escalated every 4 weeks to the next dose until the randomisation dose was achieved (3 mg, 7 mg or 14 mg). No dose escalation was required for sitagliptin. Background glucose-lowering medication was limited to concomitant metformin only, maintained at the same dose level and frequency as at enrolment, unless rescue medication was needed.
Participants with persistent and unacceptable hyperglycaemia received rescue medication if confirmatory fasting plasma glucose (FPG) tests reported values of >14.4 mmol/l from week 8 to the end of week 13, or >13.3 mmol/l from week 14 to the end of treatment at week 26 (including self-monitored plasma glucose [SMPG]), and no intercurrent cause of the hyperglycaemia could be identified. Rescue medication was prescribed at the investigators’ discretion according to ADA/EASD guidelines [22, 23]. Use of GLP-1RAs, DPP-4is or amylin analogues as rescue medication was not permitted.
Endpoints and assessments
The primary endpoint was change in HbA1c from baseline to week 26, and the confirmatory secondary endpoint was change in body weight (kg) from baseline to week 26. Supportive secondary endpoints included change from baseline to week 26 in FPG, 7-point SMPG, body weight (%), BMI, waist circumference and fasting lipid profile. Additional supportive secondary endpoints included achievement of HbA1c <53 mmol/mol (<7.0%; ADA target), HbA1c ≤48 mmol/mol (≤6.5%; American Association of Clinical Endocrinologists [AACE] target), an HbA1c reduction ≥10.9 mmol/mol (≥1 percentage point) or body weight loss ≥3%, ≥5 or ≥10% at week 26. Two composite endpoints of HbA1c <53 mmol/mol (<7.0%) without hypoglycaemia (treatment-emergent severe or blood glucose-confirmed symptomatic hypoglycaemia, confirmed by a glucose value <3.1 mmol/l with symptoms consistent with hypoglycaemia) and with no body weight gain, and an HbA1c reduction ≥10.9 mmol/mol (≥1 percentage point) with body weight loss ≥3% were also assessed at week 26. The change from baseline to week 26 in patient-reported outcomes was also evaluated using the 36-item Short Form Health Survey (Acute Version) (SF-36v2).
Safety endpoints included the number of treatment-emergent adverse events (AEs) and the number of hypoglycaemic episodes up to approximately 31 weeks, defined according to the three-tier ADA 2018 classification [23], and the change from baseline to week 26 in laboratory assessments and vital signs.
Information on protocol deviations as a result of the COVID-19 pandemic is provided in ESM Appendix 2.
Statistical analysis
A sample size of 361 participants per treatment arm was calculated to provide 85% power to jointly confirm the superiority of oral semaglutide (14 mg and 7 mg) vs sitagliptin 100 mg and the non-inferiority of oral semaglutide 3 mg vs sitagliptin 100 mg in reducing HbA1c at week 26 for the trial product estimand. The efficacy endpoint analyses were based on the full analysis set (FAS), which included all randomised participants; analyses of safety endpoints were based on the safety analysis set (SAS), which included all participants exposed to at least one dose of trial product.
Two questions relating to the efficacy objectives were addressed through the definition of two estimands. The trial product (primary) estimand evaluated the treatment effect for all randomised participants under the assumption that all participants continued taking the trial product for the entire planned duration of the trial and did not use rescue medication. The treatment policy (secondary) estimand evaluated the treatment effect for all randomised participants regardless of trial product discontinuation or use of rescue medication. A series of observation periods was defined (ESM Appendix 3).
The primary analysis for the trial product estimand was carried out using a mixed model for repeated measurements using a restricted maximum likelihood, with treatment and region as categorical fixed effects and baseline HbA1c or body weight (depending on the analysis) as covariate. For the trial product estimand, a closed testing procedure was used to control the overall type 1 error at a nominal two-sided 5% level. Overall significance of 0.05 (two-sided) was initially allocated to the HbA1c non-inferiority test of oral semaglutide 14 mg vs sitagliptin 100 mg (ESM Fig. 2). The prespecified margin for assessment of the non-inferiority of semaglutide 3 mg compared with sitagliptin was 0.3%. The statistical testing strategy ensured that non-inferiority was established in terms of HbA1c before testing for superiority and the benefits on body weight at the same dose. Superiority for HbA1c had to be established at higher doses before testing hypotheses at lower doses (ESM Fig. 2). The local significance level was to be reallocated if a hypothesis was confirmed. Further details regarding the statistical analyses performed are provided in ESM Appendix 3 and ESM Fig. 2.
All analyses of the primary and secondary endpoints described above were repeated for the Chinese subpopulation, except for the removal of region as a categorical fixed effect in the model (prespecified).
Results
Participants and baseline characteristics
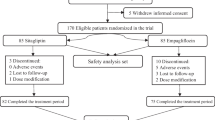
Between July 2019 and October 2021, 1839 individuals were screened, with 1441 randomly assigned to oral semaglutide 3 mg (n=361), 7 mg (n=360) or 14 mg (n=361) or sitagliptin 100 mg (n=359). All randomised participants were included in the FAS, and all except for two participants in the oral semaglutide 7 mg arm and one participant in the sitagliptin 100 mg arm were exposed to the trial product and included in the SAS (Fig. 1). The trial was completed by 95.2% (n=1372) of participants. The trial product was prematurely discontinued by 8.3% (n=30), 8.6% (n=31) and 15.0% (n=54) of participants in the oral semaglutide 3 mg, 7 mg and 14 mg arms, respectively, and by 4.2% (n=15) of participants in the sitagliptin 100 mg arm (Fig. 1). Two participants (0.1%) prematurely discontinued the trial product because of COVID-19 infection, and three participants (0.2%) prematurely discontinued the trial product because of disruption caused by the COVID-19 pandemic. Otherwise, the pandemic and related restrictions did not affect the conduct of the trial and are not considered to have impacted the trial results.

Participant flow diagram
Baseline characteristics were similar across treatment groups (Table 1). Just over half (58.3% [n=840]) of all participants were male and 75.2% were from the China region. The mean age of participants was 53 years, mean HbA1c was 66 mmol/mol (8.2%), mean duration of diabetes was 5.6 years, mean FPG was 9.1 mmol/l and mean body weight was 79.5 kg. All participants except for one in the sitagliptin arm were receiving only background metformin at randomisation.
Primary endpoint
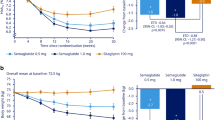
For the trial product estimand, oral semaglutide 3 mg, 7 mg and 14 mg were superior to sitagliptin 100 mg in reducing HbA1c from baseline to week 26 (Fig. 2). Estimated mean changes in HbA1c from baseline to week 26 were –9 mmol/mol, –14 mmol/mol and –17 mmol/mol (–0.8 percentage points, –1.3 percentage points and –1.6 percentage points) for oral semaglutide 3 mg, 7 mg and 14 mg, respectively, and –7 mmol/mol (–0.6 percentage points) for sitagliptin 100 mg (Fig. 2a,b). The estimated treatment differences (ETDs [95% CI]) for oral semaglutide 3 mg, 7 mg and 14 mg vs sitagliptin 100 mg were –2 (–4, –1) mmol/mol (p<0.01), –8 (–9, –6) mmol/mol and –11 (–12, –9) mmol/mol, respectively (p<0.001 for semaglutide 7 mg and 14 mg; Fig. 2b). The corresponding ETDs (95% CI) in percentage points vs sitagliptin 100 mg were –0.2 (–0.3, –0.1), –0.7 (–0.8, –0.6) and –1.0 (–1.1, –0.8), respectively. Similar results were observed for the treatment policy estimand (Fig. 2c,d) and for the Chinese subpopulation for both estimands (Fig. 3).

Change in HbA1c from baseline to week 26 (primary endpoint) in the overall trial population. Observed and estimated mean values (±SEM) and estimated mean change from baseline for HbA1c by (a, b) the trial product estimand and (c, d) the treatment policy estimand. Data are for the on-treatment without rescue medication (a, b) or for the in-trial period (c, d) for the total trial population. At baseline, mean (SD) HbA1c was 65 (10) mmol/mol (8.1% [0.9%]), 66 (10) mmol/mol (8.1% [0.9%]), 65 (9) mmol/mol (8.1% [0.9%]) and 67 (10) mmol/mol (8.2% [0.9%]) for the oral semaglutide 3 mg, 7 mg, 14 mg and sitagliptin 100 mg groups, respectively (FAS). ETDs (95% CI) for oral semaglutide 3, 7 and 14 mg vs sitagliptin 100 mg for the trial product estimand were −2 (−4, −1), −8 (−9, −6) and −11 (−12, −9) mmol/mol, respectively. ETDs (95% CI) for oral semaglutide 3 mg, 7 mg and 14 mg vs sitagliptin 100 mg for the treatment policy estimand were −2 (−4, −1), −7 (−9, −5) and −10 (−11, −8) mmol/mol, respectively. aEstimated means and corresponding error bars are from the primary analysis. **p<0.01; ***p<0.001. ETD, estimated treatment difference

Change in HbA1c from baseline to week 26 (primary endpoint) in the Chinese subpopulation. Observed and estimated mean values (±SEM) and estimated mean change from baseline for HbA1c by (a, b) the trial product estimand and (c, d) the treatment policy estimand. Data are for the on-treatment without rescue medication (a, b) or for the in-trial period (c, d) for the Chinese subpopulation. At baseline, mean (SD) HbA1c was 65 (10) mmol/mol (8.1% [0.9%]), 66 (10) mmol/mol (8.2% [0.9%]), 65 (9) mmol/mol (8.1% [0.8%]) and 66 (10) mmol/mol (8.2% [0.9%]) for the oral semaglutide 3 mg, 7 mg, 14 mg and sitagliptin 100 mg groups, respectively (Chinese subpopulation FAS). ETDs (95% CI) for oral semaglutide 3 mg, 7 mg and 14 mg vs sitagliptin 100 mg for the trial product estimand were −2 (−4, −1), −8 (−10, −7) and −10 (−12, −8) mmol/mol, respectively. ETDs (95% CI) for oral semaglutide 3 mg, 7 mg and 14 mg vs sitagliptin 100 mg for the treatment policy estimand were −2 (−4, −1), −7 (−9, −6) and −9 (−11, −7) mmol/mol, respectively. aEstimated means and corresponding error bars are from the primary analysis. *p<0.05; **p<0.01; ***p<0.001
Confirmatory secondary endpoint
For the trial product estimand, oral semaglutide 3 mg, 7 mg and 14 mg were superior to sitagliptin 100 mg in reducing body weight from baseline to week 26 (Fig. 4). Estimated mean changes in body weight from baseline to week 26 were –1.4 kg, –2.8 kg and –3.8 kg for oral semaglutide 3 mg, 7 mg and 14 mg, respectively, and –0.5 kg for sitagliptin 100 mg (Fig. 4a,b). ETDs (95% CI) vs sitagliptin 100 mg were –0.9 kg (–1.4 kg, –0.4 kg; p<0.001), –2.3 kg (–2.8 kg, –1.8 kg; p<0.001) and –3.3 kg (–3.8 kg, –2.8 kg; p<0.001), respectively (Fig. 4b). Similar results were observed for the treatment policy estimand (Fig. 4c,d) and for the Chinese subpopulation for both estimands (Fig. 5).

Change in body weight from baseline to week 26 (confirmatory secondary endpoint) in the overall trial population. Observed and estimated mean values (±SEM) and estimated mean change from baseline for body weight by (a, b) the trial product estimand and (c, d) the treatment policy estimand. Data are for the on-treatment period without rescue medication (a, b) or for the in-trial (c, d) for the total trial population. At baseline, mean (SD) body weight was 80.8 (19.3) kg, 80.1 (17.7) kg, 79.0 (16.7) kg and 78.3 (17.6) kg for the oral semaglutide 3 mg, 7 mg, 14 mg and sitagliptin 100 mg groups, respectively (FAS). ETDs (95% CI) for oral semaglutide 3 mg, 7 mg and 14 mg vs sitagliptin 100 mg for the trial product estimand were −0.9 (−1.4, −0.4), −2.3 (−2.8, −1.8) and −3.3 (−3.8, −2.8) kg, respectively. ETDs (95% CI) for oral semaglutide 3 mg, 7 mg and 14 mg vs sitagliptin 100 mg for the treatment policy estimand were −0.9 (−1.4, −0.5), −2.2 (−2.6, −1.7) and −3.0 (−3.5, −2.5) kg, respectively. aEstimated means and corresponding error bars are from the primary analysis. ***p<0.001

Change in body weight from baseline to week 26 (confirmatory secondary endpoint) in the Chinese subpopulation. Observed and estimated mean values (±SEM) and estimated mean change from baseline for body weight by (a, b) the trial product estimand and (c, d) the treatment policy estimand. Data are for the on-treatment period without rescue medication (a, b) or for the in-trial (c, d) for the Chinese subpopulation. At baseline, mean (SD) body weight was 75.3 (15.2) kg, 75.0 (13.9) kg, 74.1 (13.7) kg and 73.0 (13.2) kg for the oral semaglutide 3 mg, 7 mg, 14 mg and sitagliptin 100 mg groups, respectively (Chinese subpopulation FAS). ETDs (95% CI) for oral semaglutide 3 mg, 7 mg and 14 mg vs sitagliptin 100 mg for the trial product estimand were −0.8 (−1.3, −0.2), −2.1 (−2.6, −1.6) and −3.0 (−3.5, −2.5) kg, respectively. ETDs (95% CI) for oral semaglutide 3 mg, 7 mg and 14 mg vs sitagliptin 100 mg for the treatment policy estimand were −0.8 (−1.3, −0.3), −1.9 (−2.4, −1.4) and −2.6 (−3.1, −2.1) kg, respectively. aEstimated means and corresponding error bars are from the primary analysis. **p<0.01; ***p<0.001
Supportive secondary endpoints (trial product estimand)
The mean changes in FPG and 7-point SMPG from baseline to 26 weeks were significantly greater with oral semaglutide (all doses) than with sitagliptin 100 mg (Table 2; ESM Fig. 3). The 7-point SMPG postprandial increments were also significantly reduced with oral semaglutide 7 mg and 14 mg vs sitagliptin 100 mg (ESM Table 2). The observed proportions of participants achieving HbA1c <53 mmol/mol (<7.0%; ADA target) and HbA1c ≤48 mmol/mol (≤6.5%; AACE target) were statistically significantly greater with oral semaglutide 7 mg and 14 mg than with sitagliptin 100 mg after 26 weeks of treatment (Table 2).
Significantly greater reductions in body weight (%) at 26 weeks were observed with all doses of oral semaglutide vs sitagliptin (p<0.001; Table 2). The observed proportions of participants achieving ≥3%, ≥5% or ≥10% body weight loss at 26 weeks were greater with all oral semaglutide doses than with sitagliptin 100 mg (Table 2). The ORs of achieving ≥5% or ≥10% body weight loss were statistically significant in favour of oral semaglutide 7 mg (p<0.001 for both) and 14 mg (p<0.001 for both) vs sitagliptin 100 mg (Table 2). For oral semaglutide 3 mg, achievement of ≥5% body weight loss was significant (p<0.01) but achievement of ≥10% body weight loss was not significant (Table 2). Significantly greater reductions in BMI and waist circumference were seen with oral semaglutide (all doses) than with sitagliptin 100 mg (p<0.05 for all; ESM Table 2).
The ORs of achieving both composite endpoints (i.e. achieving HbA1c <53 mmol/mol [<7.0%] without hypoglycaemia or weight gain and achieving a ≥10.9 mmol/mol [≥1 percentage point] HbA1c reduction and ≥3% weight loss) were statistically significant in favour of oral semaglutide (all doses) vs sitagliptin 100 mg (Table 2).
Estimated treatment ratios to baseline were generally similar between treatment groups across the lipid profile, except for total cholesterol, which was significantly lower in participants treated with oral semaglutide 7 mg and 14 mg than in participants treated with sitagliptin (estimated treatment ratio [trial product estimand] 0.97 [95% CI 0.95, 1.00; p<0.05] and 0.97 [0.94, 0.99; p<0.05], respectively; Table 2).
Overall, patient-reported outcomes (assessed by the SF-36v2) improved, but there were no statistically significant differences between oral semaglutide and sitagliptin (ESM Table 2). The proportions of participants on additional concomitant and rescue glucose-lowering medication were low and similar across all treatment groups; the time from first dose to rescue medication use was not significantly different between any of the oral semaglutide doses and sitagliptin 100 mg (ESM Table 3).
The results for the treatment policy estimand are also presented in Table 2 and ESM Table 2 and are broadly similar to the results for the trial product estimand.
Adverse events and tolerability
The overall proportion of participants experiencing AEs while on treatment was slightly higher for oral semaglutide 7 mg and 14 mg than for oral semaglutide 3 mg and sitagliptin 100 mg, with most AEs being mild/moderate in severity (Table 3). Across all semaglutide treatment arms, the most frequent AEs were gastrointestinal disorders (most commonly nausea, diarrhoea and vomiting), the majority of which were mild/moderate in severity. Gastrointestinal disorders were reported by a greater proportion of participants on all doses of oral semaglutide than participants on sitagliptin 100 mg, and increased in frequency as the oral semaglutide dose increased. For sitagliptin 100 mg, the most common AEs were upper respiratory tract infections, diabetic retinopathy and diarrhoea (Table 3). In the Chinese subpopulation, the proportion of participants experiencing AEs was slightly higher across all treatment arms compared with the overall population; however, a higher proportion of total AEs was classed as mild in severity compared with the overall population.
The proportion of participants with serious adverse events (SAEs) was low in all treatment groups but was highest in the oral semaglutide 3 mg and sitagliptin 100 mg arms; this was also the case for the Chinese subpopulation (Table 3). There were five deaths among exposed participants (of which one was in the Chinese subpopulation): two in the oral semaglutide 3 mg arm from infection (one as a result of acute exacerbation of interstitial lung disease and one from COVID-19 pneumonia), two in the oral semaglutide 7 mg arm from infection and other causes (COVID-19 pneumonia and pulmonary embolism) and one in the oral semaglutide 14 mg arm (undetermined cause). All deaths were judged by the investigators as unlikely to be related to the trial product.
The proportion of participants who prematurely discontinued the trial product because of AEs in both the overall population and the Chinese subpopulation was higher in the oral semaglutide 14 mg arm (9.7% and 11.1%, respectively) than in the oral semaglutide 3 mg (4.7% and 5.9%), oral semaglutide 7 mg (4.5% and 5.6%) and sitagliptin 100 mg (1.7% and 2.2%) arms, with gastrointestinal AEs being the most common reason for discontinuation across all treatment groups. The frequency of hypoglycaemic episodes was low across groups, with 1.4%, 0.6%, 0.6% and 0% of participants experiencing a level 2 hypoglycaemic event in the oral semaglutide 3 mg, 7 mg and 14 mg and sitagliptin 100 mg arms, respectively. Only one participant in the Chinese subpopulation, who was in the oral semaglutide 14 mg arm, experienced a level 2 hypoglycaemic event. No level 3 severe hypoglycaemic events were reported (Table 3).
Eye complications (including diabetic retinopathy) were mild/moderate in severity for both the overall and the Chinese subpopulation. In the overall population they were reported by 4.7% (17/361), 3.6% (13/358), 3.3% (12/361) and 5.6% (20/358) of participants in the oral semaglutide 3 mg, 7 mg, 14 mg and sitagliptin 100 mg arms, respectively. Corresponding values in the Chinese subpopulation were 5.9% (16/272), 3.7% (10/268), 4.4% (12/271) and 7.0% (19/270) of participants, respectively.
The frequencies of event adjudication committee-confirmed events, including acute kidney injury, cardiovascular events and malignant neoplasms, were low and similar across all treatment arms in the overall population (ESM Table 4). There were no event adjudication committee-confirmed cases of acute pancreatitis or medullary thyroid cancer. Treatment with oral semaglutide resulted in mean increases in amylase and lipase levels during the initial 14 weeks of the trial, after which no further increases were observed. Increases in amylase and lipase were statistically significant for oral semaglutide 14 mg vs sitagliptin 100 mg (treatment ratio [95% CI] 1.05 [1.01, 1.09; p<0.01] for amylase; 1.11 [1.03, 1.18; p<0.01] for lipase). Other safety variables, including laboratory assessments and vital signs, are reported in ESM Table 5.
Discussion
The results of PIONEER 12 demonstrate that once-daily oral semaglutide was superior to sitagliptin 100 mg in improving glycaemic control and lowering body weight in combination with metformin in a predominantly Chinese population with type 2 diabetes inadequately controlled with metformin. Significantly greater reductions in HbA1c and body weight from baseline to week 26 were observed for all doses of oral semaglutide than for sitagliptin 100 mg using the trial product estimand. Similar reductions were observed in the Chinese subpopulation, which represented 75.2% of the overall population. For the treatment policy estimand, the observed reductions in HbA1c and body weight were dose dependent, a finding that is consistent with the results using the treatment policy estimand in the global PIONEER programme [12,13,14,15,16,17,18,19]. Reductions in HbA1c and body weight from baseline to week 26 in the overall PIONEER 12 population were greater than those observed at week 26 in the PIONEER 3 trial, which assessed oral semaglutide 3 mg, 7 mg and 14 mg vs sitagliptin 100 mg in a global population across 78 weeks [18].
Participants treated with all doses of oral semaglutide experienced greater reductions in FPG and 7-point SMPG than those treated with sitagliptin 100 mg. These greater reductions with oral semaglutide compared with sitagliptin are consistent with the results seen in the global PIONEER 3 population. Furthermore, the reductions in FPG and 7-point SMPG observed with all doses of oral semaglutide at week 26 in PIONEER 12 were greater than those observed with the same doses of oral semaglutide at week 26 during PIONEER 3 [18]. Participants treated with oral semaglutide were more likely to achieve HbA1c <53 mmol/mol (<7.0%), HbA1c ≤48 mmol/mol (≤6.5%), body weight loss of ≥5% and ≥10% and the composite endpoints than those treated with sitagliptin. Reductions in total cholesterol were significantly greater in participants treated with oral semaglutide 7 mg and 14 mg than in those treated with sitagliptin 100 mg (trial product estimand), which is also consistent with the PIONEER 3 trial results [18]. Oral semaglutide was generally well tolerated across the trial treatment period, although a greater proportion of participants experienced AEs leading to premature trial discontinuation with oral semaglutide than with sitagliptin. This was largely due to the greater incidence of gastrointestinal AEs with oral semaglutide; these were the most frequently reported AEs across all treatment arms but they were mostly transient and mild/moderate in severity, consistent with the safety profile of the GLP-1RA drug class [8, 18, 24]. A higher rate of gastrointestinal AEs was observed in PIONEER 12 than in PIONEER 3 [18]. This could potentially be explained by the greater levels of semaglutide exposure as a result of the lower mean baseline body weight in PIONEER 12 than in PIONEER 3 (79.5 kg vs 91.2 kg) [18, 25]. Additionally, PIONEER 12 required administration of sitagliptin or sitagliptin placebo 30 min after administration of oral semaglutide or oral semaglutide placebo, whereas in PIONEER 3 these were administered simultaneously [18]; it is not unreasonable to consider that co-administration of placebo with oral semaglutide could reduce exposure and may potentially explain the increased efficacy in this trial compared with PIONEER 3. Indeed, lower semaglutide exposure was observed in PIONEER 3 than in the other global PIONEER trials [26]; however, pharmacokinetic analyses were not included in the trial design of PIONEER 12 so it is not possible to confirm this hypothesis. It is important to note that PIONEER 3 was a larger trial than PIONEER 12 (1864 participants vs 1441 participants, respectively) and had a longer duration (78 weeks with 5 weeks of follow-up vs 26 weeks with 5 weeks of follow-up, respectively) [18]. Because the incidence of AEs was greatest during dose escalation, the disparity in total years of exposure between the two trials could potentially dilute the reported rate of AEs in PIONEER 3 compared with PIONEER 12. Consistent with the previous global PIONEER trials, few SAEs were reported across treatment arms and most were reported in the oral semaglutide 3 mg and sitagliptin 100 mg treatment arms. Overall, the safety and tolerability profile for oral semaglutide in PIONEER 12 was consistent with the results from the global PIONEER trials and with the general safety profile of the GLP-1RA drug class [27].
The strengths of this trial include the high number of participants enrolled and randomised, particularly in the Chinese subpopulation, and the masking of differences in the appearance of the drugs. Potential limitations of this trial include the short trial duration. At 26 weeks, PIONEER 12 was relatively short compared with most of the global PIONEER trials, except for PIONEER 1 and 5, and was considerably shorter than the 78 week PIONEER 3 trial that it mirrors. However, considering that the safety and tolerability profile of oral semaglutide in PIONEER 12 was consistent with that of the global PIONEER trials and with the safety profile of the GLP-1RA drug class, it is reasonable to assume that long-term safety in this population may be consistent with that seen in the long-term global trials. Another limitation is that no specific analyses were performed regarding the impact of sex, gender, race or ethnicity on the results of this trial. As such, caution should be taken when applying the results to a wider population. In addition, treatment adherence was not formally measured and participants were instead reminded by the trial investigators to follow the treatment protocol at each visit, with the investigators also monitoring drug accountability. A final limitation is the use of sitagliptin as a comparator. DPP-4is have modest glucose-lowering effects and minimal body weight loss effects compared with GLP-1RAs [24]; however, their use is increasing steadily in Chinese populations [28].
In conclusion, this trial demonstrates the superiority of oral semaglutide vs sitagliptin in reducing HbA1c and body weight from baseline in a predominantly Chinese population with type 2 diabetes, as well as a safety and tolerability profile for semaglutide that is consistent with that of the GLP-1RA class.
Abbreviations
- AACE:
-
American Association of Clinical Endocrinologists
- AE:
-
Adverse event
- CDS:
-
Chinese Diabetes Society
- DPP-4i:
-
Dipeptidyl peptidase-4 inhibitor
- ETD:
-
Estimated treatment difference
- FAS:
-
Full analysis set
- FPG:
-
Fasting plasma glucose
- GLP-1RA:
-
Glucagon-like peptide-1 receptor agonist
- PIONEER:
-
Peptide Innovation for Early Diabetes Treatment
- SAE:
-
Serious adverse event
- SAS:
-
Safety analysis set
- SF-36v2:
-
36-item Short Form Health Survey (Acute Version)
- SMPG:
-
Self-monitored plasma glucose
References
International Diabetes Federation (2021) IDF diabetes atlas, 10th edn. Available from: https://diabetesatlas.org/idfawp/resource-files/2021/07/IDF_Atlas_10th_Edition_2021.pdf. Accessed 9 Jun 2023
Chinese Diabetes Society (2021) Guidelines for the prevention and treatment of type 2 diabetes mellitus in China (2020 edition). Chin J Diabetes Mellitus 13(4):315–409. https://doi.org/10.3760/cma.j.cn115791-20210221-00095
Davies MJ, Aroda VR, Collins BS et al (2022) Management of hyperglycemia in type 2 diabetes, 2022. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 65:1925–1966. https://doi.org/10.1007/s00125-022-05787-2
American Diabetes Association Professional Practice Committee (2021) 6. Glycemic targets: standards of medical care in diabetes—2022. Diabetes Care 45(Supplement_1):S83–S96. https://doi.org/10.2337/dc22-S006
Wang L, Peng W, Zhao Z et al (2021) Prevalence and treatment of diabetes in China, 2013–2018. JAMA 326(24):2498–2506. https://doi.org/10.1001/jama.2021.22208
Pan XF, Wang L, Pan A (2021) Epidemiology and determinants of obesity in China. Lancet Diabetes Endocrinol 9(6):373–392. https://doi.org/10.1016/S2213-8587(21)00045-0
American Diabetes Association (2022) Standards of medical care in diabetes–2022 abridged for primary care providers. Clin Diabetes 40(1):10–38. https://doi.org/10.2337/cd22-as01
Htike ZZ, Zaccardi F, Papamargaritis D, Webb DR, Khunti K, Davies MJ (2017) Efficacy and safety of glucagon-like peptide-1 receptor agonists in type 2 diabetes: a systematic review and mixed-treatment comparison analysis. Diabetes Obes Metab 19(4):524–536. https://doi.org/10.1111/dom.12849
Khunti K, Gomes MB, Pocock S et al (2018) Therapeutic inertia in the treatment of hyperglycaemia in patients with type 2 diabetes: a systematic review. Diabetes Obes Metab 20(2):427–437. https://doi.org/10.1111/dom.13088
Gallwitz B, Giorgino F (2021) Clinical perspectives on the use of subcutaneous and oral formulations of semaglutide. Front Endocrinol (Lausanne) 12:645507. https://doi.org/10.3389/fendo.2021.645507
US Food and Drug Administration (2021) Rybelsus®: highlights of prescribing information. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213051s006lbl.pdf. Accessed 9 Jun 2023
Aroda VR, Rosenstock J, Terauchi Y et al (2019) PIONEER 1: randomized clinical trial of the efficacy and safety of oral semaglutide monotherapy in comparison with placebo in patients with type 2 diabetes. Diabetes Care 42(9):1724–1732. https://doi.org/10.2337/dc19-0749
Husain M, Birkenfeld AL, Donsmark M et al (2019) Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 381(9):841–851. https://doi.org/10.1056/NEJMoa1901118
Mosenzon O, Blicher TM, Rosenlund S et al (2019) Efficacy and safety of oral semaglutide in patients with type 2 diabetes and moderate renal impairment (PIONEER 5): a placebo-controlled, randomised, phase 3a trial. Lancet Diabetes Endocrinol 7(7):515–527. https://doi.org/10.1016/S2213-8587(19)30192-5
Pieber TR, Bode B, Mertens A et al (2019) Efficacy and safety of oral semaglutide with flexible dose adjustment versus sitagliptin in type 2 diabetes (PIONEER 7): a multicentre, open-label, randomised, phase 3a trial. Lancet Diabetes Endocrinol 7(7):528–539. https://doi.org/10.1016/S2213-8587(19)30194-9
Pratley R, Amod A, Hoff ST et al (2019) Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet 394(10192):39–50. https://doi.org/10.1016/S0140-6736(19)31271-1
Rodbard HW, Rosenstock J, Canani LH et al (2019) Oral semaglutide versus empagliflozin in patients with type 2 diabetes uncontrolled on metformin: the PIONEER 2 trial. Diabetes Care 42(12):2272–2281. https://doi.org/10.2337/dc19-0883
Rosenstock J, Allison D, Birkenfeld AL et al (2019) Effect of additional oral semaglutide vs sitagliptin on glycated hemoglobin in adults with type 2 diabetes uncontrolled with metformin alone or with sulfonylurea: the PIONEER 3 randomized clinical trial. JAMA 321(15):1466–1480. https://doi.org/10.1001/jama.2019.2942
Zinman B, Aroda VR, Buse JB et al (2019) Efficacy, safety, and tolerability of oral semaglutide versus placebo added to insulin with or without metformin in patients with type 2 diabetes: the PIONEER 8 trial. Diabetes Care 42(12):2262–2271. https://doi.org/10.2337/dc19-0898
Yabe D, Nakamura J, Kaneto H et al (2020) Safety and efficacy of oral semaglutide versus dulaglutide in Japanese patients with type 2 diabetes (PIONEER 10): an open-label, randomised, active-controlled, phase 3a trial. Lancet Diabetes Endocrinol 8(5):392–406. https://doi.org/10.1016/S2213-8587(20)30074-7
Yamada Y, Katagiri H, Hamamoto Y et al (2020) Dose-response, efficacy, and safety of oral semaglutide monotherapy in Japanese patients with type 2 diabetes (PIONEER 9): a 52-week, phase 2/3a, randomised, controlled trial. Lancet Diabetes Endocrinol 8(5):377–391. https://doi.org/10.1016/S2213-8587(20)30075-9
Davies MJ, D’Alessio DA, Fradkin J et al (2018) Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 61(12):2461–2498. https://doi.org/10.1007/s00125-018-4729-5
American Diabetes Association (2018) 6. Glycemic targets: standards of medical care in diabetes–2018. Diabetes Care 41(Suppl 1):S55–S64. https://doi.org/10.2337/dc18-S006
Tran S, Retnakaran R, Zinman B, Kramer CK (2018) Efficacy of glucagon-like peptide-1 receptor agonists compared to dipeptidyl peptidase-4 inhibitors for the management of type 2 diabetes: a meta-analysis of randomized clinical trials. Diabetes Obes Metab 20(Suppl 1):68–76. https://doi.org/10.1111/dom.13137
Overgaard RV, Navarria A, Ingwersen SH, Bækdal TA, Kildemoes RJ (2021) Clinical pharmacokinetics of oral semaglutide: analyses of data from clinical pharmacology trials. Clin Pharmacokinet 60(10):1335–1348. https://doi.org/10.1007/s40262-021-01025-x
Overgaard RV, Hertz CL, Ingwersen SH, Navarria A, Drucker DJ (2021) Levels of circulating semaglutide determine reductions in HbA1c and body weight in people with type 2 diabetes. Cell Rep Med 2(9):100387. https://doi.org/10.1016/j.xcrm.2021.100387
Thethi TK, Pratley R, Meier JJ (2020) Efficacy, safety and cardiovascular outcomes of once-daily oral semaglutide in patients with type 2 diabetes: the PIONEER programme. Diabetes Obes Metab 22(8):1263–1277. https://doi.org/10.1111/dom.14054
Yang A, Wu H, Lau ESH et al (2020) Trends in glucose-lowering drug use, glycemic control, and severe hypoglycemia in adults with diabetes in Hong Kong, 2002–2016. Diabetes Care 43(12):2967–2974. https://doi.org/10.2337/dc20-0260
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Acknowledgements
Medical writing and editorial support were provided by A. Pearson (Apollo, OPEN Health Communications, London, UK) and were funded by Novo Nordisk, in accordance with Good Publication Practice 3 (GPP3) guidelines (www.ismpp.org/gpp3). The authors thank the participants, the investigators, all trial site staff and all Novo Nordisk A/S employees involved in the trial. The authors also thank W. Liu of Novo Nordisk (China) Pharmaceuticals Co. for her role in reviewing this manuscript.
Data availability
Data will be shared with bona fide researchers submitting a research proposal approved by the independent review board. Information on requesting access to datasets can be found at www.novonordisk-trials.com. Data will be made available after research completion and approval of the product and product use in the European Union and the USA. Individual participant data will be shared in data sets in a de-identified/anonymised format.
Funding
This trial was funded by Novo Nordisk A/S, Søborg, Denmark. The study sponsor was involved in study design, data collection, analysis and interpretation and the writing of this report.
Authors’ relationships and activities
LJ reports receiving consulting and lecture fees from Abbott, AstraZeneca, Bayer, Boehringer Ingelheim, Eli Lilly, Medtronic, Merck, MSD, Novo Nordisk, Roche and Sanofi-Aventis. RMA and SG are employees and shareholders of Novo Nordisk A/S. SCB has received honoraria, teaching and research sponsorship/grants from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, MSD, Novo Nordisk, Pfizer, Sanofi and Takeda. BL and YT are employees of Novo Nordisk (China) Pharmaceuticals Co. All other authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
All authors made substantial contributions to the conception and design of the trial, acquisition of data and/or the analysis and interpretation of data. All authors had access to the study data, critically reviewed the manuscript and approved the final version to be published. LJ, RMA, SG, BL and YT are guarantors of this work and, as such, have full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A complete list of investigators in the Peptide Innovation for Early Diabetes Treatment 12 (PIONEER 12) trial is provided in the electronic supplementary material.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ji, L., Agesen, R.M., Bain, S.C. et al. Efficacy and safety of oral semaglutide vs sitagliptin in a predominantly Chinese population with type 2 diabetes uncontrolled with metformin: PIONEER 12, a double-blind, Phase IIIa, randomised trial. Diabetologia (2024). https://doi.org/10.1007/s00125-024-06133-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00125-024-06133-4