
Abstract
The use of precise, rationally designed gene-editing nucleases allows for targeted genome and transcriptome modification, and at present, four major classes of nucleases are being employed: zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), meganucleases (MNs), and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9. Each reagent shares the ability to recognize and bind a target sequence of DNA. Depending on the properties of the reagent, the DNA can be cleaved on one or both strands, or epigenetic changes can be mediated. These novel properties can impact hematological disease by allowing for: (1) direct modification of hematopoietic stem/progenitor cells (HSPCs), (2) gene alteration of hematopoietic lineage committed terminal effectors, (3) genome engineering in non-hematopoietic cells with reprogramming to a hematopoietic phenotype, and (4) transcriptome modulation for gene regulation, modeling, and discovery.
Similar content being viewed by others

Introduction
The accessibility of progenitor and terminal effector cells of the hematopoietic system for isolation and the ability to culture and manipulate them ex vivo has placed hematological disorders at the forefront of gene-based therapeutic medicine. Classically, this has involved isolation of the cells from the marrow or peripheral blood and the delivery of a functional copy of the defective, disease-causing gene via integrating viral or non-viral gene transfer vectors and delivery back to the patient. In this strategy, gene regulation is typically governed by exogenous regulatory elements that mediate sustained and supraphysiological levels of the cargo. The integrating properties of the vector result in transmission to progeny cells that allows for life-long therapy. Moreover, the ability to correct a patient’s own cells mitigates the incidence of graft-versus-host disease (GVHD) that can result in significant morbidity and mortality when allogeneic hematopoietic cell transplantation (HCT), the standard of care for multiple disorders, is employed. However, the integrating and sustained gene expression properties of gene therapy vectors can result in significant genotoxicity and/or dysregulation of endogenous gene(s) due to integration of the therapeutic cassette [1, 2]. Furthermore, genes not subject to the regulatory cues that govern normal gene expression may be undesirable [3]. Gene editing is a powerful approach that maintains the advantage of hematopoietic compartment-based therapies (cell availability, isolation, and ex vivo modification), while drastically improving safety. This approach refers to the precision alteration of a sequence of DNA at the genome level making it a permanent event that maintains gene control under endogenous elements subject to the regulatory mechanisms of the cell. Presented here are the options, considerations, and applications to date for gene editing in cells of the hematopoietic system.
At present, four major classes of nucleases are available for human genome engineering: zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), meganucleases (MNs), and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 (Fig. 1). Each reagent shares the ability to recognize and bind a target sequence of DNA. Depending on the properties of the reagent, the DNA can be cleaved on one or both strands, or epigenetic changes can be mediated. These novel properties can impact hematological disease by allowing for: (1) direct modification of hematopoietic stem/progenitor cells (HSPC), (2) gene alteration of hematopoietic lineage committed terminal effectors, (3) genome engineering in non-hematopoietic cells with reprogramming to a hematopoietic phenotype, and (4) transcriptome modulation for gene regulation, modeling, and discovery (Fig. 2).

Engineered nuclease architecture. a Zinc finger nucleases. ZFNs are dimeric arrays with DNA binding fingers (blue) that are tethered to the nuclease domains of FokI (yellow) that mediate the DNA cleavage event. The coordinating zinc ion required for DNA binding is shown in orange. b Transcription activator-like effector nucleases. TALENs act as heterodimers that are linked to a FokI nuclease domain. The TAL binding domains (colored ovals) interact with a single base of DNA by virtue of the repeat variable diresidues that recognize the four bases. The RVD HD (blue) binds DNA base C, NN (black) recognizes G, NI (green) interacts with A, and NG (red) binds T. c MegaTAL nuclease. TAL RVD units are fused to a custom engineered homing endonuclease (orange) that generates the DNA break. d Clustered regularly interspaced short palindromic repeats/Cas9. Watson–Crick base pairing of the gRNA (purple) complexed with the Cas9 protein (light blue) results in unwinding and cleavage of each strand of the DNA helix by the HNH and RuvC nuclease domains. In other configurations, the nuclease domains of Cas9 can be inactivated, and alternative functional domains included. Bases are all color-coded: A (green), C (blue), G (black), T (red), U (gold)

Hematopoietic system and gene editing. a The hematopoietic system is derived from a hematopoietic stem/progenitor cell that is self-renewing and differentiates into the common lymphoid (CLP) and myeloid progenitors (CMP). The CL and CMPs give rise to the effector cells of the lymphoid and myeloid compartments, respectively. These cells include T- and B- lymphocytes, NK cells, plasma cells, and mononuclear, red blood and dendritic cells and thrombocytes. Gene editing. b Homology-directed repair. A single- or double-stranded DNA donor can be utilized as a repair template for the correction of disease-causing mutations (shown as a reversion of the red base to green). c Non-homologous end joining. DNA lesions can be repaired by direct ligation of free ends by the error prone NHEJ pathway that results in insertions and deletions. These can cause frameshift mutations that disrupt the open reading frame of genes and cause loss of protein activity. d Autologous cellular engineering. Terminally differentiated cells can be obtained and modified by gene editing nucleases before or after reprogramming to pluripotency. The multipotent progenitors can then be employed for therapeutic lineage commitment. e Epigenome modification. The DNA binding properties of engineered proteins can be employed for transcriptome modulation for increased or decreased gene expression
The critical considerations for gene-editing strategies are summarized in Table 1 and include: reagent choice, cost, availability of starting material reagents, engineering process complexity/length of time, ability to be packaged in efficient gene delivery vehicles, and potential for broad applicability (e.g., ability to be multiplexed, activate, or repress genes). Moreover, the ability to isolate gene edited cells is an important consideration that will be impacted by the end user’s cell type of choice, disease model, and purity required for infusion to achieve therapeutic effect based on the pathobiology of the individual and unique disorders broadly characterized as hematological diseases.
ZFNs
The Zif268 transcription factor has served as the template for generating custom Cys2–His2 ZFNs, with each zinc finger (ZF) comprised of approximately 30 amino acids in a ββα configuration that typically binds three nucleotides [4]. ZFNs are structured as dimeric left and right arrays, each containing between 3 and 6 ZF modules capable of targeting 18–36 bases of genomic sequence (Fig. 1a). In their basal state, ZFs lack intrinsic nuclease activity, necessitating their linkage to a non-specific catalytic domain of the type IIS restriction enzyme FokI [5]. The FokI nuclease functions only as a dimer, and thus a pair of ZFNs can be engineered, whereby they flank a target genomic sequence, allowing dimerization and cleavage of the intervening DNA, resulting in a double-strand break (DSB). To maximize specificity and prevent activity of the left–left or right–right arrays, the FokI domain has been engineered to function as an obligate dimer, such that only the left–right arrays create a DSB [6]. The DNA binding property of ZFs is such that adjacent finger modules appear to influence neighboring ZF recognition and binding, and this context specificity has frustrated approaches to construct ZFNs [7, 8]. Therefore, screening procedures were developed to identify three ZF arrays that accurately target their intended sequence, thus surmounting the problem of context-specificity [9–14]. Oligomerized pool engineering [9] and context-dependent assembly [13] allowed designer ZFNs to be obtained with greater affinity and higher specificity for the desired target sequence. Using these approaches, ZFNs are predicted to have a targeting capacity of approximately one site per 500 bp of human genomic DNA. This restricted targeting capacity, coupled with complicated generation procedures, makes ZFNs limiting [13, 15]. However, a valuable attribute of ZFNs is their small size that allows for simplified delivery as DNA, RNA, or in any viral expression/delivery vehicle.
TALENs
The pathogenic bacterial genus Xanthomonas uses specific DNA recognition proteins to regulate the transcription of target genes in the genomes of the plants they infect [16, 17]. These transcription activator-like (TAL) effectors are DNA binding proteins containing between 13 and 28 repeats, each of which is composed of 33–35 amino acids. The 12th and 13th amino acids of each repeat are known as the repeat variable diresidues (RVDs) and are responsible for DNA binding. Each RVD interfaces with a single nucleotide: RVD NI recognizes adenosine, HD interacts with cytosine, and NG binds thymine, while NN, NH, and NK all have varying specificities for guanine [18–20]. TAL effector nucleases (TALENs) utilize the modularity of these TAL effector RVDs to create novel DNA binding domains that are then linked to the nuclease domain of FokI. Akin to ZFNs, TALENs are designed in pairs that flank the target sequence, allowing the two FokI monomers to dimerize and create a DSB (Fig. 1b).
Because of the highly repetitive nature of the TAL effector, construction of each TALEN pair via PCR is challenging. To remedy this, a Golden Gate assembly procedure utilizes plasmid templates with the desired RVD building block flanked by type IIS restriction enzyme sites. This allows for ordered assembly of repeats to create the desired TAL effector DNA binding sequence [21]. These specifically designed sequences are then attached to the FokI nuclease domain to create the final TALEN construct [22–24]. Like ZFNs, specificity is increased due to the necessity of the TALEN pair to dimerize to cleave the desired genomic DNA sequence. The DNA binding capability of TAL arrays has been exploited further by fusing them to transcriptional activators to allow for gene expression modulation [25]. The ability to expand functionality beyond DNA cleavage, the greatly simplified engineering process using context-independent modules, and the higher frequency of target sites in the human genome (approximately one TAL site per 35 bp of DNA [15, 22]) has made the use of TALENs widespread. Furthermore, TALENs are readily deliverable as mRNA; however, their large, repetitive nature limits their use in some DNA and viral vectors [26].
MNs
Multiple MN families exist, and the class containing the LAGLIDADG protein motif has been primarily used for reengineering to recognize unique target sites [27]. These monomeric enzymes are derived from bacterial homing endonucleases and are constructed around a ‘central 4’ base pair recognition sequence [28, 29]. Recently, a hybrid MN-TAL (megaTAL) protein has been employed for mammalian genome modification (Fig. 1c) [30–32]. The small size and robust activity of MNs and megaTALs are a decided advantage; however, the complicated engineering procedure, requirement for the presence of the ‘central 4’ base composition in the target gene of interest, and potential for promiscuous activity at other genomic loci represent hurdles for expansive application [33, 34].
CRISPR/Cas9
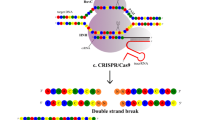
The most recent addition to the genome engineering toolbox is derived from a so-called bacterial adaptive immune system that utilizes an RNA/protein complex [35]. This endogenous system operates in response to phage infection, resulting in the integration of short stretches (approximately 20 nucleotides) of invading phage DNA into the host genome between palindromic repeats [36–38]. These palindromic repeats provide for the ability of the bacterium to differentiate self from non-self to direct DNA cleavage of phage DNA while preserving the integrity of the bacterial genome [39]. The endogenous CRISPR/Cas9 system consists of two RNAs, one that targets the Cas9 protein to the desired DNA sequence (crRNA), and the other that hybridizes to the crRNA and helps to stabilize the Cas9 protein (tracrRNA) [40, 41]. Upon RNA/DNA hybridization, the HNH nuclease domain of Cas9 cleaves the complementary target DNA strand, whereas the RuvC nuclease domain cleaves the opposite strand (Fig. 1d). This cleavage requires the presence of a three nucleotide sequence termed the protospacer adjacent motif (PAM), the sequence of which is 5′-NGG-3′ in the CRISPR/Cas9 system from Streptococcus pyogenes (Sp.), which to date has been utilized the most frequently for mammalian gene editing [42–45]. Shortly after delineation of how the CRISPR/Cas9 system functioned, it was shown that the crRNA and the tracrRNA could be shortened and combined into a single hybrid RNA, called a guide RNA (gRNA) [40]. This minimal RNA can be expressed as a PolIII transcript from a DNA vector or delivered as an in vitro-transcribed RNA species [43, 46]. A major power of the CRISPR/Cas9 system is that the Cas9 component is constant and can recognize multiple, independent gRNAs. Such a multiplexed targeting approach that is not easily achievable with ZFNs, MNs or TALENs, which require protein engineering for each genomic target [47–49]. Limitations of the CRISPR/Cas9 system include the PAM requirement and the potential for off target (OT) effects. Toward expanding the targeting capacity of Cas9, new variants with alternate PAMs have been described [50, 51], including a Cas9 system from Staphylococcus aureus, whose functional components are able to be packaged into the size-restricted adeno-associated virus (AAV) platform [52]. Rationale engineering of the existing Cas9 proteins and the discovery of new candidates, such as the recently described Cpf [53], have further expanded the PAM recognition repertoire and improved specificity, allowing for access to larger portions of the genome of interest [54–58].
Due to initial concerns related to high-level OT activity of CRISPR/Cas9 reagents [56], further modifications to the Cas9 or gRNAs have been undertaken. Truncated gRNAs appear to result in a more targeted activity profile [59, 60]. Direct alteration of Cas9 [57, 58, 61] or the generation of paired Cas9 nickase mutants capable of cleaving a single strand of DNA have been developed, such that only one of the nuclease domains is catalytically active [62]. Then, by designing two gRNAs complementary to opposite DNA strands, two single-strand breaks are generated to make a structure analogous to a staggered DSB [63, 64]. In addition, complete nuclease mutants lacking all catalytic activity can be fused to the nuclease domain of FokI, such that they function much like ZFNs and TALENs [65]. In addition, engineering studies have shown that Cas9 is amenable to modifications that retain its DNA binding ability allowing different domains to be fused to the complex to mediate effects, such as transcriptional activation, repression, and histone acetylation [66–68].
Hematopoietic cell genome engineering
Each of the major classes of gene editing proteins has been employed in hematopoietic lineage cells, primarily for HSPC and/or T-cells, and work to date is summarized in Table 2. HSPC gene modification is highly desirable, as these cells are the source of all lineages of the immune system over the lifespan of an individual (Fig. 2a). Furthermore, allogeneic HCT is a major therapeutic option for a multitude of disorders that are due to defects in the hematopoietic cell compartment (e.g., leukemia, Fanconi anemia, immunodeficiencies, Wiskott–Aldrich syndrome, and hemoglobinopathies) as well as those without an overt blood cell developmental defect (e.g., lysosomal storage disease, Wilms’ tumor, and hemophilia). As such, the ability to precisely modify HSPC in an autologous manner would result in life-long therapy that greatly mitigates the side effects related to GVHD, and transcriptional- and integrating vector-related genomic perturbation.
Following nuclease-mediated DNA cleavage, two competing DNA repair pathways are employed for resolution of the break that left untended is highly toxic: homology-directed repair (HDR) or non-homologous end joining (NHEJ). HDR is an exquisitely precise repair pathway utilizing a homologous sequence of endogenous or exogenous DNA as the repair template–the latter of which allows for the acquisition of a user-defined sequence at the targeted locus (Fig. 2b). NHEJ is a mutagenic DNA repair pathway where broken DNA ends are processed and ligated back to one another in a manner that commonly results in insertions/deletions (indels) that can disrupt the targeted open reading frame, resulting in a loss of function mutation (Fig. 2c).
Initial efforts to mediate HDR in HSPC showed low overall rates of <1 % using ZFNs specific for the CCR5 locus and a donor, each delivered as integrase-deficient lentiviral vector (IDLV) particles [69]. In 2014, members of this same group improved HDR rates in HPSCs from both healthy donors and a patient with X-linked severe combined immunodeficiency caused by an IL2RG gene mutation [70]. To accomplish this, nuclease/donor delivery and HSC culture conditions were optimized to allow for the HDR donor derived insertion of exons 5–8 into the IL2RG locus [70]. This resulted in restoration of the proper reading frame, with transcription mediated by the endogenous promoter, and is a broadly applicable strategy for the correction of SCID-X1 mutations that occur downstream of exon 4 [70]. The IDLV donor was delivered first followed by ZFN mRNA to cells cultured in the presence of 16,16-dimethyl-prostaglandin E2 (PGE2) [71, 72] with or without StemRegenin 1 (SR1) [73]. PGE2 promotes HSC survival, homing, and expansion [74], and SR1 is an aryl hydrocarbon receptor antagonist that facilitates ex vivo expansion and retention of engraftment potential [73]. The use of both agents was additive in the resultant ability to recover the highest levels of gene-corrected cells that were used for primary and secondary engraftment of immunodeficient mice [70].
A second study by Wang assessed HDR in CD34+ HSPCs derived from fetal liver or cord blood using ZFNs delivered as mRNA and a donor template borne on an AAV serotype 6 vector [75]. HDR-driven insertion of a GFP expression cassette was observed at rates of 17 % at CCR5 and 26 % at the AAVS1 loci in G-CSF mobilized peripheral HSPCs and 19 % (CCR5) and 43 % (AAVS1) in HSPCs from fetal liver [75]. Importantly, this group showed that the CD34+ cells treated in bulk resulted in modification of the subfraction of cells with a CD34+CD133+CD90+ phenotype that represents a primitive population of highly engraftable precursors [75]. In addition, using a donor template packaged in AAV-6, Sather et al. further employed electroporation of megaTAL nuclease mRNA that targeted the CCR5 locus in mobilized CD34s cultured with SR1 and achieved maximal HDR rates of 35 % [76]. In this same study, AAV-6 was also highly capable of mediating HDR in primary T-cells, utilizing an HDR targeting construct containing homology arms to the CCR5 locus flanking cargo comprised of: the C46 gene that encodes an HIV restriction protein that confers resistance to HIV, the PGT145 anti-HIV chimeric antigen receptor (CAR), or the CD-19 CAR, respectively [76]. As such, HDR-mediated insertion of the exogenous expression cassettes is paired with loss of CCR5 to confer greater resistance to HIV infection by CXCR4 HIV strains, HIV target lysis mediated by PGT145, or dual HIV resistance and ability to abolish secondary CD19+ B cell tumors [76]. These studies complement another study reported by Wang and colleagues that showed robust HDR in CD4 and CD8 cells, further supporting nuclease and AAV-based cellular engineering for infections, autoimmunity or malignancies [77].
The ability to selectively delete receptors/genes that contribute to the primary or secondary disease process holds tremendous potential for engineering cells for therapy. At present, two disorders with a hematological component are at the leading edge for translational application of engineered nucleases for mutagenic NHEJ-based cellular therapy. The first in human use of gene-editing nucleases employed a CCR5-targeted ZFN and is being utilized as part of an autologous therapy in HIV patients [78]. The next-generation strategies utilizing tandem CCR5 and CXCR4 ZFNs, as well as the above HDR-based strategies, have been used preclinically to establish proof of principle for engineering pan HIV resistant T-cells [76, 79].
B-cell leukemia is a common adult and childhood malignancy with a poor prognosis with relapse. The treatment option of choice at present is allogeneic HCT with the rationale that HLA-mismatched donor cells will recognize tumor cells as foreign as part of a graft-versus-leukemia (GVL) effect. However, due to off-tumor GVHD-associated pathology, autologous-based therapies employing CARs for the CD19 epitope on malignant (and normal) B-cells have been used as part of an individualized treatment strategy that has resulted in some extraordinary successes [80]. However, this personalized approach can result in variability in transduction efficiencies and cell numbers [80] that can be delivered to patients and thus is impractical for wider spread use. As a solution to this, engineered nucleases specific for components of the T-cell receptor (TCR), a multiunit complex that is responsible for antigen recognition has been developed. This strategy has allowed for the generation of cells that are greatly reduced in their GVHD-based reactivity toward healthy tissue and represents an off the shelf, universal donor therapy option.
Provasi et al. in 2012 employed ZFNs to selectively disrupt the genes that encode the alpha and beta chains of the TCR to render the cells TCR null [81]. The TCR negative cells were then paired with a CAR for Wilms’ tumor to mediate anti-tumor effects [81]. In the same year, Torikai et al. employed ZFNs for the alpha chain of the TCR to generate non-alloreactive cells that express the CD19 CAR [82]. Robust TCR disruption rates have also been reported employing megaTALs and TALENs, respectively [31, 83]. Extending this approach to confer greater therapeutic benefits, Poirot et al. employed a multiplex strategy utilizing TALENs for the TCR-alpha chain and CD52 genes, respectively [32]. CD52 encodes a receptor that can be targeted by the monoclonal antibody alemtuzumab that is used therapeutically for leukemia [84]. However, because CD52 is present on both T- and B-cells, T-cells can be depleted and TALEN-mediated disruption of the gene confers a survival advantage to the engineered cells, following administration of alemtuzumab, which may potentiate their therapeutic value [32]. An important finding from this study was that the simultaneous generation of DNA DSBs at the TCR and CD52 loci resulted in translocation events [32]. An extension of this consideration is the potential for overlapping areas of sequence homology to the intended target site, giving rise to OT DNA DSBs. Genome-level screens have shown that these OT events can result in translocations and/or indels [34, 85, 86]. Previous work has shown that the CCR5 ZFN, TALEN, and CRISPR/Cas9 reagents can have OT activity at the CCR2 locus that shares close sequence homology to CCR5 [87–89]. In an effort to define the potential for OT effects at the TCR-alpha locus, members of this group performed a genome-level comparative analysis for a TCR-alpha megaTAL, TALEN, and CRISPR/Cas9 reagent, and observed OT effects for the megaTAL but not the other classes of reagents [34]. Comprehensive OT mapping is of great importance, as it will aid in safer cellular engineering and in the development/re-engineering of more target-restricted reagents for translational application. Despite the presence of OT sites, both ZFNs and TALENs have been utilized in humans. The use of TALENs maximized safety considerations due to the engineering process by including a suicide gene to allow for targeted deletion of infused cells. Using the co-delivery/targeting approach that was validated in a murine model [32], Qasim and colleagues engineered allogeneic T-cells to be TCR-alpha and CD52 null that expressed a CD19 CAR. The cells were infused into a pediatric patient with relapsed B-cell leukemia, who has subsequently shown cytogenetic and molecular remission [90]. As such, the ability to deliver genome-editing reagents to hematopoietic progenitors and effectors represents a translational pipeline for both individualized and universal approaches for hematological disorders.
Future directions
While a major advantage for hematopoietic cell modification is their relative accessibility for explant, modification, and reinfusion, the extended manipulation and culture ex vivo have the possibility of negatively impacting cellular phenotype and engraftability rates [91]. Advances in culture techniques with small molecule regulators that maintain and sustain primitive phenotypes (e.g., SR1 [73]) aid in circumventing this obstacle. However, for disorders such as the bone marrow failure syndromes, where progenitor cells are in short supply, future therapies may include in vivo gene editing or the development of the next-generation cellular engineering-based differentiation methodologies.
Blood development occurs within a complex milieu of progenitor cells and a microenvironment with instructive and supportive signals that are a crucial part of the multimodal maturation process. Hematopoiesis occurs in two waves: embryonic and definitive. The embryonic phase is characterized by the generation of transient cell populations and occurs in the yolk sac giving rise to myelo-erythroid effectors [92]. The definitive wave results in the self-renewing hematopoietic stem/progenitor cell that develops in the aorta/gonad/mesonephros region of the embryo and gives rise to all of the mature blood lineages (erythroid, myeloid, and lymphoid) [92, 93]. These cells then migrate and expand in the fetal liver before transiting to and taking up residence in the bone marrow cavity where they remain through life [94]. This latter developmental process involves endothelial and hematopoietic precursors ultimately giving rise to the definitive HSPC [95].
A decided advantage of in vivo gene editing would be that HSPCs could be targeted in situ, with the native bone marrow stroma providing the proper developmental programming instructions. Indeed, murine models of non-hematopoietic in vivo genome modification have been reported, whereby a gene-specific nuclease and a donor template are delivered to a target organ(s), and the modification is done at the organ and DNA levels [96]. However, modification of HSPCs at sufficient frequency may be limiting in the context of gene delivery, HDR rates, and therapeutic effect related to whether the targeted/corrected cell possesses a preferential selective outgrowth profile. Therefore, the generation of primitive or terminally differentiated hematopoietic effector cells from an induced pluripotent stem cell (iPSC) intermediary holds tremendous promise for future regenerative medicine therapies.
The introduction of the iPSC reprogramming cocktail of c-myc, Oct3/4, Klf4, and Sox2 into terminally differentiated cell types results in a population of renewable cells with multilineage potential [97]. Megakaryocytes are able to be derived from iPSCs under GMP conditions [98] and such a strategy would allow for a renewable population of cells for a bank of off-the-shelf platelet products. Employing an allogeneic approach for T-cell immunotherapy, Themeli et al. [99] generated iPSCs from T-cells, redifferentiated them into T-cells that resulted in a γδ T-cell phenotype, and transduced them with a CAR for tumor therapy in a murine model. Such an approach would result in the standardization of cell doses and transduction levels that have been variable in trials to date [80, 100, 101]. Furthermore, this procedure could be optimized to produce T-cells with a stem memory phenotype, a cell that is normally at low levels in the peripheral blood and is highly desirable due to its ability to self-renew, and give rise to memory and effector cells [102, 103]. Toward developing red blood cells (RBCs) in vitro, Giani and colleagues used a population-based genetics analysis and discovered a naturally occurring loss of function variant of the SH2B3 gene that results in increased RBC counts [104]. Using a CRISPR/Cas9 gene deletion strategy, they were subsequently able to inhibit SH2B3 expression and recapitulate the variant phenotype resulting in enhanced RBC precursor formation in vitro [104].
The ability to correct disease-causing mutations in terminally differentiated cells with the subsequent capacity to differentiate them into repopulating blood progenitors would be a paradigm shift for treating disorders of the hematopoietic system. Gene correction of fibroblast cells from patients with Fanconi anemia has been reported using ZFNs and CRISPR/Cas9 reagents [105, 106], as has their ability to be induced to pluripotency, albeit with no in vivo blood development potential [106, 107]. Using iPSCs as a starting point for HSPC generation, however, has largely resulted in progenitors of the primitive type that do not allow for long-term repopulation and engraftment [95, 108]. Toward recapitulating the critical supportive bone marrow microenvironment, groups led by Amabile [109] and Suzuki [110] injected human iPSCs into immune-deficient animals resulting in teratoma-derived CD34+CD45+ that arose from a complex, multilineage formative environment with the cells being capable of serial engraftment. Due to the clinical hurdles of a teratoma-based approach, a highly promising avenue of investigation is the use of in vitro vascular cell co-culture to provide the instructive niche required for HSPC development [95]. Sandler and colleagues utilized stable adenovirus E4ORF1 gene expression to generate VeraVec™ cells that serve as a supportive matrix of cells. This group further defined a transcriptional cocktail of four genes FOSB, GFI1, RUNX1, and SPI1 (FGRS) that when upregulated in primary endothelial cells that were cultured with VeraVec™ resulted in progenitors capable of multihematopoietic lineage engraftment with the ability to be serially transferred [111]. As such, gene correction/modification of endothelial cells, either primary or derived from iPSCs, could be potentially used as a foundational template for autologous HSPC generation.
The modulation of the FGRS gene set observed to drive HSPC development represents an ideal platform for the application of genome-level screens for genes that can be expressed/repressed to enhance the further/refined development of therapeutically viable HSPCs in vitro (Fig. 2d). The most powerful system for such an approach is the CRISPR/Cas9 system by virtue of its multiplex ability, whereby multiple different gRNAs can simultaneously target genes and the Cas9 can act as a nuclease, transcriptional activator, or repressor to modulate the intended genes [41, 47, 112]. In 2014, Shalem et al. generated over 64,000 gRNAs capable of targeting more than 18,000 genes and performed genome-level knockout studies to discover genes previously unknown in melanoma drug resistance [113]. In 2015, a study to achieve saturating mutagenesis of the BCL11A erythroid enhancer that is a direct regulator of hemoglobin F (HbF) levels [114] was undertaken to define and disrupt critical sequences to mediate HbF induction [115]. As such, the ability to precisely target this non-coding sequence may facilitate threshold levels of functional HbF for treating β-hemoglobinopathies [115].
Less permanent, transcriptome/epigenome modifications may also be employed to determine gene candidates that drive HSPC development, and this platform may hold direct therapeutic value for primary patient-derived cells. Konermann et al. designed more than 70,000 gRNAs that target upstream of every human coding isoform and performed a genome-level screen with a Cas9 transcriptional activator system to discover genes that were upregulated resulting in resistance to a BRAF inhibitor involved in melanoma drug resistance [49]. Specific to hematopoietic therapeutic intent, Hilton and colleagues used the epigenome modifying properties of a Cas9-acetyltransferase fusion complex to an enhancer region of the β-globin locus control region and observed significant upregulation of the downstream HBE, HBG, and HBD genes [116].
Permanent (HDR and NHEJ) and less permanent (gene/chromatin regulation) modifications represent powerful tools for improving existing, developing, and discovering new treatment approaches in cells of the hematopoietic lineage. These methodologies will intersect with novel stem cell development and differentiation approaches and will synergize for the development of the next-generation cell- and genome-based therapies. Guiding principles for the design and use of gene editing reagents for hematopoietic cell modification and therapy will encompass multiple points of emphasis: the cell and disease model will inform the choice of reagent whose activity, specificity, and deliverability properties will dictate their application in the cell and model of choice. Each reagent has attributes and limitations that must be considered in the context of the cell target and/or disease model. For instance, ZFNs and MNs can be readily delivered to most cell types using a multitude of delivery vehicles; however, their complex engineering process can make their application limiting. The more easily generated TALEN and CRISPR/Cas9 reagents level the playing field by placing the technology in the hands of the entire scientific community; however, their size and the target sequence requirements can make delivery and access to certain portions of the genome challenging. Importantly, the rapidly evolving area of study with discovery of new gene-editing reagents is additive to the field and increases the flexibility of the applications for scientists but does not relegate previous classes of nucleases to obscurity.
The transition from bench to bedside for the application of gene editing reagents has been described for T-cells in the context of HIV therapy and leukemia treatment [78, 90]. As such, the developmental pipeline has been established, and the continued application of gene editing in novel ways in cells of the hematopoietic system will aid in more accurate disease modeling that will facilitate more effective autologous, gene-based, individualized therapies for more uniform therapeutic responses. This process will be further augmented by the largely freely available starting materials and enhanced culture techniques that allow the hematological community at large to consider, plan, and execute novel gene editing strategies.
References
Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–42.
Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–9.
Cumming RC, Liu JM, Youssoufian H, Buchwald M. Suppression of apoptosis in hematopoietic factor-dependent progenitor cell lines by expression of the FAC gene. Blood. 1996;88:4558–67.
Beerli RR, Barbas CF 3rd. Engineering polydactyl zinc-finger transcription factors. Nat Biotechnol. 2002;20:135–41.
Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA. 1996;93:1156–60.
Doyon Y, Vo TD, Mendel MC, et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods. 2011; 8:74–79
Ramirez CL, Foley JE, Wright DA, et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods. 2008;5:374–5.
Lam KN, van Bakel H, Cote AG, van der Ven A, Hughes TR. Sequence specificity is obtained from the majority of modular C2H2 zinc-finger arrays. Nucleic Acids Res. 2011;39:4680–90.
Maeder ML, Thibodeau-Beganny S, Osiak A, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31:294–301.
Maeder ML, Thibodeau-Beganny S, Sander JD, Voytas DF, Joung JK. Oligomerized pool engineering (OPEN): an ‘open-source’ protocol for making customized zinc-finger arrays. Nat Protoc. 2009;4:1471–501.
Reyon D, Kirkpatrick JR, Sander JD, et al. ZFNGenome: a comprehensive resource for locating zinc finger nuclease target sites in model organisms. BMC Genom. 2011;12:83.
Hermann M, Maeder ML, Rector K, et al. Evaluation of OPEN zinc finger nucleases for direct gene targeting of the ROSA26 locus in mouse embryos. PLoS One. 2012;7:e41796.
Sander JD, Dahlborg EJ, Goodwin MJ, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods. 2011;8:67–9.
Osborn MJ, DeFeo AP, Blazar BR, Tolar J. Synthetic zinc finger nuclease design and rapid assembly. Hum Gene Ther. 2011;22:1155–65.
Voytas DF, Joung JK. Plant science. DNA binding made easy. Science. 2009;326:1491–2.
Kay S, Hahn S, Marois E, Hause G, Bonas U. A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science. 2007;318:648–51.
Romer P, Hahn S, Jordan T, Strauss T, Bonas U, Lahaye T. Plant pathogen recognition mediated by promoter activation of the pepper Bs3 resistance gene. Science. 2007;318:645–8.
Boch J, Scholze H, Schornack S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–12.
Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333:1843–6.
Streubel J, Blucher C, Landgraf A, Boch J. TAL effector RVD specificities and efficiencies. Nat Biotechnol. 2012;30:593–5.
Engler C, Gruetzner R, Kandzia R, Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One. 2009;4:e5553.
Cermak T, Doyle EL, Christian M, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82.
Li L, Piatek MJ, Atef A, et al. Rapid and highly efficient construction of TALE-based transcriptional regulators and nucleases for genome modification. Plant Mol Biol. 2012;78:407–16.
Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc. 2012;7:171–92.
Maeder ML, Linder SJ, Reyon D, et al. Robust, synergistic regulation of human gene expression using TALE activators. Nat Methods. 2013;10:243–5.
Holkers M, Maggio I, Liu J, et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41:e63.
Silva G, Poirot L, Galetto R, et al. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther. 2011;11:11–27.
Baxter S, Lambert AR, Kuhar R, et al. Engineering domain fusion chimeras from I-OnuI family LAGLIDADG homing endonucleases. Nucleic Acids Res. 2012;40:7985–8000.
Stoddard BL. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure. 2011;19:7–15.
Baxter S, Lambert AR, Kuhar R, et al. Engineering domain fusion chimeras from I-OnuI family LAGLIDADG homing endonucleases. Nucleic Acids Res. 2012;40:7985–8000.
Boissel S, Jarjour J, Astrakhan A, et al. megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res. 2014;42:2591–601.
Poirot L, Philip B, Schiffer-Mannioui C et al. Multiplex genome edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75:3853–64.
Baxter SK, Lambert AR, Scharenberg AM, Jarjour J. Flow cytometric assays for interrogating LAGLIDADG homing endonuclease DNA-binding and cleavage properties. Methods Mol Biol. 2013;978:45–61.
Osborn MJ, Webber BR, Knipping F, et al. Evaluation of TCR Gene Editing achieved by TALENs, CRISPR/Cas9 and megaTAL nucleases. Mol Ther. 2016;24:570–81.
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169:5429–33.
Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–61.
Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–82.
Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–63.
Marraffini LA, Sontheimer EJ. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010;463:568–71.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
Qi LS, Larson MH, Gilbert LA, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–83.
L. Cong, F. A. Ran, D. Cox et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science.
Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6.
Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–2.
Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471.
Hendel A, Bak RO, Clark JT, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. 2015;33:985–9.
Cheng AW, Wang H, Yang H, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23:1163–71.
Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23.
Konermann S, Brigham MD, Trevino AE, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–8.
Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10:1116–21.
Hou Z, Zhang Y, Propson NE, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA. 2013;110:15644–9.
Ran FA, Cong L, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–91.
Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–71.
Kleinstiver BP, Prew MS, Tsai SQ, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–5.
Kleinstiver BP, Prew MS, Tsai SQ, et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 2015;33:1293–8.
Fu Y, Foden JA, Khayter C, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6.
Kleinstiver BP, Pattanayak V, Prew MS, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–5.
Kleinstiver BP, Prew MS, Tsai SQ, et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 2015;33:1293–8.
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–84.
Dahlman JE, Abudayyeh OO, Joung J, Gootenberg JS, Zhang F, Konermann S. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat Biotechnol. 2015;33:1159–61.
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–8.
Shen B, Zhang W, Zhang J, et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods. 2014;11:399–402.
Fujii W, Onuma A, Sugiura K, Naito K. Efficient generation of genome-modified mice via offset-nicking by CRISPR/Cas system. Biochem Biophys Res Commun. 2014;445:791–4.
Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–9.
Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32:577–82.
Farzadfard F, Perli SD, Lu TK. Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth Biol. 2013;2:604–13.
Perez-Pinera P, Kocak DD, Vockley CM, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10:973–6.
Gilbert LA, Larson MH, Morsut L, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–51.
Lombardo A, Genovese P, Beausejour CM, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–306.
Genovese P, Schiroli G, Escobar G, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–40.
Goessling W, Allen RS, Guan X, et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell. 2011;8:445–58.
North TE, Goessling W, Walkley CR, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447:1007–11.
Boitano AE, Wang J, Romeo R, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–8.
Hoggatt J, Singh P, Sampath J, Pelus LM. Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood. 2009;113:5444–55.
Wang J, Exline CM, DeClercq JJ, et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol. 2015;33:1256–63.
Sather BD, Romano Ibarra GS, Sommer K, et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med. 2015;7:307ra156.
Wang J, DeClercq JJ, Hayward SB, et al. Highly efficient homology-driven genome editing in human T cells by combining zinc-finger nuclease mRNA and AAV6 donor delivery. Nucleic Acids Res. 2016;44:e30.
Tebas P, Stein D, Tang WW, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–10.
Didigu CA, Wilen CB, Wang J, et al. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood. 2014;123:61–9.
Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17.
Provasi E, Genovese P, Lombardo A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012;18:807–15.
Torikai H, Reik A, Liu PQ, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–705.
Berdien B, Mock U, Atanackovic D, Fehse B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. 2014;21:539–48.
Alinari L, Lapalombella R, Andritsos L, Baiocchi RA, Lin TS, Byrd JC. Alemtuzumab (Campath-1H) in the treatment of chronic lymphocytic leukemia. Oncogene. 2007;26:3644–53.
Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol. 2015;33:179–86.
Tsai SQ, Zheng Z, Nguyen NT, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33:187–97.
Mussolino C, Alzubi J, Fine EJ, et al. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014;42:6762–73.
Cho SW, Kim S, Kim Y, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–41.
Gabriel R, Lombardo A, Arens A, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29:816–23.
Qasim W, Amrolia PJ, Samarasinghe S, Ghorashian S, Zhan H, Stafford S, Butler K, Ahsan G, Gilmour K, Adams S, Pinner D, Chiesa R, Chatters S, Swift S, Goulden N, Peggs K, Thrasher AJ, Veys P, Pule M. First clinical application of Talen engineered universal CAR19 T cells in B-ALL. Blood. 2015;126:2046.
Hofmeister CC, Zhang J, Knight KL, Le P, Stiff PJ. Ex vivo expansion of umbilical cord blood stem cells for transplantation: growing knowledge from the hematopoietic niche. Bone Marrow Transpl. 2007;39:11–23.
Medvinsky A, Rybtsov S, Taoudi S. Embryonic origin of the adult hematopoietic system: advances and questions. Development. 2011;138:1017–31.
Tavian M, Biasch K, Sinka L, Vallet J, Peault B. Embryonic origin of human hematopoiesis. Int J Dev Biol. 2010;54:1061–5.
Arora N, Wenzel PL, McKinney-Freeman SL, et al. Effect of developmental stage of HSC and recipient on transplant outcomes. Dev Cell. 2014;29:621–8.
Vo LT, Daley GQ. De novo generation of HSCs from somatic and pluripotent stem cell sources. Blood. 2015;125:2641–8.
Li H, Haurigot V, Doyon Y, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–21.
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72.
Moreau T, Colzani M, Arumugam M, et al. In vitro production of megakaryocytes and platelets from human induced pluripotent cells by GMP compatible methods. Blood. 2013;122:2401.
Themeli M, Kloss CC, Ciriello G, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. 2013;31:928–33.
Lee DW, Barrett DM, Mackall C, Orentas R, Grupp SA. The future is now: chimeric antigen receptors as new targeted therapies for childhood cancer. Clin Cancer Res. 18:2780–2790.
Maude SL, Shpall EJ, Grupp SA. Chimeric antigen receptor T-cell therapy for ALL. Hematol Am Soc Hematol Educ Progr. 2014;2014:559–64.
Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–84.
Lugli E, Dominguez MH, Gattinoni L, et al. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest. 2013;123:594–9.
Giani FC, Fiorini C, Wakabayashi A, et al. Targeted application of human genetic variation can improve red blood cell production from stem cells. Cell Stem Cell. 2016;18:73–8.
Osborn MJ, Gabriel R, Webber BR, et al. Fanconi anemia gene editing by the CRISPR/Cas9 system. Hum Gene Ther. 2015;26:114–26.
Rio P, Banos R, Lombardo A, et al. Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol Med. 2014;6:835–48.
Muller LU, Milsom MD, Harris CE, et al. Overcoming reprogramming resistance of Fanconi anemia cells. Blood. 2012;119:5449–57.
Keller G, Kennedy M, Papayannopoulou T, Wiles MV. Hematopoietic commitment during embryonic stem cell differentiation in culture. Mol Cell Biol. 1993;13:473–86.
Amabile G, Welner RS, Nombela-Arrieta C, et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood. 2013;121:1255–64.
Suzuki N, Yamazaki S, Yamaguchi T, et al. Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol Ther. 2013;21:1424–31.
Sandler VM, Lis R, Liu Y, et al. Reprogramming human endothelial cells to haematopoietic cells requires vascular induction. Nature. 2014;511:312–8.
Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6.
Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7.
Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–7.
Canver MC, Smith EC, Sher F, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–7.
Hilton IB, D’Ippolito AM, Vockley CM, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–7.
Acknowledgements
MJO and JT appreciate funding support from The Children’s Cancer Research Fund, the Epidermolysis Bullosa Research Partnership, Epidermolysis Bullosa Medical Research Fund, Sohana Research Fund, Richard M Schulze Family Foundation, DebRA, the Lindahl Family, and the Corrigan Family. JT is supported in part by R01 AR063070, R01 AR059947, DOD W81XWH-12-1-0609, and P01 CA065493. MJO is supported by 8UL1TR000114-02. JB is supported by a grant from the National Science Foundation to DV (IOS-1339209). Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health Award Number UL1TR000114 (MJO). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Osborn, M.J., Belanto, J.J., Tolar, J. et al. Gene editing and its application for hematological diseases. Int J Hematol 104, 18–28 (2016). https://doi.org/10.1007/s12185-016-2017-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-016-2017-z