
Abstract
Since the publication of the JUPITER trial, attention has been focused on the adverse glycemic effects of statin therapy. Although the modest increase in the risk of new diabetes mellitus is outweighed by the reduction in cardiovascular events for statins, emerging biochemical and genetic links between lipid metabolism and glycemic control raise the prospect of a broader diabetogenic effect of lipid-lowering therapies. For the novel and powerful PCSK9-inhibitor class available evidence does not support a major glycaemic effect with the results of large scale trials awaited although preliminary genetic data does suggest a link. In contrast, there is clear evidence of a diabetogenic effect for the now outdated but well-studied niacin. For ezetimibe and fibrates, evidence is scarce but currently broadly unconcerning. For now, the glycemic effects of lipid-lowering therapies should have a limited influence on clinical decision-making. Further study in this topical area is needed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypercholesterolaemia is one of the most important modifiable risk factors for cardiovascular disease. Efficacious for primary prevention, even in individuals with only modest cardiovascular risk, statins are some of the most widely used medicines worldwide. Despite this, a very small proportion of individuals do not sufficiently respond or are unable to tolerate these drugs. Although the risks of liver derangement, myositis, and even rhabdomyolysis, have long been recognised, it was not until the publication of the 2008 randomised JUPITER trial that focus turned to modest but significant increases in the new development of diabetes mellitus [1]. Individuals receiving maximal doses of rosuvastatin had a 28 % excess rate of diabetes (mainly confined to those with preexisting risk factors) compared to the placebo arm. Subsequent randomised trial meta-analyses showed a 9–11 % increased risk of diabetes amongst statin users [2 3•] with evidence of a higher rates amongst those on intensive statin therapy [4].
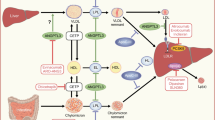
In vitro and population studies have revealed possible mechanistic explanations. Firstly, ‘off-target’ effects potentiating insulin resistance (inhibition of GLUT4 translocation to the cell membrane and of adipocyte differentiation) or decreased insulin secretion (inhibiting calcium uptake into pancreatic beta cells and induction of beta cell apoptosis) have been proposed [5]. It is suggested that these effects are concentrated amongst the lipophilic statins—which may account for the reduced risk of new-onset diabetes originally seen amongst users of the hydrophilic pravastatin in the pivotal West of Scotland Coronary Prevention Study [6]. However, this line of argument remains speculative and diabetes risk was clearly elevated in the pravastatin arm of the PROSPER trial [7]. Secondly, there appear to be downstream ‘on-target’ effects of HMG-CoA reductase inhibition which alter insulin metabolism and weight and parallel clinical evidence that HMG-CoA reductase inhibition leads to minor weight gain amongst those who take statins [4]. In other words, it appears that at least part of the diabetogenic potential of statins is mediated via weight change although the precise mechanisms are yet to be elucidated but speculatively could include a a very small effect on exercise capacity sufficient to tip the balance towards very minor weight gain in some.
Finally, and perhaps most interestingly, there is emerging evidence of an association between alleles influencing lipid metabolism and the risk of diabetes mellitus. At one extreme, individuals with familial hypercholesterolaemia have a lower incidence of diabetes mellitus compared to unaffected family members—despite early and aggressive treatment with statins [8]. Broader mendelian randomisation studies (the use of genetic polymorphisms to help dissect causality) including tens of thousands of individuals have suggested an inverse relationship between single nucleotide polymorphisms (SNPs) predisposing to elevated LDL cholesterol and the risk of developing diabetes [9•], with particular alleles for the biological target of statin therapy implicated [4]. Whilst a number of potential confounding factors prevent firm conclusions (including survivor bias, pleiotropy, the greater use of statin therapy amongst individuals with high LDL), the foregoing observations raise the possibility of an inherent diabetogenic effect of lipid lowering therapies, especially LDL cholesterol-lowering therapies.
In this review, we summarise the evidence regarding the glycemic effects of non-statin lipid-lowering therapies, with results summarised (Table 1).
PCSK9 Inhibitors
The PCSK9 inhibitors (including alirocumab, evolocumab and bococizumab, although the latter now being withdrawn) are an emerging drug class of monoclonal antibodies against PCSK9—a binding protein potentiating the degradation of hepatocyte LDL-R, which can be delivered subcutaneously at monthly or bimonthly intervals and offer potent reductions in LDL cholesterol (up to 61 %). Colhoun et al. examined the influence of alirocumab on transition to diabetes amongst 10 randomised control trials (n = 3448) with a high proportion of prediabetic subjects (39.6 %). They found no clear evidence of increased development of diabetes compared to placebo (HR 0.64 (0.36–1.14)) or ezetimibe (HR 0.55 (0.22–1.41)) [10••]. Furthermore, there was no significant change in HbA1c over time. However, the follow-up period was relatively short (6–18 months), and the total number of new cases of diabetes was limited (n = 218). Similarly, there was no significant glycemic effect of monthly evolocumab over a 52-week period in a randomised comparison with placebo (n = 901) [11] nor any glycemic effect amongst participants in a randomised trial against standard care who had preexisting dysglycaemia or metabolic syndrome over 52 weeks [12]. In a recent meta-analysis, we did not find any significant difference in lipid effects of evolocumab on diabetic patients versus controls [13]. Further work on the glycaemic effects of evolocumab is currently ongoing.
Although the data for PCSK9 inhibitors is thus far reassuring, a more subtle effect cannot be excluded and it is notable that widespread acceptance of the link between statins and diabetes came only after large scale meta-analysis including thousands of new cases of diabetes. Furthermore, most trials have examined the addition of PCSK9 inhibitors to statins which may partially mask a diabetogenic effect if mechanisms are shared between classes. Further work in this area is proceeding rapidly especially regarding SNPs related to PCSK9 function and glycaemic control as well as to weight changes. Such work requires the integration of results from multiple large studies to yield sufficient power and is now readily achievable in an era of both major genetic studies and distributed information technology. Indeed, a recent relevant study exploiting genetic data has examined this issue and reported genetic variants in PCSK9 were associated with a 19 % (95 % CI 2 to 38 %) higher risk for diabetes per mmol/l lower LDL-c levels [14••]. This is the first large genetic study linking PCSK9 snps to diabetes risk and reports a significant higher risk, albeit modestly so, but with stronger associations with diabetes for genes encoding the molecular targets for ezetimibe. Nevertheless, the results of ongoing cardiovascular end-point trials are needed to determine the true effect of short term PCSK9 inhibition on diabetes risks. Fortunately, we will not have to wait too long for these to report and all have pre-specified assessment of diabetes risk and HbA1c from the outset.
One final comment deserves mention. Since these drugs are currently being targeted to those at very high risk of cardiovascular events—i.e. patients with either familial hypercholesterolaemia or preexisting cardiovascular disease with uncontrolled LDL cholesterol despite maximally tolerated statin and/or ezetimibe therapy—it is unlikely that a modest diabetogenic effect would alter treatment decisions [15•].
Niacin
The B vitamin niacin initially showed promise, with a reduction in 17 % reduction in MI seen in results from the Coronary Drug Project [16], but despite beneficial effects on serum LDL no clear benefit in cardiovascular events or mortality was seen in addition to statin therapy [17, 18]. As well as the well-recognised flushing effects of niacin, one notable side effect is a worsening of glycemic control. Although early work concluded this effect was modest and unlikely to lead to the onset of diabetes [19], post hoc analysis, new trials and a well conducted meta-analysis of both published and unpublished work are concordant in suggesting a significant effect. Over a 5-year follow-up, in the Coronary Drug Project’s randomised comparison of niacin to placebo, patients with impaired fasting glucose tolerance at baseline progressed to diabetes at a rate of 19.8 % on niacin versus 15.2 % on placebo (HR 1.34 (1.00–1.80)) with a similar (although non-significant) relative risk increase amongst those with normal glucose tolerance (HR 1.41 (0.97, 2.05)) [20]. In the HPS2-THRIVE comparison of the addition of niacin or placebo to those already on statins there was a 55 % increase in serious disturbances in glycemic control (for which many patients were hospitalised), affecting 11.2 % of diabetics assigned to niacin [7]. Furthermore, there was a 30 % increase in the risk of new-onset diabetes in the treatment group. Goldie et al. performed a rigorous meta-analysis on the effect of niacin on the risk of new-onset diabetes [21•]. Overall, they found a 34 % increased risk of new diabetes associated with the use of niacin (although this may be a slight overestimate as trials with no new cases of diabetes were excluded). Niacin is no longer in widespread clinical use but these results do add to concerns of an implicit diabetogenic effect of lipid-lowering therapies.
Ezetimibe
Ezetimibe inhibits enterocyte absorption and increases hepatocyte uptake of cholesterol and currently is the mainstay of therapy in individuals who do not tolerate statins or require further LDL cholesterol reductions. Added to statin therapy, it offers both a 15 % absolute reduction in LDL cholesterol [22] and a modest reduction in cardiovascular events over a 7-year post-acute coronary syndrome ((HR 0.94 (0.89, 0.99)) [23], but data regarding its glycemic effects are rather limited. Saito et al. randomised diabetics to ezetimibe or placebo and did not find any significant effect on glycemic control (assessed by either HbA1c or fasting plasma glucose) or exacerbations of diabetes over a short follow-up period of 24 weeks [24]. However, 9 % of diabetics in the ezetimibe group had their drug regime altered due to hyperglycaemia versus 4 % of the placebo group. Although the trial was well conducted, its very limited sample size (n = 152), single nationality population (Japanese) and exclusion of those with poor diabetic control, preexisting cardiovascular disease or concurrent statin use means wider interpretation of its results is fraught. In the IMPROVE-IT randomised comparison of additional ezetimibe or placebo for subjects on statins with recent acute coronary syndrome a preliminary report, not yet peer-reviewed, suggests ezetimibe resulted in a small, non-statistically significant, increase in the relative risk of new-onset diabetes (9 % per 1 mmol/L reduction in LDL cholesterol) [25•]. These clinical data should be contrasted with recent genetic data [14••] which suggests a much larger effect on diabetes risk of lifelong inhibition of the genes encoding the molecular targets of ezetimibe: a 2.42 (95 % CI 1.70 to 3.43) odds ratio per mmol/l lower LDL-c. Therefore, longer term data on diabetes risks with ezetimibe treatment are needed to rule out a diabetes risk.
Fibrates
Fibrates interact with peroxisome proliferator-activated receptors to reduce plasma LDL cholesterol and triglycerides. Although they reduce the rate of cardiovascular events, they do not affect all-cause mortality or rates of cardiovascular death [26]. Overall there is very limited data on the interaction between fibrates and glycemic control. The FIELD study randomised diabetic subjects not on any statin therapy at baseline (n = 9795) to either fenofibrate or placebo. Over 6 years, the change in HbA1c was negligible regardless of randomisation but formal statistical testing was not reported [27]. Lee et al. examined the effects of fibrates on the rate of new-onset diabetes amongst Taiwanese patients aged 55–59 with dyslipidaemia (n = 3815) over a 5-year period [28]. Although the overall rate of new-onset diabetes was unconcerning (3.8 %), the number of new diabetes cases was limited (n = 145) and the lack of a comparison group means no firm conclusions can be drawn. Tennabaum et al. found a reduced rate of diabetes over a 6-year follow-up amongst subjects with impaired fasting glucose and known coronary artery disease assigned to benzafibrate rather than placebo [29]. There are also mixed data on the association of PPAR-alpha SNPs and diabetes risk, and thus, overall, no clear conclusions can yet be drawn [30]. Newer classes of fibrate drugs are currently being tested with respect to cardiovascular outcome risks and it is likely that such studies will also include measures of glycaemia as pre-specified outcomes.
Conclusions
It is striking that both genetic studies and large scale meta-analysis for statins and niacin (the two lipid-lowering therapies for which there is a reasonable body of data) point towards an inverse link between LDL cholesterol and glycemic control, although niacin also influences HDL-cholesterol levels which may have separate links to diabetes. However, it is not yet clear that all lipid-lowering therapies are implicitly diabetogenic, or that all statins are equally diabetogenic [31]).
There are a number of gaps in the literature. Further studies delineating the mechanisms by which statins cause diabetes are clearly needed. Both ezetimibe and PCSK9 inhibitors lack sufficient clinical trial data to exclude a diabetogenic effect, although, as noted, large scale trials of PCSK9 inhibitors are underway, and will also examine diabetes outcomes, important given emerging evidence of a link between PCSK9 genetic variants and diabetes risk.
For PCSK9 inhibitors, any diabetogenic effect would have to be very significant to further limit their use beyond individuals at high risk of cardiovascular events without other proven options. For statins, the benefits clearly outweigh the risks, even for primary prevention. Potential diabetogenic effects can be easily monitored, managed and mitigated by lifestyle changes. Indeed, a positive consequence of this area is that more patients at risk of, or with existing cardiovascular disease, are screened for diabetes, receive preventative advice and have earlier diagnosis and treatment.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ridker PM, Danielson E, Fonseca FAH, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. NEJM. 2008;359:2195–207.
Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–42.
•Swerdlow DI, Preiss D, Kuchenbaecker KB, et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet. 2015;385:351–61. This paper demonstrates the power to link genetics and clinical trial data to improve causal understanding of the effects of drug therapy on adverse effects; in this case, examining the diabetogenic effects of statins.
Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA. 2011;305:2556–64.
Ostrowska M, Adamski P, Kosinski M, et al. Diabetogenic effect of statins: a comprehensive review on the clinical relevance, underlying pathomechanisms and rationale for tailored statin therapy. Folia Med Copernicana. 2015;3:145–53.
Ford I, Murray H, Packard C, et al. Long-term follow-up of the West of Scotland Coronary Prevention Study. NEJM. 2007;357:1477–86.
Shepherd J, Blauw GJ, Murphy MB, PROSPER Study Group, et al. PROspective Study of Pravastatin in the Elderly at Risk. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623–30.
Besseling J, Kastelein JJP, Defesche JC, et al. Association between familial hypercholesterolaemia and prevalence of type 2 diabetes mellitus. JAMA. 2015;313:1029–36.
•Fall T, Xie W, Poon W, et al. Using Genetic variants to assess the relationship between circulating lipids and type 2 diabetes. Diabetes. 2015;64:2676–84. Demonstrates the power of genetics to improve understanding of complex biological links, but also the potential caveats one must consider.
Coulhoun HM, Ginsberg HN, Robinson JG et al.: No effect of PCSK9 inhibitor alirocumab on the incidence of diabetes in a pooled analysis from 10 ODYSSEY phase 3 studies. European Heart Journal. 2016; ehw292 [Epub ahead of print]. The first study to examine whether there is any obvious and immediate effect of PCSK9 inhibition (alirocumab in this case) on diabetes risk and glycaemia levels.
Blom DJ, Hala T, Bolognese M, et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidaemia. NEJM. 2014;370:1809–19.
Henry RR, Holman RR, Giugliano RP, et al. Effects of long-term, monthly administration of the PCSK9 inhibitor evolocumab in patients with dysglycemia or metabolic syndrome. Can J Diabetes. 2014;38:S18.
Sattar N, Preiss D, Robinson JG, et al. Lipid-lowering efficacy of the PCSK9 inhibitor evolocumab (AMG 145) in patients with type 2 diabetes: a meta-analysis of individual patient data. Lancet Diabetes Endocrinol. 2016;4:403–10.
••Lotta LA, Sharp SJ, Burgess S, et al. Association Between low-density lipoprotein cholesterol-lowering genetic variants and risk of type 2 diabetes: A meta-analysis. JAMA. 2016;4;316(13):1383-1391. This is the first large genetic study of PCSK9 snps and diabetes risk and shows a significant higher risk, albeit modestly so, but with stronger associations with diabetes for genes encoding the molecular targets for ezetimibe.
•Sattar N. PCSK9 inhibitors and diabetes risk: a question worth asking? Eur Heart J. 2016. doi:10.1093/eurheartj/ehw346. This commentary pulls together the existing evidence for statins and PCSK9 and risks of diabetes and gives clinical implications as well as important areas for future research.
Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in coronary drug project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8:1245–55.
HPS2-THRIVE collaborative group, Landray MJ, Haynes R, et al. Effects of extended-release niacin with laropiprant in high-risk patients. NEJM. 2014;371:203–12.
AIM-HIGH investivators, Boden WE, Probstfield JL, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. NEJM. 2011;365:2255–67.
Goldberg RB, Jacobson TA. Effects of niacin on glucose control in patients with dyslipidemia. Mayo Clin Proc. 2008;83:470–80.
Sazonov V, Maccubbin D, Sisk CM, et al. Effects of niacin on the incidence of new onset diabetes and cardiovascular events in patients with normoglycaemia and impaired fasting glucose. Int J Clin Pract. 2013;67:297–302.
•Goldie C, Taylor AJ, Nguyen P, et al. Niacin therapy and the risk of new-onset diabetes: a meta-aaalysis of randomised controlled trials. Heart. 2016;102:198–203. A well-conducted meta-analysis of trials of Niacin and its risk for diabetes.
Morrone D, Weintraub WS, Toth PP, et al. Lipid-altering efficacy of ezetimibe plus statin and statin monotherapy and identification of factors associated with treatment response: a pooled analysis of over 21,000 subjects from 27 clinical trials. Atherosclerosis. 2012;223:251–61.
Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe added to statin therapy after acute coronary syndromes. NEJM. 2015;372:2387–97.
Saito I, Azuma K, Kakikawa T, et al. A randomized, double-blind, placebo-controlled study of the effect of ezetimibe on glucose metabolism in subjects with type 2 diabetes mellitus and hypercholesterolaemia. Lipids Health Dis. 2015;14:40.
•Blazing MA, on behalf of the IMPROVE-IT Investigators. Incidence of new onset diabetes in the IMPROVE-IT trial: does adding ezetimibe to simvastatin increase risk compared to simvastatin alone? London: Presented at: European Society of Cardiology Congress; 2015. An early examination of the risks of diabetes with ezetimibe.
Jun M, Foote C, Licheng LV, et al. Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet. 2010;375:1875–84.
Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–61.
Lee CY, Huang KH, Lin CC, et al. A neutral risk on the development of new-onset diabetes mellitus (NODM) in Taiwanese patients with dyslipidaemia treated with fibrates. Scientific World Journal. 2012;2012:392734.
Tenenbaum A, Motro M, Fisman EZ, et al. Peroxisome proliferator-activated receptor ligand bezafibrate for prevention of type 2 diabetes mellitus in patients with coronary artery disease. Circulation. 2004;109:2197–202.
Silbernagel G, Stefan N, Hoffmann MM, et al. The L162V polymorphism of the peroxysome proliferator activated receptor alpha gene (PPARA) is not asocialted with type 2 diabetes, BMI or body fat composition. Exp Clin Endocrine Diabetes. 2009;117:113–8.
Baker WL, Talati R, White CM, et al. Differing effect of statins on insulin sensitivity in non-diabetics: a systematic review and meta-analysis. Diabetes Res Clin Pract. 2010;87:98–107.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
Patrick D. Collins declares that he has no conflict of interest. Naveed Sattar has consulted for Amgen, Sanofi, Boehringer Ingelheim, Eli Lilly, Novo Nordisk, Janssen, and Merck, and received research funding from AstraZeneca.
Human and Animal Rights and Informed Consent
All procedures performed in studies involving human participants by the authors were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Additional information
This article is part of the Topical Collection on Diabetes and Cardiovascular Disease
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
