
Abstract
Ecological conditions shape natural distribution of plants. Populations are denser in optimal habitats but become more fragmented in the areas of suboptimal environmental conditions. Usually, fragmentation increases towards the limits of species distribution. Fragmented populations are often characterised by decreased genetic variation, and this effect is frequent in peripheral populations, mostly due to the reduced effective population size. Interestingly, the genetic consequences of fragmentation seem to be relatively weak in forest trees. Using microsatellite markers, we assessed the impact of population fragmentation on the genetic structure of a European tree species Acer campestre. Within the study area, this medium-size wind-dispersed and insect-pollinated tree reveals a gradual decrease in population density towards the northern range limit. Over the distance of 150 km, we detected the significant decrease in allelic richness, heterozygosity as well as an increase in the rate of population divergence along with latitude. On the other hand, we failed to show that the observed patterns of genetic structure result from the variation in population densities. Moreover, inbreeding levels revealed no association with both density and geographic location, suggesting that pollen limitation does not occur, even at the range margin. As we showed that there is no difference in a dispersal scale between low- and high-density populations in the study species, we argue that the genetic structure is a result of postglacial recolonization. However, unlike many other forest trees, A. campestre showed the sharp latitudinal genetic pattern at a very restricted spatial scale. Limited dispersal and high fragmentation are likely the reasons.
Similar content being viewed by others
Introduction
Besides the climate, one of the most critical factors of the current distribution of plants is historical and current landscape use by humans. Exploitation of natural resources often causes habitat fragmentation, limiting plant distribution and increasing the risk of local extinction. Along with human impact, ecological conditions shape natural distribution of plants (Brown 1984; Austin 2007). Generally, populations are denser in ecologically suitable habitats and become more fragmented towards ecological margins. In fact, ecological conditions often co-vary with geographic location (Holt & Keitt 2000; Eckert et al. 2008), leading to geographic patterns of species abundance and diversity (Martin & McKay 2004; Eo et al. 2008; Kunin et al. 2009; Guo 2012).
Population fragmentation often leads to reduced effective population size (Young et al. 1996; Vucetich & Waite 2003). In effect, fragmented populations are generally characterised by decreased genetic variation (Leimu et al. 2006). In particular, it is well recognised in peripheral populations as compared with central ones (the ‘central-marginal’ genetic pattern) (reviewed in Eckert et al. 2008; but see, e.g. Munwes et al. 2010). According to the theoretical predictions, less genetically diverse populations are often characterised by lower viability (Frankham 2003; Reed and Frankham 2003; Aguilar et al. 2008) and/or adaptability (Young et al. 1996, Willi et al. 2006). However, the risk of negative consequences of fragmentation is related primarily to the dispersal capability (Thomas 2000). Dispersal is a means for both colonisation of new habitats and gene exchange among populations (Howe and Smallwood 1982; Travis and Dytham 1998). Therefore, dispersal influences rates of extinction and recolonisation, as well as genetic drift within existing populations. Additionally, rates of dispersal determine mating patterns and spatial genetic structure of a population. Limited dispersal accompanied by low density of pollen donors can lead to pollen limitation syndrome, which often drives inbreeding through increased self-fertilisation (in self-compatible species) or mating between relatives (Kalisz et al. 1999; Rajora et al. 2002; Johnson et al. 2009). In consequence, a decreased heterozygosity (due to inbreeding) facilitates expression of deleterious genes, having negative impact on the fitness and, consequently, increasing extinction risk (Newman and Pilson 1997; Herlihy and Eckert 2002; Reed and Frankham 2003; Vilas et al. 2006; Gargano et al. 2009). Thus, it is expected that the species characterised by low dispersal capabilities are more prone to negative consequences of fragmentation (White et al. 2002; Jump and Penuelas 2006; O'Connell et al. 2007; Montoya et al. 2008) and, they should also exhibit more apparent central-marginal patterns.
Interestingly, the genetic consequences of fragmentation seem to be generally weak in forest trees (Kramer et al. 2008), although more recent reviews indicated that the effect size may be related with the pollination mechanism (Vranckx et al. 2011). However, forest trees are often characterised by intensive gene flow and strong human impact through the forest management (including translocation of forest reproductive material), which make studies of the effects of population fragmentation upon the distribution of genetic diversity difficult. Therefore, more data are needed to evaluate the actual susceptibility of this group of species to fragmentation.
European tree Acer campestre L. (field maple) can serve as a valuable example of tree species living at different levels of population fragmentation. The natural species distribution covers most of Europe, excluding northern parts. Across its natural range, field maple does not form pure stands, but instead it is often a subdominant species in many plant communities. Given its low commercial importance, field maple does not experience a silvicultural treatment and often grows in spontaneously established, semi-natural populations. In Poland, the distribution of A. campestre reaches a north-eastern limit, with the apparent latitudinal reduction of species abundance within several hundred kilometres. This makes excellent opportunity to study the impact of natural population fragmentation on the genetic structure of a tree species over a relatively short distance.
So far, Acer species were found to reveal high genetic variation (Rusanen et al. 2003; Beletti et al. 2007; Pandey et al. 2012) and a local spatial genetic structuring, mostly due to limited seed dispersal (Geburek 1993; Young and Merriam 1994; Pandey et al. 2012). On the contrary, at metapopulation scale, spatial genetic structure is not so evident (Young et al. 1993; Rusanen et al. 2000; Beletti et al. 2007; but see Guarino and Cipriani 2013), suggesting weak barriers to gene flow between populations. As compared with the other Acer species, however, population genetic data on field maple are extremely scarce. Two instances include the phylogenetic study on European Acer species (including A. campestre; Guarino et al. 2008) and the unpublished dissertation on reproduction system of A. campestre (Bendixen 2001). However, no systematic data exist on genetic variation and population genetic structure. Because field maple exhibits natural fragmentation increasing northwards (i.e. towards the natural limit of the distribution) in the study area, our goal was to verify whether genetic variation (as a result of changes in effective population size) and inbreeding (as a result of changes in pollen availability) co-varies with the species’ density. Additionally, because the decrease in species density overlaps with the direction of historical recolonisation wave after the last glacial period, we tested whether fragmentation had an effect on the scale of gene dispersal, and thus whether contemporary dispersal rather than historical colonisation was a true factor of the observed genetic structure.
Materials and methods
Study species
Field maple is a medium-size European tree, typically reaching 15 m tall (exceptionally 25 m) and 60–70 cm in trunk diameter. The species is fairly resistant to drought and often exists in xerothermic communities. Nonetheless, it is very often found in wet-ground forests. Thus, moisture or precipitation is not the main factor limiting the natural distribution. On the other hand, field maple seems to be quite susceptible to frost, especially at the beginning of a vegetative season. In Poland, the frequency of late frosts reveals sharp latitudinal gradient (Woś 1999), having potentially impact on the distribution of the species. As a shade-tolerant middle-size tree, field maple is typically present under canopy of forest stand. It produces hermaphrodite flowers, but usually individuals show complex temporal patterns of sex expression during a flowering season (De Jong 1976). Consequently, field maple is pre-dominantly allogamous, although individual trees can be capable of self-fertilisation (Bendixen 2001). Interestingly, pollen of A. campestre is less sticky as compared with other European Acer species (Hesse 1979). Hence, typically entomophilous A. campestre is supposedly capable of dispersing some portion of pollen by wind. Winged seeds (samaras) are dispersed by wind in autumn. In forests, seed dispersal is restricted mostly to the neighbourhood of a tree. On the other hand, in open areas samaras can be dispersed over larger distances, allowing for occasional colonisation of new habitats and gene flow between populations.
Study populations and sampling
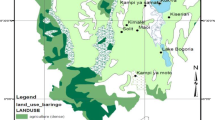
In order to minimise the risk of human impact, all study sites were located within nature reserves. After careful inspection within two neighbouring provinces (voivodeships), 13 representative nature reserves were chosen (Table 1). Generally, the selected sites represent wet-ground forests such as riparian forests (mostly in the north) and hornbeam-oak forests. According to the species’ abundance, localities were divided into the northern and the southern metapopulation (Table 1; Fig. 1). The northern metapopulation was composed of marginal, highly fragmented populations. Conversely, the southern metapopulation represented a region of high species abundance and low fragmentation. For each locality, bioclimatic data were extracted from WorldClim database (Hijmans et al. 2005) using DIVA GIS 7.5 software (Hijmans et al. 2001). Out of 19 climatic variables in total, we retained only annual mean temperature (MAT) and total annual precipitation (TAP), as the two basic components of the climate. Both MAT and TAP were significantly correlated with latitude (p value <0.001 and 0.032, respectively; based on the product moment correlation coefficient) but revealed no association with longitude (p value 0.462 and 0.201, respectively). In total, 431 samples (a single leaf per individual) were taken. After transportation to the lab, leaves were left to dry out at room temperature and then grinded into powder for DNA extraction.

Distribution of the study species in Poland and location of the study sites. Small circles show the distribution of Acer campestre in Poland in the form of the presence-absence data (Zając and Zając 2001). The heat map shows the relative abundance of Acer campestre in Poland estimated using kernel density estimation based on the presence-absence data. The line shows how the high- (southern) and low-density (northern) metaopopulations were delimited
Molecular methods
Total genomic DNA was extracted using GeneMatrix Plant and Fungi DNA Purification Kit (EURx, Gdańsk, Poland), according to the manufacturer's protocol. Genetic variation was investigated based on nuclear (nSSR) microsatellite loci. We initially tested 16 primers developed for European Acer species (MAP series developed by Pandey et al. 2004, Aop series developed by Segarra-Moraguez et al. 2008). Although all 16 primers gave positive amplification results, unequivocal interpretation of PCR products was possible for seven markers only. Interestingly, MAP40 showed two independent amplification zones, with one of them being highly polymorphic while the second one being monomorphic. Using the Multiplex PCR Kit (QIAGEN) two PCR reactions were designed in order to amplify four (MAP46, MAP9, Aop132 and Aop943) and two (Aop116 and Aop 450) markers in a single reaction. In addition, MAP40 was amplified in a separate PCR reaction. PCR programme started with initial denaturation at 95° for 5 min. It was followed by nine touch-down cycles (94° for 30 s, 56° (−1°/cycle) for 90 s and 72° for 1 min) and 25 regular cycles (94° for 30 s, 48° for 90 s and 72° for 1 min). Finally, samples were kept at 72° for 10 min in order to finalise reactions. PCR products were sized using the automatic sequencer ABI3130XL (Applied Biosystems/Hitachi).
Data analysis
Species distribution, local fragmentation intensity and effective population size
Based on the presence-absence data reflecting the distribution of the species in Poland (Fig. 1; based on Zając and Zając 2001), the kernel density estimation was used to obtain a generalised density map of the species′ distribution in Poland (Fortin et al. 2005). For this purpose, we used the function ‘heatmap’ available in QGIS 2.2 (http://qgis.org), setting the kernel function to Epanechnikov and choosing the radius of 40 km. Then, for the ith population, we extracted the point density value ρ i using point sampling tool plug-in (http://hub.qgis.org/projects/pointsamplingtool) working under QGIS. Although the value of ρ i estimated in that way did not represent a density expressed in biologically meaningful units (e.g. populations/ha), it was used as a surrogate of (relative) local fragmentation of a metapopulation. The two-sample t test was applied for testing the difference in the mean density ρ between the two metapopulations, with p value determined by permutation.
For each population, the effective population size N i was estimated using the linkage disequilibrium-based approach (ldne computer programme; Waples and Do 2008). Because the method is sensitive to low-frequency alleles, we used 0.02 as a minimum threshold for allele frequencies. In order to compare the rate of genetic drift at a local level between the two metapopulations, the average of N i was computed for each metapopulation. For this purpose, we used the harmonic mean of the estimates for individual populations because N i estimates are in fact inverses of the true estimates (coefficients of linkage disequilibrium, r 2). Based on the inverses of N i values, the two-sample t test was used to test the difference in the mean N between the two metapopulations.
Genetic variation and inbreeding
Genetic variation was investigated using standard genetic indices calculated per locus/population, including a number of alleles (A), allelic richness (AR; estimated using rarefaction), observed and expected heterozygosity (Ho and He, respectively). To describe the overall genetic variation Nei's statistics (expected within-population heterozygosity (Hs) and total heterozygosity (Ht), Gst = 1 – Hs/Ht) were computed using FSTAT (Goudet 1995). Inbreeding coefficients F IS were estimated using the Bayesian procedure implemented in INEST 2.0 software (using the individual inbreeding model; Chybicki & Burczyk 2009), which is robust to the presence of null alleles. Posterior distribution was approximated based on 100,000 MCMC samples (disregarding first 10,000 samples). In order to assess the statistical significance of inbreeding we compared the full model with the random mating model (i.e. when F IS is fixed at 0) using the Bayesian procedure based on the Deviance Information Criterion (DIC; cf. Chybicki et al. 2011).
Genetic structuring
Using hierarchical analysis of molecular variance (AMOVA), we quantified the partitioning of genetic variance within and among the hierarchal levels (populations within the two metapopulations, metapopulations and total). The analysis was performed in Arlequin 3.5.1.3 (Excoffier and Lischer 2010).
Using STRUCTURE 2.3.4 software (Pritchard et al. 2000; Falush et al. 2003, 2007), we tested whether any discrete genetic structure exists among populations. Genotypes were clustered assuming correlated allele frequencies and admixture. Because null alleles were likely to be present at some loci (see ‘Results’), the ‘Recessive Alleles’ model was used. The posterior distribution was approximated with 1,000,000 samples (but first 500,000 samples were disregarded for burn-in). The analysis was performed assuming the number of clusters K = 1 to 26, with 25 repetitions per each K. Then, the optimum K was determined based on the Delta K approach (Evanno et al. 2005) using STRUCTURE HARVESTER Web application (Earl & vonHoldt 2012). Given that the optimum K > 1 (see ‘Results’), for each population admixture level was assessed using the Gini-Simpson index computed as \( {D}_j=1-{\displaystyle \sum_{k=1}^K}{\pi}_{jk}^2 \) (Simpson 1949), where π jk denotes the probability that a random individual in the jth population is assigned to the kth genetic cluster (out of K). In other words, D j equals the probability that two random individuals in the jth population are assigned to different genetic clusters.
Patterns of genetic divergence, genetic variation and inbreeding
Although care was taken to ensure that sampled populations represented stable effects of long-lasting natural genetic processes, due to the lack of historical records, we were not certain of population establishment histories. Among possible scenarios, especially recent bottlenecks might have a strong (and destructive) impact on our inferences about factors of the genetic structure. Therefore, based on expected heterozygosities we tested the hypothesis about recent bottleneck using BOTTLENECK software (Cornuet and Luikart 1996). The analysis was performed under TPM model (with default settings), and the heterozygosity excess was verified using the Wilcoxon test.
Rates of genetic divergence were estimated through population-specific F ST indices (here denoted by F i ; i = 1, 2, …), as formulated in the F-model of allele frequencies (Gaggiotti and Foll 2010). In order to verify the null hypothesis that divergence rates are not associated with longitude, latitude or species density, we used the Bayesian approach implemented in GESTE 2.0 software (Foll and Gaggiotti 2006). Briefly, the method uses the specific prior distribution for F i making the parameter to be a linear function of explanatory variables (e.g. environmental data). GESTE performs cross-validation of the alternative models underlying the observed divergence rates F i . In result, population-specific F i estimates are provided along with the posterior probabilities for competing models enabling straightforward model comparison and thus selection of the significant (environmental) factors of genetic divergence observed in the sample. For the purpose of the analysis, we set the regression model to be a function of density (ρ i ), latitude, longitude and a free term. GESTE programme was run using the default settings.
Using the same logic, we also performed a multiple regression analysis to verify the null hypothesis of no association between AR, He, D i or F IS and the species’ density or geographic variables. The regression model was of the form θ i = b 0 + b 1 x i + b 2 y i + b 3 ρ i , where θ i is a given genetic parameter for the ith population, x i , y i and ρ i refer to longitude, latitude and the species’ density estimated for the ith population, b 1, b 2 and b 3 are the slopes of the effects of longitude, latitude and density, respectively and b 0 is the intercept. Because He, D i and F IS represent probability measures, their values were logit-transformed before the analysis. For a given genetic parameter, aside from the full regression model, a series of reduced models were analysed. Then, the final parameter estimates were obtained using the procedure for model averaging (Burnham and Anderson 2002), as \( \hat{Y}={\displaystyle \sum_i}{w}_i{\widehat{Y}}_i \), where \( {\widehat{Y}}_i \) − the estimate of a given parameter under the ith model, w i − the Akaike weight for the ith model equal to \( \exp \left(-\frac{\Delta_i}{2}\right)/{\displaystyle \sum_k} \exp \left(-\frac{\Delta_k}{2}\right) \). In the latter formulae, Δ i was calculated as the difference between Akaike Information Criterion (AIC) for the ith model and AIC for the best model (i.e. that with the smallest AIC). For the averages obtained across models, unconditional standard errors were estimated according to \( \widehat{\mathrm{SE}}={\displaystyle \sum_i}{w}_i\sqrt{\mathrm{VAR}\left({\widehat{Y}}_i\Big|{\mathrm{Model}}_i\right)+{\left({\widehat{Y}}_i-\widehat{Y}\right)}^2} \), where \( \mathrm{VAR}\left({\widehat{Y}}_i\Big|{\mathrm{Model}}_i\right) \) is the variance conditioned on the ith model. Finally, the model-averaged estimates were used to assess the statistical significance of a particular explanatory variable using the Z-test. Multiple regression analysis was performed using MuMIn 1.7.2 (CRAN) library working under the R-project environment (R Development Core Team 2011).
Difference in gene dispersal between metapopulations
Under the isolation-by-distance theory, the slope of the regression function (b) obtained for pairwise F ST vs. log-distance can be used to assess the axial variance of dispersal distance σ 2 (or its square root) solving the equation b = (4πDσ 2)− 1, where D is the (effective) population density (Rousset 1997). We followed this approach in order to test if there is any difference in the scale of dispersal between the northern (σ N ) and the southern (σ S ) metapopulation. However, rather than assessing σ N and σ S separately, we focused on their ratio. For this purpose, we first expressed the density of the northern metapopulation as D N = αD S (N and S subscripts used for northern and southern metapopulation, respectively). Then, the ratio of dispersal distances can be computed as \( \frac{\sigma_N}{\sigma_S}=\sqrt{\frac{4\pi {D}_S{b}_S}{4\pi {D}_N{b}_N}}=\sqrt{\frac{b_S}{b_N\alpha }} \), where b S and b N are the slopes of regression function obtained for the northern and the southern metapopulation, respectively. Unlike the standard approach (Rousset 1997), our procedure did not require real (i.e. biologically meaningful) population densities but only the coefficient of proportionality α to be known.
The α coefficient was assessed as follows. Under the isolation-by-distance theory, if gene dispersal occurs within a two-dimensional habitat, D is equal to N/ϵ 2, where N is the effective size of a population and ϵ is the distance to the nearest population (Rousset 1997). Therefore, in order to obtain α, we required N and ϵ for the two metapopulations. In the case of N, we used the harmonic mean of the estimates N i for individual populations. The distance to the nearest population ϵ was computed based on the mean densities ρ N and ρ S (estimated from the presence-absence data), based on the theoretical expectation for the distance to the nearest neighbour under a random distribution, i.e. \( \epsilon =\frac{1}{2\sqrt{\rho }} \) (Clark and Evans 1954). We need to stress that ϵ N and ϵ S estimated in that way were not expressed in a meaningful scale (e.g. in metres). However, because we dealt with their ratio, the impact of the scale vanished. Finally, having ϵ N and ϵ S as well as N N and N S , α was computed as \( \frac{N_N{\epsilon}_S^2}{N_S{\epsilon}_N^2} \).
In order to test whether metapopulations differ in dispersal range the following procedure was applied. First, six datasets were prepared by successively removing one locus. For each data set, the matrix of pairwise F ST values was obtained using the ENA procedure implemented in FREE-NA software (Chapuis and Estoup 2007). Based on each F ST matrix and the invariant matrix of geographic distances, the ratio \( \frac{\sigma_N}{\sigma_S} \) was estimated as \( \sqrt{\frac{b_S}{b_N\alpha }} \). A series of six values of \( \frac{\sigma_N}{\sigma_S} \) was used to compute the jackknife standard error (SE). Finally, the approximate 95 % confidence limits were computed as \( \frac{\sigma_N}{\sigma_S}\pm 2\times \mathrm{SE} \). We treated σ N and σ S as significantly different when the confidence interval did not cover unity.
Results
Local species density and effective population size
Local species density ρ i extracted from the generalised distribution (Fig. 1), as well as estimates of the effective population size are shown in Table 2. Values of ρ i varied from 0.0018 (Kuźnik) to 0.0136 (Las Liściasty w Promnie). Averages computed for the high- and the low-density region equalled 0.0122 and 0.0063, respectively. The t test revealed that the difference in the mean species density between regions was statistically significant (p value = 0.0012). The effective population size N spanned from 6.3 (Kępa Bazarowa) to 1,700.3 (Czeszewski Las). We observed slightly lower N in the northern metapopulation (15.7 vs. 17.8). However, there was no difference between metapopulations in a local effective population size (t test; p value = 0.808).
Genetic variation and inbreeding
Among microsatellite markers used in this study, six were polymorphic. Consequently, all genetic parameters were computed disregarding the monomorphic locus Aop450. The total per-locus number of alleles varied from 3 (MAP46) to 26 (MAP40) (Table 3), with the average being 12.5. Null allele frequencies were generally low (0.062 on average), except for Aop943. The overall heterozygosity within populations was equal to 0.529 (from 0.106 to 0.805). However, the total population exhibited apparently higher heterozygosity level, reaching on average 0.606. According to the estimate of Nei’s G ST, the deficiency of diversity at the population level varied from 0.080 to 0.184 (0.126 on average).
The average number of alleles within populations ranged from 4.5 (Las Liściasty w Promnie) to 8.2 (Czeszewski Las), with the mean 6.1 (Table 4). Allelic richness measured after rarefaction ranged from 3.8 (Łęgi na Ostrowiu Panieńskim) to 5.7 (Czeszewski Las). It is worth noting that populations were ranked somewhat differently according to AR and A. However, because the latter one is strongly influenced by a sample size, as a measure of polymorphism we always used AR. The average expected heterozygosity varied from 0.509 (Las Mariański) to 0.699 (Czeszewski Las). Populations revealed deficiency of heterozygotes (Ho) as compared with Hardy-Weinberg proportions (He), except for Las Liściasty w Promnie. The inbreeding coefficient ranged from 0.015 to 0.300, with the grand average of 0.107. However, inbreeding coefficient (F IS) was significantly different from zero in five populations only, indicating that the deficiency of heterozygotes is mostly due to the presence of null alleles (see the previous section). Based on the Wilcoxon test for the expected heterozygosity excess we detected no population having significant signatures of recent bottleneck (data not shown).
Genetic structuring
The AMOVA showed that the most variation (85.1 %) is captured within populations (Table 5). About 11.8 % variation was among populations within regions, while only 3.2 % variation was between regions. Nonetheless, fixation indices at all levels were significantly different from zero.
The optimal number of genetic clusters was found to be 4 (Fig. 2). Generally, populations in the southern metapopulation appeared to be a mixture of different gene pools (clusters), except for Bielawy population, which was relatively homogeneous. In the case of the northern metapopulation, two clusters can be distinguished. The first cluster was composed of two the most southern populations (Kuźnik and Kępa Bazarowa). The remaining three populations (Las Mariański, Łęgi na Ostrowiu and Ostrów Panieński) formed roughly the second cluster. Interestingly, Łęgi na Ostrowiu and Ostrów Panieński were not completely homogeneous, despite the fact that they are located in the same forest patch (1.4 km apart). Admixture levels (D i ) estimated based on individual assignment probabilities ranged from 0.134 (Kępa Bazarowa) to 0.687 (Czeszewski Las) (Table 4) and were significantly negatively correlated with F i (Spearman’s ρ = −0.724; p value = 0.005).

Clustering of individual genotypes. Top, the change in the probability of data and the Evanno′s Delta K measure as a function of the number of clusters (K). Bottom, the results of the model-constrained (Hardy-Weinberg and linkage equilibrium) clustering obtained using STRUCTURE software for K = 2 and 4. Note that, according to the Evanno′s method, K = 4 is the optimal model
Patterns of genetic divergence, genetic variation and inbreeding
Divergence rates (F i ) for individual populations ranged from 0.040 (Czeszewski Las) to 0.226 (Kępa Bazarowa) with the mean of 0.143 (Table 4). When plotted against density, longitude and latitude, F i values increased systematically with increasing latitude, while any apparent association was found neither with density nor longitude (Fig. 3a). In total, eight competing models were analysed using GESTE software (Fig. 3b). Among these alternatives, the model containing latitude only appeared to be the most likely, with the posterior probability of 0.516. The second best model, in which F i was factor-independent (the null model), had the posterior probability of 0.282, while the remaining alternatives had the cumulative posterior probability of 0.202. Among the three explanatory variables, latitude had the highest posterior probability of 0.634 (Fig. 3c). Thus, GESTE revealed that the genetic divergence was significantly explained by latitude, while the impact of the other variables was relatively low.

Results of the Bayesian estimation of the effect of longitude and latitude on genetic divergence rates (expressed as F i /(1 – F i )). a Individual divergence rates as a function of density, longitude and latitude. b The posterior probability of eight competing models (Dens density, Long longitude, Lat latitude). c The posterior probability, and thus the impact, of explanatory variables
The geographic pattern, but the opposite to that for divergence rates, was also identified for allelic richness and genetic diversity (Fig. 4). Based on the multiple regression analysis (Table 6), latitude appeared to be the only significant factor of genetic structure, explaining the decrease in polymorphism (AR), genetic diversity (He) and, marginally significantly, admixture level (D i ). Nonetheless, latitude did not explain inbreeding levels. Interestingly, although significantly correlated with latitude, density (and thus population fragmentation) appeared to be insignificant explanatory variable for the observed genetic structure.

Geographic patterns of genetic polymorphism (a), genetic diversity (b), inbreeding (c) and admixture level (d). Lines show the best-fitting linear regression functions
Difference in gene dispersal between metapopulations
Given the mean values of ρ for each metapopulation (Table 2), the (unitless) values of ϵ computed for the southern and northern regions were 4.529 and 6.325, respectively. Taking the mean effective sizes, the ratio of densities of the two metapopulations equalled α = 0.452. The slope of regression function, computed to fit the relationship between genetic and geographic distance, equalled 0.031 (±0.008) and 0.012 (±0.003) for the northern and the southern metapopulations, respectively (Fig. 5). Consequently, the ratio of dispersal scales \( \frac{\sigma_N}{\sigma_S} \) equalled 0.949 with the 95 % confidence interval between 0.596 and 1.303. Thus, the test showed no difference in dispersal scales between the two metapopulations.

Spatial genetic structure measured using pairwise (linearized) F ST coefficients between populations in the northern and the southern metapopulation
Discussion
The results obtained in this study showed that populations of A. campestre located closer to the northern margin of the natural distribution are characterised by lower genetic variation and higher divergence rates. This pattern is in line with the theoretical predictions for the ‘central-marginal’ theory, that peripheral populations experience more genetic drift leading to reduced genetic variation as compared with more central populations (Vucetich & Waite 2003; Eckert et al. 2008). This kind of spatial pattern is generally found in tree species (e.g. Tollefsrud et al. 2009; Grivet et al. 2009), although such studies often require large spatial scales (Comps et al. 2001; Coart et al. 2005). This is because trees are often characterised by wide distribution, high potential for gene flow and strong human impact, including forest management and translocation of seed material (Petit and Hampe 2006). Interestingly, some authors propose the existence of ‘the paradox of forest fragmentation genetics’, suggesting that trees may not follow the standard consequences of population fragmentation (Kramer et al. 2008; but see Piotti 2009). On the contrary, our study demonstrated that the significant spatial genetic patterns can be detected for a tree species even at relatively restricted spatial scale.
Theoretically, fragmented populations tend to show elevated inbreeding (Young et al. 1996; Jump and Penuelas 2006; Aguilar et al. 2008). When fragmentation leads to isolation (i.e. dispersal between fragments is rare), populations experience pollen limitation in effect of reduced number of pollen donors (Bierzychudek 1982; Fox 1992; Scobie and Wilcock 2009). In such conditions recurrent consanguineous mating can lead to genetic purging (Pujol et al. 2009), weakening inbreeding depression and opening potential for further increase of inbreeding. When fragmentation is due to ecological conditions, like in marginal habitats, inbreeding can be especially adaptive because it can help avoiding decreased fecundity (Fischer and Matthies 1997; Busch 2005; Mimura and Aitken 2007; Michalski and Durka 2007; Johnson et al. 2009; Tollefsrud et al. 2009). Nonetheless, because empirical studies provided a mixed support for this prediction, it was argued that in many cases populations experienced fragmentation only recently, so that the observed inbreeding level still reflects more the historical conditions rather than the reduced population size (Aguilar et al. 2008). Alternatively, fragmentation did not lead to increased inbreeding because of long-distance gene flow (Wang et al. 2011; Leonardi et al. 2012). In our study, however, A. campestre represents a stable distribution, with northern populations being naturally fragmented long enough to assure inbreeding to accumulate. However, in the study species, we found no support for the theoretical prediction, as inbreeding did not increase together with the population fragmentation (i.e. towards northern periphery). In fact, inbreeding levels were not associated with geographic location, and populations were rather outbred, except for five populations with significant F IS values. Extremely high values of F IS (>>0.1) were estimated for two populations. In the case of microsatellite markers, null alleles often cause overestimation of the inbreeding coefficient (Van Oosterhout et al. 2006). Nonetheless, in this study F IS estimates were obtained with the method robust to null alleles (Chybicki and Burczyk 2009; Campagne et al. 2012), so that other reasons were required. In the case of Czeszewski Las (located in the region of higher species abundance), the genetic clustering revealed the highest admixture level. As the probability model underlying the estimation procedure assumes a single gene pool (Chybicki & Burczyk 2009), at least in this case F IS could be biased. Similar reasons could lead to the elevated inbreeding estimate in the case of Las Mariański, which represented the most admixed gene pool in the region of lower species abundance. Alternative explanation would be a specific colonisation history, if a few (related) adult trees gave a rise to the population (Pujol et al. 2009; Chybicki et al. 2012). However, because no signatures of a strong bottleneck were revealed, the hypothesis of bottleneck has no support from the data. Another possibility is that self-fertilisation contributed substantially to the observed generation. Bendixen (2001) showed that A. campestre can reproduce through self-fertilisation, but the ability to selfing revealed high individual variation ranging from 0 to 100 %, with the average 16.7 %. Because she observed significant correlation in selfing rates between two successive seasons, it cannot be excluded that this feature is partly genetically determined. If so, the ability to selfing may follow the high differentiation between populations observed for the neutral genetic variation, but this needs to be verified empirically. If this was the case, however, the ‘central-marginal’ prediction for inbreeding levels would not be applicable in the study species.
Besides assessing spatio-genetic patterns, we also tested whether fragmentation had an impact on the scale of gene dispersal. However, we failed to show that increased fragmentation leads to decreased gene dispersal. In addition, we showed that the estimates of effective population size appeared practically the same in low- and high-density regions. However, we showed that the increased fragmentation or decreased species density shaped the steeper relationship between F st and the geographic distance, implying that, given an invariant dispersal scale and equal effective population sizes, a species density influences only the effective number of migrants (Rousset 1997). In fragmented populations, the scale of effective gene dispersal tends to be reduced compared with the potential gene dispersal if the radius of local population (clump) is shorter than the scale of potential dispersal. It is because only short-distance (relatively frequent) and long-distance (relatively rare) dispersal takes effectively place, while intermediate-distance dispersal events are missing (Robledo-Arnuncio and Austerlitz 2006). On the other hand, if the scale of potential dispersal is much shorter than the radius of local population, potential and effective dispersal should be equal (Cuartas-Hernández et al. 2010). Thus, the result of no difference in dispersal between the two metapopulations would be reasonable only, if gene dispersal in the study species was very restricted. Interestingly, both AMOVA and low effective size estimates suggest low gene flow between populations. On the other hand, the model-based clustering showed rather low genetic structuring, suggesting a possibility for long-distance dispersal. Thus, the questions remains: what is the actual potential for gene dispersal in the study species?
Field maple produces winged seeds (samara), which have rather limited dispersal capabilities (Young and Merriam 1994; Clark et al. 1998), except for specific situations, like dispersal in open areas (Johnson 1988). Although no data exist on seed dispersal of the study species, the study on Acer opalus, an analogous species inhabiting southern and western Europe, revealed that the average distance of seed dispersal is between 2.3 and 4.2 m but does not exceed 12.5 m (Gomez-Aparicio et al. 2007). Phylogenetic data showed that genetic differentiation of A. campestre at maternally inherited chloroplast genome is as high as that of heavy-seeded tree species (Petit et al. 2003), providing support for the restricted seed dispersal. No data on pollen dispersal are available for A. campestre. However, the parentage analysis showed that the average distance between mates in A. opalus was as short as 1.99 m only (estimated based on the scale parameter of the exponential dispersal kernel) (Gleiser et al. 2008). Also, pollination distance of 2.08 m only was estimated (based on the same approach) for Acer pictum, a bee-pollinated Asian congener (Shang et al. 2012). In both cases, mating was restricted mainly to close neighbours. Generally, studies on seed and pollen dispersal suggest that gene dispersal in the study species can occur mostly locally, i.e. within isolated populations. Nonetheless, as mentioned in the introduction, field maple is likely capable of (at least incidental) wind-pollination, which is generally known to facilitate long pollen flow (Ashley 2010 and references therein). Also, insect-pollination can be very effective in terms of dispersal distances, as shown in the case of many fragmented tropical species (Nason and Hamrick 1997; White et al. 2002). In Poland, A. campestre starts flowering relatively early in the season (in April–May) and because even a single tree can produce a large number of flowers, it may be attractive food resource for pollinating insects. However, we believe that due to low density, it does not allow for optimal foraging of pollinators so that long-distance pollen transport is rather infrequent. Overall, although the species has some potential for long-distance gene dispersal, a majority of gene flow occurs at short distances.
In many fragmented species, a common confounding factor of genetic structure is the colonisation process (Pannell and Dorken 2006). For example, in many European trees the geographic distribution of genetic diversity reflects the process of postglacial recolonisation (Comps et al. 2001; Coart et al. 2005; Tollefsrud et al. 2009; Grivet et al. 2009). In consequence, genetic diversity tends to decrease with the distance from refugia (generally northwards), even if no sharp differences in a species’ current density can be found. Interestingly, our study showed that the study species’ density did not influence both a dispersal scale and genetic structure parameters. On the other hand, both genetic variation and divergence rates co-varied with latitude. Because we excluded recent bottlenecks as well as the impact of dispersal (no differences between low- and high-density regions), it seems that the observed genetic structure likely reflects the historical process of the species’ postglacial recolonisation. In the recolonisation, the founder effect plays the key role in shaping genetic variation (Austerlitz et al. 2000; but see Born et al. 2008). It is especially expected for species with low dispersal capabilities, such as the study species, where every new population is likely established by only a few migrants (Austerlitz and Garnier-Géré 2003). In this way, together with the distance from the ancestral population (measured both in space and time), genetic variation decreases and divergence increases.
Final remarks
Our study demonstrated that the species abundance decreasing towards range limit concurs with sharp genetic patterns in A. campestre. However, we rejected the hypothesis of the impact of species’ density on the genetic variation and dispersal scale. We also showed that inbreeding levels were not associated with the species abundance. This result contradicts with the expectation for pollen limitation and may suggest that a number of pollen donors is large enough even at the range margin. Also, our study demonstrated that the latitudinal genetic variation, observed frequently in European trees, is also present in the study species. However, as compared with the other species, the pattern is observed even at a local spatial scale. We speculate that this may be mostly due to high fragmentation and low dispersal capabilities. However, these hesitations can be only resolved through in-depth insights into matting patterns. Therefore, we recommend that future studies should focus on mating patterns through paternity or parentage analyses, addressing levels of selfing, biparental inbreeding and effective number (or density) of pollen and seed parents.
References
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J (2008) Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17:5177–5188
Ashley MV (2010) Plant parentage, pollination, and dispersal: how DNA microsatellites have altered the landscape. Crit Rev Plant Sci 29:148–161
Austerlitz F, Garnier-Géré PH (2003) Modelling the impact of colonisation on genetic diversity and differentiation of forest trees: interaction of life cycle, pollen flow and seed long-distance dispersal. Heredity 90:282–290
Austerlitz F, Mariette S, Machon N, Gouyon P-H, Godelle B (2000) Effects of colonisation processes on genetic diversity: differences between annual plants and tree species. Genetics 154:1309–1321
Austin M (2007) Species distribution models and ecological theory: a critical assessment and some possible new approaches. Ecol Model 200:1–19
Beletti P, Monteleone I, Ferrazzini D (2007) Genetic variability at allozyme markers in sycamore (Acer pseudoplatanus) populations from northwestern Italy. Can J For Res 37:395–403
Bendixen K (2001) Reproductive system of field maple (Acer campestre): flowering phenology and genetic investigations. Universität Göttingen, Germany, Dissertation
Bierzychudek P (1982) Pollinator limitation of plant reproductive effort. Am Nat 117:838–840
Born C, Kjellberg F, Chevallier MH, Vignes H, Dikangadissi JT, Sanguié J, Wickings EJ, Hossaert-McKey M (2008) Colonization processes and the maintenance of genetic diversity: insights from a pioneer rainforest tree, Aucoumea klaineana. Proc R Soc B 275:2171–2179
Brown JH (1984) On the relationship between abundance and distribution of species. Am Nat 124:255–279
Burnham KP, Anderson DR (2002) Model selection and multimodel inference: a practical information-theoretic approach, 2nd edn. Springer, New York
Busch JW (2005) The evolution of self-compatibility in geographically peripheral populations of Leavenworthia alabamica (Brassicaceae). Am J Bot 92:1503–1512
Campagne P, Smouse PE, Varouchas G, Silvain JF, Leru B (2012) Comparing the van Oosterhout and Chybicki-Burczyk methods of estimating null allele frequencies for inbred populations. Mol Ecol Resour 12(6):975–982
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113
Chybicki IJ, Oleksa A, Burczyk J (2011) Increased inbreeding and strong kinship structure in Taxus baccata estimated from both AFLP and SSR data. Heredity 107:589–600
Chybicki IJ, Oleksa A, Kowalkowska K (2012) Variable rates of random genetic drift in protected populations of English yew: implications for gene pool conservation. Conserv Genet 13:899–911
Clark PJ, Evans FC (1954) Distance to nearest neighbor as a measure of spatial relationship in populations. Ecology 35:445–453
Clark JS, Macklin E, Wood L (1998) Stages and spatial scales of recruitment limitation in southern Appalachian forests. Ecol Monogr 68:213–235
Coart E, Van Glabeke S, Petit RJ, Van Bockstaele E, Roldán-Ruiz I (2005) Range wide versus local patterns of genetic diversity in hornbeam (Carpinus betulus L.). Conserv Genet 6:259–273
Comps B, Gömöry D, Letouzey J, Thiébaut B, Petit RJ (2001) Diverging trends between heterozygosity and allelic richness during postglacial colonization in the European Beech. Genetics 157:389–397
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cuartas-Hernández S, Núñez-Farfán J, Smouse PE (2010) Restricted pollen flow of Dieffenbachia seguine populations in fragmented and continuous tropical forest. Heredity 105:197–204
De Jong PC (1976) Flowering and sex expression in Acer L.: a biosystematic study. Wageningen, Nederland
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a Website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Eckert CG, Samis KE, Lougheed SC (2008) Genetics variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Mol Ecol 17:1170–1188
Eo SH, Wares JP, Carroll JP (2008) Population divergence in plant species reflects latitudinal biodiversity gradients. Biol Lett 4:382–384
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7:574–578
Fischer M, Matthies D (1997) Mating structure and inbreeding and outbreeding depression in the rare plant Gentianella germanica (Gentianaceae). Ann Bot 84:1685–1692
Foll M, Gaggiotti O (2006) Identifying the environmental factors that determine the genetic structure of populations. Genetics 174:875–891
Fortin M-J, Keitt TH, Maurer BA, Taper ML, Kaufman DM, Blackburn TM (2005) Species’ geographic ranges and distributional limits: pattern analysis and statistical issues. Oikos 108:7–17
Fox JF (1992) Pollen limitation of reproductive effort in willows. Oecologia 90:283–287
Frankham R (2003) Genetics and conservation biology. CR Biologies 326:22–29
Gaggiotti OE, Foll M (2010) Quantifying population structure using the F-model. Mol Ecol Resour 10:821–830
Gargano D, Gullo T, Bernardo L (2009) Do inefficient selfing and inbreeding depression challenge the persistence of the rare Dianthus guliae Janka (Caryophyllaceae)? Influence of reproductive traits on a plant's proneness to extinction. Plant Species Biology 24:69–76
Geburek T (1993) Are genes randomly distributed over space in mature populations of sugar maple (Acer saccharum Marsh.)? Ann Bot 71:217–222
Gleiser G, Verdú M, Segarra-Moragues JG, González-Martínez SC, Pannell JR (2008) Disassortative mating, sexual specialization, and the evolution of gender dimorphism in heterodichogamous Acer opalus. Evolution 62:1676–1688
Gomez-Aparicio L, Gomez JM, Zamora R (2007) Spatiotemporal patterns of seed dispersal in a wind-dispersed Mediterranean tree (Acer opalus subsp. granatense): implications for regeneration. Ecography 30:13–22
Goudet J (1995) FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Grivet D, Sebastiani F, González-Martínez SC, Vendramin GG (2009) Patterns of polymorphism resulting from long-range colonization in the Mediterranean conifer Aleppo pine. New Phytol 184:1016–1028
Guarino C, Cipriani G (2013) Landscape discontinuities influence the population structure of Acer opalus ssp. obtusatum Waldst. & Kit. ex Willdenow. Plant Biosystems 147:1029–1042
Guarino C, Santoro S, De Simone L, Cipriani G, Testolin R (2008) Differentiation in DNA fingerprinting among species of the genus Acer L. in Campania (Italy). Plant Biosystems 142:454–461
Guo Q (2012) Incorporating latitudinal and central-marginal trends in assessing genetic variation across species ranges. Mol Ecol 21:5396–5403
Herlihy CH, Eckert CG (2002) Genetic cost of reproductive assurance in a self-fertilizing plant. Nature 416:320–323
Hesse M (1979) Ultrastruktur und Verteilung des Pollenkitts in der insekten- und windbliitigen Gattuug Acer (Aceraceae). Plant Syst Evol 131:277–289
Hijmans RJ, Guarino L, Cruz M, Rojas E (2001) Computer tools for spatial analysis of plant genetic resources data: 1. DIVA-GIS. Plant Genetic Resources Newsletter 127:15–19
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978
Holt RD, Keitt TH (2000) Alternative causes for range limits: a metapopulation perspective. Ecol Lett 3:41–47
Howe HF, Smallwood J (1982) Ecology of seed dispersal. Annu Rev Ecol Syst 13:201–228
Johnson WC (1988) Estimating dispersibility of Acer, Fraxinus and Tilia in fragmented landscapes from patterns of seedling establishment. Landsc Ecol 1:175–187
Johnson SD, Torninger E, Agren J (2009) Relationships between population size and pollen fates in a moth-pollinated orchid. Biol Lett 23:282–285
Jump AS, Penuelas J (2006) Genetic effects of chronic habitat fragmentation in a wind-pollinated tree. Proc Natl Acad Sci U S A 103:8096–8100
Kalisz S, Vogler D, Fails B, Finer M, Shepard E, Herman T, Gonzales R (1999) The mechanism of delayed selfing in Collinsia verna (Scrophulariaceae). Am J Bot 86:1239–1247
Kramer AT, Ison JL, Ashley MV, Howe HF (2008) The paradox of forest fragmentation genetics. Conserv Biol 22:878–885
Kunin WE, Vergeer P, Kenta T, Davey MP, Burke T, Woodward FI, Quick P, Mannarelli ME, Watson-Haigh NS, Butlin R (2009) Variation at range margins across multiple spatial scales: environmental temperature, population genetics and metabolomic phenotype. Proc R Soc B 276:1495–1506
Leimu R, Mutikainen P, Koricheva J, Fischer M (2006) How general are positive relationships between plant population size, fitness and genetic variation? J Ecol 94:942–952
Leonardi S, Piovani P, Scalfi M, Piotti A, Giannini R, Menozzi P (2012) Effect of habitat fragmentation on the genetic diversity and structure of peripheral populations of Beech in Central Italy. J Hered 103:408–417
Martin PR, McKay JK (2004) Latitudinal variation in genetic divergence of populations and the potential for future speciation. Evolution 58:938–945
Michalski SG, Durka W (2007) High selfing and high inbreeding depression in peripheral populations of Juncus atratus. Mol Ecol 16:4715–4727
Mimura M, Aitken SN (2007) Increased selfing and decreased effective pollen donor number in peripheral relative to central populations in Picea sitchensis (Pinaceae). Am J Bot 94:991–998
Montoya D, Zavala MA, Rodríguez MA, Purves DW (2008) Animal versus wind dispersal and the robustness of tree species to deforestation. Science 320:1502–1504
Munwes I, Geffen E, Roll U, Friedmann A, Daya A, Tikochinski Y, Gafny S (2010) The change in genetic diversity down the core-edge gradient in the eastern spadefoot toad (Pelobates syriacus). Mol Ecol 19:2675–2689
Nason JD, Hamrick JL (1997) Reproductive and genetic consequences of forest fragmentation: two case studies of neotropical canopy trees. J Hered 88:264–276
Newman D, Pilson D (1997) Increased probability of extinction due to decreased genetic effective population size: experimental populations of Clarkia pulchella. Evolution 51:354–362
O'Connell LM, Mosseler A, Rajora OP (2007) Extensive long-distance pollen dispersal in a fragmented landscape maintains genetic diversity in white spruce. J Hered 98:640–645
Pandey M, Gailing O, Fischer D, Hattemer HH, Finkeldey R (2004) Characterization of microsatellite markers in sycamore (Acer pseudoplatanus L.). Mol Ecol Notes 4:253–255
Pandey M, Gailing O, Hattemer HH, Finkeldey R (2012) Fine-scale spatial genetic structure of sycamore maple (Acer pseudoplatanus L.). Eur J For Res 131:739–746
Pannell JR, Dorken ME (2006) Colonisation as a common denominator in plant metapopulations and range expansions: effects on genetic diversity and sexual systems. Landsc Ecol 21:837–848
Petit RJ, Hampe A (2006) Some evolutionary consequences of being a tree. Annu Rev Ecol Evol Syst 37:187–214
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, Mohanty A, Müller-Starck G, Demesure-Musch B, Palmé A, Martín JP, Rendell S, Vendramin GG (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565
Piotti A (2009) The genetic consequences of habitat fragmentation: the case of forests. iForest 2: 75–79.
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pujol B, Zhou SR, Sanchez Vilas J, Pannell JR (2009) Reduced inbreeding depression after species range expansion. Proc Natl Acad Sci U S A 106:15379–15383
Rajora OP, Mosseler A, Major JE (2002) Mating system and reproductive fitness traits of eastern white pine (Pinus strobus) in large, central versus small, isolated, marginal populations. Can J Bot 80:1173–1184
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237
Robledo-Arnuncio JJ, Austerlitz F (2006) Pollen dispersal in spatially aggregated populations. Am Nat 168:500–511
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Rusanen M, Vakkari P, Blom A (2000) Evaluation of the Finnish gene-conservation strategy for Norway maple (Acer platanoides L.) in the light of allozyme variation. For Genet 7:155–165
Rusanen M, Vakkari P, Blom A (2003) Genetic structure of Acer platanoides and Betula pendula in northern Europe. Can J For Res 33:1110–1115
Scobie AR, Wilcock CC (2009) Limited mate availability decreases reproductive success of fragmented populations of Linnaea borealis, a rare, clonal self-incompatible plant. Ann Bot 103:835–846
Segarra-Moraguez JG, Gleiser H, González-Candelas F (2008) Isolation and characterization of microsatellite loci in Acer opalus (Aceraceae), a sexually-polymorphic tree, through an enriched genomic library. Conserv Genet 9:1059–1062
Shang H, Luo YB, Bai WN (2012) Influence of asymmetrical mating patterns and male reproductive success on the maintenance of sexual polymorphism in Acer pictum subsp. mono (Aceraceae). Mol Ecol 21:3869–3878
Simpson EH (1949) Measurement of diversity. Nature 163:688
Thomas CD (2000) Dispersal and extinction in fragmented landscapes. Proc R Soc B 267:139–145
Tollefsrud MM, Sønstebø JH, Brochmann C, Johnsen Ø, Skrøppa T, Vendramin GG (2009) Combined analysis of nuclear and mitochondrial markers provide new insight into the genetic structure of North European Picea abies. Heredity 102:549–562
Travis JMJ, Dytham C (1998) The evolution of dispersal in a metapopulation: a spatially explicit, individual-based model. Proc R Soc B 265:17–23
Van Oosterhout C, Weetman D, Hutchinson WF (2006) Estimation and adjustment of microsatellite null alleles in nonequilibrium populations. Mol Ecol Notes 6:255–256
Vilas C, San Miguel E, Amaro R, Garcia C (2006) Relative contribution of inbreeding depression and eroded adaptive diversity to extinction risk in small populations of shore campion. Conserv Biol 20:229–238
Vranckx G, Jacquemyn H, Muys B, Honnay O (2011) Meta-analysis of susceptibility of woody plants to loss of genetic diversity through habitat fragmentation. Conserv Biol 26:228–237
Vucetich JA, Waite TA (2003) Spatial patterns of demography and genetic processes across the species’ range: null hypothesis for landscape conservation genetics. Conserv Genet 4:639–645
Wang R, Compton SG, Chen X-Y (2011) Fragmentation can increase spatial genetic structure without decreasing pollen-mediated gene flow in a wind-pollinated tree. Mol Ecol 20:4421–4432
Waples RS, Do C (2008) ldne: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8:753–756
White GM, Boshier DH, Powell W (2002) Increased pollen flow counteracts fragmentation in a tropical dry forest: an example from Swietenia humilis Zuccarini. Proceeding of the National Academy of Sciences of the United States of America 99:2038–2042
Willi Y, Van Buskirk J, Hoffmann AA (2006) Limits to the adaptive potential of small populations. Annu Rev Ecol Evol Syst 37:433–458
Woś A (1999) Klimat Polski. Wydawnictwo Naukowe PWN, Warszawa
Young AG, Merriam HG (1994) Effects of forest fragmentation on the spatial genetic structure of Acer saccharum Marsh. (sugar maple) populations. Heredity 72:201–208
Young AG, Merriam HG, Warwick SI (1993) The effect of forest fragmentation on genetic variation in Acer saccharum Marsh. (sugar maple) populations. Heredity 71:277–289
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418
Zając A, Zając M (2001) Distribution atlas of vascular plants in Poland. Edited by Laboratory of Computer Chorology. Jagiellonian University, Cracow, Institute of Botany
Acknowledgements
This study was supported by the Polish Ministry of Science and The Higher Education (IUVENTUS Plus programme, grant no. IP2011 007071 to IJC). The authors would like to thank Jaroslaw Burczyk for helpful comments on the first version of the manuscript, Andrzej Oleksa for his help with preparing the distribution graph as well as the two anonymous reviewers for their constructive suggestions on the manuscript.
Conflict of interest
The authors declare no conflict of interest
Data archiving
Genotype data are available at Research Gate (www.researchgate.net) (doi:10.13140/2.1.3724.2243).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. N. Aitken
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Chybicki, I.J., Waldon-Rudzionek, B. & Meyza, K. Population at the edge: increased divergence but not inbreeding towards northern range limit in Acer campestre . Tree Genetics & Genomes 10, 1739–1753 (2014). https://doi.org/10.1007/s11295-014-0793-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-014-0793-2