
Abstract
Protecting populations in their natural habitat allows for the maintenance of naturally evolved adaptations and ecological relationships. However, the conservation of genetic resources often requires complementary practices like gene banks, translocations or reintroductions. In order to minimize inbreeding depression and maximize the adaptive potential of future populations, populations chosen for ex situ conservation should be selected according to criteria that will result in a reduction of global coancestry in the population. Generally, large populations should reveal lower coancestry and higher genetic variation than small populations. If detailed knowledge about coancestry is lacking, census population number (N c ) can be used as a proxy for required characteristics. However, a simple measure of N c may be misleading in particular cases as genetic processes rely on effective population size (N e ) rather than N c and these two measures may differ substantially due to demographic processes. We used an example of English yew to address whether N c can be a good predictor of genetic parameters when used in conservation programs. Using microsatellite markers, we estimated allelic richness, inbreeding and coancestry coefficients of six relatively large yew populations in Poland. Each population was characterized by N e using the linkage disequilibrium method. Our results showed that populations of English yew were subject to substantial divergence and genetic drift, with both being inversely proportional to the effective subpopulation size (N e ). Additionally, allelic richness appeared proportional to N e but not to N c . However, the N e /N ratio differed greatly among populations, which was possibly due to different population histories. From the results we concluded that choosing source populations based only on their census size can be fairly misleading. Implications for conservation are briefly discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The conservation of endangered and threatened plant species is focused on in situ practices. Protecting populations in their natural habitat allows the maintenance of natural ecological and evolutionary processes, as well as evolved adaptations and ecological relationships. However, conservation of genetic resources usually requires additional forms of conservation, so called ex situ practices such as gene banks, translocations or reintroductions (Pierson et al. 2007). If possible, choosing populations for ex situ conservation should follow some objective criterions which include genetic parameters such as inbreeding coefficients and levels of genetic polymorphism (Mistretta 1994). High genetic variation allows a population to better respond to unpredictable environmental changes in the future (Frankham et al. 2002; Willi et al. 2006). In addition, inbreeding depression can be avoided by maintaining low levels of inbreeding (Charlesworth and Willis 2009 and the literature therein). The generally acknowledged strategy for ex situ conservation is to minimize the global coancestry in the population (Montgomery et al. 1997; Caballero and Toro 2000). This requires kinship structures among individuals to be known in advance before their contribution to the next generation can be predicted (Saura et al. 2008). If the kinship structure is unknown, census population size may be the only proxy available to determine required characteristics. Generally, small populations should reveal higher coancestry and lower genetic variation than large populations. However, as genetic processes rely on effective (N e ) rather than census (N c ) population size and as these two measures may differ substantially due to demographic processes (e.g. biased reproductive success or sex ratios) (Frankham 1995), a simple measure of N c may be misleading in particular cases.
English yew (Taxus baccata L) serves as a good example of a species that needs both in situ and ex situ conservation. The present-day occupancy of this understory species is limited mainly to small and highly discontinuous remnants (Thomas and Polwart 2003). At a metapopulation scale English yew exhibits typically substantial levels of genetic differentiation (Hilfiker et al. 2004; Myking et al. 2009; Zarek 2009; Dubreuil et al. 2010; González-Martínez et al. 2010) driven by low population size and restricted gene flow (Chybicki et al. 2011). Due to similar reasons, increased inbreeding and kinship structure can build-up within yew populations (Lewandowski et al. 1995; Myking et al. 2009; Dubreuil et al. 2010; Chybicki et al. 2011).
English yew is slow growing, dioecious, late maturating conifer. It produces airborne pollen and fleshly fruits that are often dispersed by birds (Thomas and Polwart 2003). Although high shade tolerance allows young yew recruits to outcompete other, less shade-tolerant plants (Iszkuło and Boratyński 2006), it is known to regenerate poorly within a site. This is primarily attributed to grazing and damage by fungi or insect pests (Hulme 1996; Svenning and Magård 1999; Mysterud and Østbye 2004; Thomas and Polwart 2003). Poor regeneration results in ageing of natural populations that puts them under risk of extinction. Therefore, long-term in situ conservation of the natural genetic resources may fail in the case of English yew, at least in some populations. For example, the largest Polish reserve “Cisy Staropolskie” in Wierzchlas (see Materials and methods) has been exhibiting continuing decline over the last 100 years, with less than 3,000 individuals remaining from an initial population of roughly 5,500 individuals. Given the late maturity of this species (ca. 70–120 years; Thomas and Polwart 2003), the rate of reduction of population size amounts to approximately 35 % per generation. To curb such a rapid decline, ex situ conservation employing gene bank collections and restitution programs is required. Restitution of English yew is especially important as it can play a crucial role in the conservation of genetic resources of the species. Firstly, restituted populations could constitute a natural long-term repository of genetic variation. Secondly, a net of reintroduced populations could form natural corridors for gene exchange between fragmented populations. As a consequence, the destructive process of genetic drift occurring within small yew fragments could potentially be counterbalanced by increased gene immigration.
The long-term goals of ex situ programs will be achieved given appropriately chosen source populations. Nevertheless, in many cases census population numbers are the only information available prior to administrative decisions. Because knowledge about genetic structure and demography of English yew is only starting to accumulate (Lewandowski et al. 1995; Hilfiker et al. 2004; Dubreuil et al. 2010; González-Martínez et al. 2010; Chybicki et al. 2011), it remains unclear whether N c reflects N e and other genetic structure parameters enough to make a decision about the usefulness of particular populations for ex-situ conservation. Our study aimed to address this issue using SSR data collected for six populations of English yew in Poland. Specific goals included the assessment of levels of genetic polymorphism, inbreeding and coancestry, as well as N e . We also tested if a bottleneck had occurred in the recent population history. Finally, we verified whether census population size correlates with genetically derived parameters.
Materials and methods
Study sites and sampling
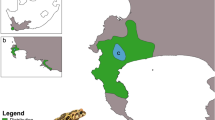
Studies were conducted within six selected populations of English yew (Fig. 1). The criterion of choice was the potential utility of the populations in ex situ conservation programs. Therefore, the study sites represented the largest yew populations in a region. The selected sites could be considered virtually independent remnants of the ancestral population as they are separated by large distances (ca. 260 km on average). All the sites are located within nature reserves established to protect yew populations. Except for Cisy Staropolskie, the study populations are characterized by a low density (as for a temperate tree species), reaching about 20 individuals per hectare (Table 1). Cisy Staropolskie is a unique site because of its large size and relatively high density. Nonetheless, it can be considered as a declining population as there is an absence of natural regeneration (absolute mortality among seedlings within two successive seasons) as well as a continuous reduction in population number. On the contrary, successful regeneration is particularly evident in Cisy nad Czerską Strugą as well as Cisy w Czarnem, with the latter seeming the most stable out of all six populations. It should be mentioned that little is known about the origin of the study sites. In consequence, we a priori assumed that all the populations are natural remnants of the larger population, with a possibility of bottleneck in the recent history. Nonetheless, other scenarios (incl. artificial origin) cannot be definitely excluded.

Map of location of the study populations of English yew in Poland (populations names coded as in Table 1)
For the analysis of DNA polymorphism, 10 cm long twigs were sampled from selected individuals at each site. In order to reflect stabilized characteristics of genetic structure, the oldest possible individuals were sampled at a particular site.
Molecular methods
A total genomic DNA was extracted using a slightly modified CTAB protocol (Doyle and Doyle 1990). For genetic typing five nuclear SSR loci were analyzed: Tax26, Tax31, Tax36 (Dubreuil et al. 2008), TS09 (Huang et al. 2007) and Tax362 (Chybicki et al. 2011) according to the protocol described elsewhere (Chybicki et al. 2011).
Statistical methods
Genetic structure
To describe genetic structure, standard per-locus genetic structure parameters were computed (observed and effective number of alleles, expected and observed heterozygosity). Because sample sizes differed among populations (Table 1), which affected levels of detected polymorphism, an added measure of allelic richness was computed using the rarefaction method (Petit et al. 1998). To study concordance with Hardy–Weinberg proportions, the MCMC approximation of the exact test was employed as implemented in GENEPOP 4.0 software (Rousset 2008). To test the overall homogeneity of genetic structure parameters across populations (a population as a factor) we used the Friedman rank test (Sheskin 2000), a nonparametric analog of a repeated measure ANOVA.
To assess the contribution of the study populations to the within- and between-population allelic richness, we used the rarefaction-based method of partitioning of allelic diversity introduced recently by Caballero and Rodríguez-Ramilo (2010). Decomposition of allelic richness into within- (A S ) and between-population (D B ) measures allows the computation, by analogy to F ST , of a global allelic diversity A ST = D B /A T , where A T = A S + D B . The relative contribution of a given population to a global within- and between-population allelic diversity was estimated by disregarding this population in the re-estimation of A S , D B and A T parameters (Petit et al. 1998; Caballero and Rodríguez-Ramilo 2010).
Inferences about inbreeding levels were conducted using the method (IIM) implemented in INEst software (Chybicki and Burczyk 2009), which allows the calculation of unbiased estimates for a multilocus average inbreeding coefficient (F IS ) in the presence of null alleles (p n ) (that are typically present in SSR markers used in this study; Dubreuil et al. 2008; González-Martínez et al. 2010; Chybicki et al. 2011).
Each population was also characterized by the coancestry coefficient (θ), which corresponds to the probability that two random genes in a subpopulation are identical by descent due to shared ancestry within a subpopulation (Reynolds et al. 1983). In this study θ was estimated using a model introduced firstly by Balding and Nichols (1995) and then adapted to various applications (e.g. Falush et al. 2003; Beaumont and Balding 2004). The model used here, which follows that in Beaumont and Balding (2004), allows subpopulations to diverge from a common ancestral population with different rates of genetic drift. The rates of genetic drift are measured with the subpopulation-specific parameters that can be interpreted as the coancestry coefficients θ (Balding 2003). The estimation procedure was based on the multinomal-Dirichlet likelihood for the allele counts, which for the i-th subpopulation at the j-th locus equals.
where a ijk denotes the count of the k-th allele at the j-th locus in the i-th population, p jk denotes the frequency of the k-th allele at the j-th locus in the ancestral population, θ i denotes the coancestry coefficient for the i-th population (Balding 2003) and Γ() is the standard gamma function (Abramowitz and Stegun 1965). To estimate θ i we used Metropolis–Hastings algorithm, choosing a uniform (0,1) distribution as a prior for {p jk }, while a truncated exponential distribution X ∈ (0,1) with a mean μ as a prior for {θ i }. We subsequently set μ = 0.01, 0.1, and 0.25. Among these values μ = 0.1 corresponds roughly to the average F ST reported for Taxus species (e.g. Senneville et al. 2001; Myking et al. 2009; Mohapatra et al. 2009; González-Martínez et al. 2010), while two remaining values may represent two possible extremes for the species. The algorithm (described in Appendix) used to estimate θ i was implemented in the Pascal/Delphi computer program BayeF (available from IJC).
It is worth noting that the across-populations average coancestry coefficient estimated as described above is closely related to a global F ST parameter (Holsinger 1999), whereas under the scenario of completely isolated subpopulations, each population-specific θ has a useful interpretation in terms of population-scaled divergence time (t) through −ln(1 − θ) ≈ t /2N e (Reynolds et al. 1983). Although the study populations were assumed to be highly isolated, to verify whether gene flow occured among the study populations we used a Bayesian clustering method. Generally, such methods provide estimates for the number of distinct populations and their genetic homogeneity (through the identification of immigrants) based genetic structure alone. In this tudy we used an algorithm implemented in GENELAND ver. 3.2.4 (Guillot et al. 2008). This method was chosen because, unlike similar methods, it treats a number of clusters (K) as an estimable parameter, enabling straightforward hypothesis testing. The estimation procedure was based on the F-model (assuming dependency of allele frequencies among sub-populations). Because markers are affected by null alleles, the Null Allele model was used. Estimates were obtained after 100,000 iterations (saving every 50th).
Effective population size
To test whether a bottleneck had occurred in the recent population history, BOTTLENECK software (Cornuet and Luikart 1996) was used. Although the step-wise mutation model (SMM) seems theoretically the most relevant for microsatellites, recent studies suggested that the exact SMM may be not optimal in the case of Taxus baccata (González-Martínez et al. 2010). Therefore, the bottleneck test was performed under the assumption of the infinite allele model (IAM), the step-wise mutation model and the intermediate model (30 % multiple-steps mutations and 70 % single-step mutations), so-called the two-phase mutation model (TPM).
Rates of genetic drift were scaled with effective population size (N e ). N e was estimated using two different approaches, both being the single-generation estimators. First we use the estimator based on a linkage disequilibrium measure (r 2, a coefficient of determination reflecting a level of nonrandom associations among alleles from different loci) using the methods implemented in LDNe software (Waples and Do 2008). We denoted the LD-based estimator by N e(LD). As N e(LD) can be biased due to low-frequency alleles (Waples and Do 2010), the values of r 2 were estimated after omitting alleles with a sample frequency lower than three copies. Approximate confidence intervals around the estimated N e were obtained after jackknifing over pairs of loci. Obtained N e estimates are valid under the assumption that linkage disequilibrium is solely an effect of genetic drift. However, because LD can arise due to physical linkage or epistatic selection, marker pairs were checked for homogeneous r 2 across different populations using the Friedman rank test across pair-wise r 2 values (a pair of markers as a factor). N e was also assessed according to the method proposed by Wang (2009), based on the reconstructed proportion of full-, half-sib and unrelated pairs among a cohort of genotyped individuals (hereafter referred to as N e(SA)). These proportions were estimated with a simulated annealing algorithm implemented in Colony 2 (Jones and Wang 2009). Unlike N e(LD), N e(SA) accounts for non-random mating and genotyping errors (Wang and Santure 2009). N e(SA) was estimated with the following settings: the full-likelihood-based analysis (medium likelihood precision and medium length of run), the assumption of polygamy and dioecy. Rates for allelic dropout were substituted with locus-specific estimates of null allele frequencies provided with INEst software (see Genetic structure section). Rates of other error types were arbitrarily fixed at 0.025. Allele frequencies were updated during optimisation. No pair of individuals was a priori excluded as a possible full- or half-sib. Also, sampled individuals were assumed to form a single generation, therefore parent-offspring relations were not allowed. The analysis was initiated without a prior distribution for the paternal and maternal sib-ship sizes of the offspring.
Finally, we studied the relationship between census and effective population sizes and genetic structure parameters: allelic richness, inbreeding and coancestry. The expected number of alleles in a population generally increases nonlinearly with a population size under both the step-wise mutation model (Kimura & Ohta 1975) and the infinite alleles model (Watterson 1975). However, because our estimates of N e were within a relatively narrow range (15–175, see Results), we assumed direct proportionality between a population size and allelic richness. In the case of both coancestry and inbreeding we used N −1 c or N −1 e as a predicting variable, because both quantities are inversely proportional to population size (Wright 1931; Reynolds et al. 1983).
Results
Genetic structure
The average number of alleles detected at five SSR loci equaled 14 per population. However, alleles were mostly detected in low copy numbers, resulting in deflated effective numbers of alleles of only 5.1 on average (Table 2). As expected, the number of alleles detected correlated with sample size. Therefore, to better reflect polymorphism levels in the study populations, allelic richness (AR) values were of primary interest. AR values spanned from 7.8 to 14 with the average of 10.9. The Friedman test revealed that single-locus AR values were not homogeneously distributed across populations (\( \chi_{r}^{2} \) = 15.17, df = 5, p-value = 0.010). When treating single-locus AR values as independent replicates, the approximate 95 % jackknife confidence bounds did not include the average AR value for two populations (i.e. Cisy nad Liswartą and Radomice). Thus, both Cisy nad Liswartą and Radomice can be considered as negatively outlying populations in terms of polymorphism levels. Yet using the same procedure, no population could be identified as a positive outlier.
The contribution of the study populations to within- and between-population allelic richness is shown on Fig. 2. Among studied populations, only Cisy nad Liswartą and Radomice had a negative contribution to the total allelic diversity. This was mainly because of a negative contribution to the within-population component (A S ). Cisy w Czarnem had, by far, the largest contribution, of which both between- and within-population components exceeded the estimates for remaining populations. Cisowa Góra and Cisy Staroploskie also contributed positively to both components of the overall allelic diversity. However, the latter population had a minor impact only. Interestingly, in spite of its positive contribution to A T , Cisy nad Czerską Strugą had a slightly negative impact on between-population allelic diversity. This suggests that the population had relatively small pair-wise differences in allelic composition with respect to the others. Finally, the partitioning method allowed the estimation of the global allelic diversity at the level A ST = 0.297. Thus, about 30 % of the total allelic diversity is between populations.

Partitioning of allelic diversity into within- (A S ) and between-population (D B ) components (note that a total allelic diversity A T = A S + D B ). Bars show relative contribution (in %) of each population into the global A S and D B , while open circles—to the global A T . Positive values indicate a loss of global allelic diversity at a particular level when disregarding the population, and vice versa
Generally, genotypic proportions deviated significantly (p < 0.05) from panmictic expectations, except in three single-locus cases, namely for Tax31 in Cisowa góra, Tax362 in Cisy staropolskie, and Tax26 in Radomice (data not shown). In all cases a deficiency of heterozygotes was found (Table 2). Using INEst software, the analysis revealed that two phenomena contributed unevenly to the excessive homozogosity: null alleles and inbreeding. The average null allele frequency was about 13.3 %. Tax31 appeared the least affected by null alleles (4.2 % on average), while for the others the average null allele frequency ranged between 12.1 and 17 %. ‘Null-free’ estimates of F IS laid within 0.017–0.116 (Table 2), with the grand average 6.7 %. Among the study populations, Cisowa Góra, Cisy nad Czerską Strugą and Cisy nad Liswartą were characterized by higher than average F IS . It is worth mentioning that multilocus F IS estimates obtained with INEst appeared much lower than the classically derived values (i.e. on average 1−H o /H e = 0.33). Hence, inbreeding explained about 20 % of the large heterozygote deficiency observed in the study populations suggesting that it resulted primarily from the presence of null alleles.
The estimates for coancestry coefficients were only weakly dependent on the assumed prior distribution (Table 3), though the results were generally lower for the prior with a mean of 0.01. Individual θ values varied much, with the coefficient of variation between 53.8 and 63.4 %. For the most representative prior (μ = 0.1, see Materials and Methods) θ spanned between 0.04 and 0.222, with the average equal 0.115. Again, two populations were clearly distinguishable from the rest, namely Cisy nad Liswartą and Radomice, both having θ ≈ 0.2 (twice the average).
Using GENELAND we found that six study populations form K = 5 distinct genetic pools. The posterior probability that K > 5 was P = 0.072, while for K < 5 the probability was P ≈ 0. Based on individual ancestry probabilities (Fig. 3) for each population we estimated the overall frequency of genes belonging to one of five clusters. Except for Cisy nad Czerską Strugą, the study populations appeared clearly distinguishable from each other, with >84 % genes belonging to one of five clusters. Cisy nad Czerską Strugą, on the other hand, had the most ambiguous genetic pool with 64 % genes belonging to the cluster most likely referring to the Cisowa Góra population.

Population genetic structure estimated with GENELAND software. Each individual is represented by a vertical line, which is partitioned into five segments reflecting the individual’s assignment to a given cluster. The study populations are separated with black thick lines
Effective population size
The deviance of the expected heterozygosity (H e ) levels from their theoretical values under the drift-mutation equilibrium depended on the mutation model assumed. Under IAM all populations revealed significant heterozygosity excess (the Wilcoxon test; p-value < 0.05). In contrast, neither TPM- nor SMM-based tests showed heterozygosity excess. Under TPM model, however, some signatures of bottleneck were noted for Cisy nad Liswartą and Radomice, where three out of five loci were characterized by excessive H e levels, yet in these cases the overall H e excess was not significant.
Pair-wise linkage disequilibrium coefficients across populations did not deviate from homogeneity as revealed by the Friedman test (\( \chi_{r}^{2} \) = 10.95, df = 9, p-value = 0.279). Hence, no marker combination was specifically suspected for tight linkage or epistatic selection. The LD-based effective population sizes (N e(LD)) ranged from 15.1 to 175.7 (Table 4), with the extremes significantly different from the (harmonic) overall mean 42.5. As compared with N e(LD), the N e(SA) values estimated from reconstructed pedigrees were on average ca. 20 % lower and revealed more homogeneity among populations, ranging between 32 (LI) and 113 (CZ). Nonetheless, N e(LD) and N e(SA) were significantly positively correlated (0.94, p-value = 0.002). Generally, the lowest N e s (averaged for the two estimators) were estimated for those populations, which also exhibited more pronounced bottleneck signatures (RA and LI). The study populations differed strikingly in N e /N c ratio, ranging from 0.03 to 0.4 (with an average of 0.13) and from 0.02 to 0.256 (with an average of 0.11), for N e(LD) and N e(SA), respectively. Thus, association between N c and N e was poor in the study populations regardless the N e estimator.
Population sizes were associated with estimated genetic structure parameters. The majority of variation in the genetic structure parameters was explained by N e (from 51 to 90 %, Fig. 4). A significant positive correlation was found between number of alleles and estimates of effective population size (r = 0.95, p-value = 0.002 and r = 0.94, p-value = 0.003 for N e(LD) and N e(SA), respectively). A similar, yet not significant, relationship was found between N e and allelic richness (r = 0.76, p-value = 0.081 and r = 0.71, p-value = 0.123 for N e(LD) and N e(SA), respectively). We also found a systematic increase in genetic identity, measured with coancestry (θ) and inbreeding (F IS ), as 1/N e increased. In the case of N e × θ relationship correlation coefficients equalled r = 0.81 (p-value = 0.052) and r = 0.79 (p-value = 0.063) for N e(LD) and N e(SA), respectively. In the case of N e × F IS relationship correlation coefficients equaled r = 0.78 (p-value = 0.070) and r = 0.92 (p-value = 0.005) for N e(LD) and N e(SA), respectively. In contrast, census population size (N c ) poorly explained all the above genetic structure characteristics with the values of R 2 ranged from 0.002 to 0.151.

The relationship between census population size (a–b) or effective population size (c–f) and levels of polymorphism (A, average number of alleles; AR, average allelic richness) and genetic identity (FIS, inbreeding coefficient; θ, coancestry). Lines show the best fitting linear regression functions
Discussion
The study populations of English yew were found to differ in genetic structure parameters. For example, allelic richness spanned from 3.8 to 7.4, while inbreeding coefficient and average coancestry reached values from 0.017 to 0.116 and from 0.04 to 0.222, respectively. We found that all these features were poorly related with census numbers. In contrast, marker-assisted characteristics such as effective population size and bottleneck signatures were found to explain the observed genetic structure.
Up to now several studies showed that some inbreeding might be present in yew populations, although F IS estimates varied much among populations (Lewandowski et al. 1995; Myking et al. 2009; Dubreuil et al. 2010; Chybicki et al., 2011). Our results with the average F IS = 0.067 were close to those reported for Norway populations (Myking et al. 2009) but lower than those reported by Dubreuil et al. (2010). This suggests that southern yew populations, at least in the Montseny Mountains, might be subject to more intense inbreeding. However, F IS estimates reported for populations in the Montseny Mountains did not account for null alleles and thus might be severely overestimated (Chybicki and Burczyk 2009). Generally, given dioecy of the species, even low inbreeding levels suggest that strong bi-parental inbreeding occurs routinely in this species. A clear kinship structure found within populations may facilitate mating between closely related individuals (Chybicki et al., 2011), given that pollen dispersal is limited. Nonetheless, to our knowledge, rates of pollen-mediated gene flow and mating patterns have not yet been directly studied in English yew. The only results are probably those by Lewandowski et al. (1995), based on seeds collected from 15 female trees in the Cisy Staropolskie reserve. However, they do not provide a strong support for the hypothesis of non-random mating, highlighting the need for additional studies that address the issue more directly.
The study populations appeared to have elevated coancestry coefficients. It is generally acknowledged that coancestry results from limited gene immigration, when the probability of sharing a common ancestry evolves over generations (Reynolds et al. 1983). Results of GENELAND analysis revealed that all the study populations, except Cisy nad Czerską Strugą, are characterized by distinct genetic pools. This finding supports the hypothesis about strong isolation of yew populations. In the case of Yew, the isolation is directly related with the fact that their populations in Poland are typically separated by large distances. Pollen might experience difficulty in traveling large distances in spite of being wind-dispersed and possesing a relatively low terminal velocity (Dyakowska 1959). For pollen to be dispersed over long distances, movement to higher altitudes is generally required (Di-Giovanni et al. 1996). This poses a significant obstacle for typical understory species, such as English yew, as the presence of a closed canopy can substantially reduce wind speed and convective currents needed for long distance pollen dispersal. Moreover, one can predict that together with an increase of forest density, wind speed can decrease enough to severely restrict pollination within a population. Cisy Staroploskie is a particularly good example of such a situation. The study on pollen dispersal conducted there with the use of pollen traps revealed that pollen grain density decreases rapidly together with distance from a male individual (Noryśkiewicz 2006). This phenomenon certainly facilitates a development of inbreeding by reduction of effective population size, as discussed in our previous paper (Chybicki et al., 2011).
In contrast with remaining populations, Cisy nad Czerską Strugą had a relatively low estimate of coancestry, given that it has the lowest census number among all study populations. However, GENELAND analysis showed that individuals belonging to Cisy nad Czerską Strugą did not cluster as a separate genetic pool, and instead they formed a mixture of different populations. Interestingly, about 64 % of genes clustered with Cisowa Góra located about 370 km away. Such a high admixture would be difficult to explain in this case unless we assume that Cisy nad Czerską Strugą was established artificially with a use of seedlings of various origin. In this way we could address the issues of low coancestry in the population coupled with relatively high allelic richness, which has support in neither census nor even effective population size. High heterogeneity of the genetic pool of Cisy nad Czerską Strugą was also in agreement with the partitioning of allelic diversity. The contribution of between-population allelic diversity was in this case negative because the population shares many alleles with the others.
In the face of strong fragmentation, high isolation of yew populations allow them to maintain many unique alleles. Our results showed that four out of six study populations had a positive contribution to the total allelic diversity (see Fig. 2). Interestingly, the two populations that had a negative contribution (RA and LI) also displayed signatures of a possible bottleneck in their recent history. This rapid reduction in population size could explain these partitioning patterns. However, using genetic markers alone, we could not verify whether bottlenecked populations experienced a natural process of rapid reduction in size or rather they were established with human assistance from seeds derived from a few female individuals in the recent past. Unfortunately, the history of these populations before the present generation is not known precisely, making factors of a putative bottleneck effect rather speculative.
Wind-pollination can generally facilitate the attainment of a large population size, though it can be influenced by a level of population fragmentation (Bucci et al. 1997; De-Lucas et al. 2009) interfering with a subpopulation density (Grivet et al. 2009). Nonetheless, the values of effective population size estimated in this study were generally lower than those observed for trees forming large continuous populations (e.g. Bucci et al. 1997; Chybicki et al. 2008). We also found a poor correspondence between census and effective population size suggesting that different populations have different demographic processes, or different demographic histories. For example, incidental bottlenecks and different sex proportions might be responsible for N e /N c heterogeneity. Under random mating the expected N e /N c ratio equals 4r f (1−r f ), where r f is the proportion of females. For the study population, one might then expect N e /N c between 88.6 and 99.6 % (based on data in Table 1), while our N e /N c estimates were within 3.2–40.2 % or even 2.4–25.9 %, depending on the estimation method. This clearly shows that biased sex proportions alone cannot explain the poor relationship between census and effective number. One reason for such discrepancy would be some variation in a fraction of sexually active individuals in populations. It is known that the production of reproductive organs increases strongly with tree size and canopy openness (Svenning & Magård 1999). Also, femaleness is dominant in the open, while maleness in shade. This could explain relatively high N e /N c for Cisy w Czarnem, which occurs at the most open habitat compared with the others. For comparison, Cisy Staropolskie represents the opposite extreme, with a high density and a well developed canopy layer. Among additional factors, non-random mating patterns (as discussed earlier), fluctuating population size, population substructure (see Dubreuil et al. 2010, Chybicki et al. 2011) or overlapping generations could be attributed to deflated N e values. Overlapping generations needs particular attention, as this feature might potentially influence both a demographic parameter (i.e. N e ) and its estimate (both N e(LD) and N e(SA) relied on the assumption of a single-generation cohort). English yew is know for longevity, that makes a possibility of mixing generations in natural populations. Therefore, although our study is based on samples of adult individuals, we cannot definitely exclude a possibility that, though to limited degree, our samples are a mixture of two generations. Although the two methods gave well correlated estimates, the values of N e(SA) were on average lower than those of N e(LD). Generally, the above-discussed factors could be responsible for discrepancies between the two N e estimators. Nonetheless, it should be noted that N e(SA) was found to be negatively biased when information content from genetic markers is low (here only five loci) (Wang 2009). On the other hand, low frequency alleles could cause overestimated N e(LD) (although we applied the heuristic correction, as suggested by Waples and Do 2010, see M&M). Interestingly, similar trends were also noted in the recent study of ranid frogs (Phillipsen et al. 2011), suggesting that further attention is needed to determine susceptibility of a particular method to an information content of genetic markers.
We found high correlation between coancestry and 1/N e estimates. It is worth mentioning that the correlation is not explicitly obvious. Actually, these two measures were estimated from very different data features, with θ being of single-locus (standardized) variances of allele frequencies, while N e —a function of between—loci allelic correlations (LD) or a function of inter-individual relationships (SA) (however, all these features are related to the probability of identity by descent, Sved 1971, Balding 2003). Furthermore, both N e estimators refer to inbreeding effective population size, while θ has affinity with variance effective population size (Lindgren and Mullin 1998). These two differently defined N e s can vary to a large extent unless a population is of constant size (Crow and Kimura 1970). Thus, the lack of complete correlation between 1/N e and θ might be accountable (besides the low precision of estimates) to differences in demographic history of the populations, including migration rates, divergence times and even initial coancestry and linkage disequilibrium of the isolates.
Finally we would like to address the result of allelic diversity through A ST . Recently, simulation studies have revealed that allelic diversity and gene differentiation measures can provide complementary information about demographic processes (Caballero and Rodríguez-Ramilo 2010). In particular, A ST and F ST behave differently in the case of low and high migration rates. For the study populations A ST was about three times higher than the average coancestry coefficient (which can serve as an estimate of global F ST , Holsinger 1999). Such a pronounced difference was due to the presence of many private alleles at low frequencies, which have a strong impact on A ST while negligible on F ST . Asymptotically, an A ST greater than F ST is predicted for high migration rates (Caballero and Rodríguez-Ramilo 2010). In our case, however, it is a questionable scenario, because populations seem almost completely disconnected due to separation by large distances. One reasonable explanation could be related to the observation, that A ST approaches stationarity much faster than F ST (Caballero and Rodríguez-Ramilo 2010), especially when migration is incidental. Another possible explanation could be high mutation rates of microsatellite markers (μ > 0.0001), that would strongly enhance allelic diversity but only weakly gene differentiation.
Perspectives for conservation
Currently, conservation biologists in Poland face the problem of the restitution of English yew as a component of the forest understory. However, because existing populations are strongly fragmented and typically small, the question remains, which populations should serve as main seedling sources? Two factors should be considered here. Firstly, population characteristics can determine present-day seedling viability, which is important for the effectiveness of re-establishment. Secondly, population characteristics can determine genetic quality of seedlings in terms of polymorphism, inbreeding, and genetic diversity levels. All these features are crucial from the perspective of long-term conservation of species’ gene pools (Mistretta 1994; Ellstrand and Elam 1993; Frankham et al. 2002; Willi et al. 2006; Charlesworth and Willis 2009).
Our study suggested that choosing source populations for ex situ conservation based on their size only can be fairly misleading. For example, Radomice and Cisy nad Liswartą, two relatively large yew populations, both had a negative contribution to the total allelic richness, which means that excluding these populations in ex situ conservation programs would cause a gain in the total allelic richness in the next generation. Interestingly, the largest Polish population Cisy Staropolskie had almost null contribution to all components of allelic diversity. In contrast, the relatively small population of Cisy nad Czerską Strugą had a positive contribution to the total allelic richness. These conclusions could also be extended to coancestry or inbreeding. It is evident that inbreeding would not be minimized efficiently when seedlings were derived from populations chosen based on the census number only. It is worth noting that current-generation coancestry coefficients can shed light on future inbreeding levels within a population. It is known that, under random mating, the total (or group) coancestry of a population becomes inbreeding of the progeny. Thus, based on the results we can predict that all the study populations, except Cisy nad Czerską Strugą, will probably be subject to an increase of inbreeding in the next generation, but the rates of increase will be poorly related with census population numbers.
Expectedly, allelic richness and inbreeding were functions of effective and not census population size. Although, generally the N e /N c ratio was about 0.1 and hence corresponded well with the findings for natural populations (Frankham 1995), N e /N c appeared to vary much from population to population, making indirect inferences about N e problematic. Incorporating data on sex ratio or population density was ineffective and did not improve inferences, as discussed above. Further studies are required to resolve whether more precise predictions of the N e /N c ratio could be obtained with a number of additional characteristics, such as age structure and fecundity distribution. However, one can anticipate that information on the current demographic structure would be useful if the population is demographically stable. If not, historical factors could dominate over present-day processes making predictions invalid. Our study demonstrated that the use of neutral genetic markers would be helpful to determine demographic parameters of conservation units. However, neutral markers could not enable direct insights into adaptive potential of populations, which might do not correspond with polymorphism and inbreeding levels. Hence, ex situ conservation based solely on the predictions drawn from neutral markers might lead to negative consequences. For example, if isolated populations are well adopted through specific gene combinations, progeny populations derived as a mixture might suffer from outbreeding depression. This need to be taken into account in conservation programs. Nevertheless, the use of neutral genetic markers in ex situ programs should not be underestimated.
References
Abramowitz M, Stegun IA (1965) Handbook of mathematical functions with formulas, graphs, and mathematical tables. Dover Publications, New York
Balding DJ (2003) Likelihood-based inference for genetic correlation coefficients. Theor Pop Biol 63:221–230
Balding DJ, Nichols RA (1995) A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica 96:3–12
Beaumont MA, Balding DJ (2004) Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol 13:969–980
Bucci G, Vendramin GG, Lelli L, Vicario F (1997) Assessing the genetic divergence of Pinus leucodermis Ant. endangered populations: use of molecular markers for conservation purposes. Theor Appl Genet 7:1138–1146
Caballero A, Rodríguez-Ramilo ST (2010) A new method for the partition of allelic diversity within and between subpopulations. Conserv Genet 11:2219–2229
Caballero A, Toro MA (2000) Interrelations between effective population size and other pedigree tools for the management of conserved populations. Genet Res 75:331–343
Charlesworth D, Willis JH (2009) The genetics of inbreeding depression. Nat Rev Genet 10:783–796
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113
Chybicki IJ, Dzialuk A, Trojankiewicz M, Slawski M, Burczyk J (2008) Spatial genetic structure within two contrasting stands of scots pine (Pinus sylvestris L.). Silvae Genet 57:193–202
Chybicki IJ, Oleksa A, Burczyk J (2011) Increased inbreeding and strong kinship structure in Taxus baccata estimated from both AFLP and SSR data. Heredity 107:589–600
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Crow JF, Kimura M (1970) An introduction to population genetics theory. Harper & Row Publishers, New York
De-Lucas AI, González-Martínez SC, Vendramin GG, Hidalgo E, Heuertz M (2009) Spatial genetic structure in continuous and fragmented populations of Pinus pinaster Aiton. Mol Ecol 18:4564–4576
Di-Giovanni F, Kevan PG, Arnold J (1996) Lower planetary boundary layer profiles of atmospheric conifer pollen above a seed orchard in northern Ontario, Canada. For Ecol Manag 83:87–97
Doyle J, Doyle J (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Dubreuil M, Sebastiani F, Mayol M, González-Martínez S, Riba M, Vendramin G (2008) Isolation and characterization of polymorphic nuclear microsatellite loci in Taxus baccata L. Conserv Genet 9:1665–1668
Dubreuil M, Riba M, González-Martínez SC, Vendramin GG, Sebastiani F, Mayol M (2010) Genetic effects of chronic habitat fragmentation revisited: strong genetic structure in a temperate tree, Taxus baccata (Taxaceae), with great dispersal capability. Am J Bot 97:303–310
Dyakowska J (1959) Podręcznik palinologii. Wydawnictwa Geograficzne, Warszawa (in Polish)
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: implications for plant conservation. Annu Rev Ecol Syst 24:217–242
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Frankham R (1995) Conservation genetics. Annu Rev Genet 29:305–327
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
González-Martínez SC, Dubreuil M, Riba M, Vendramin GG, Sebastiani F, Mayol M (2010) Spatial genetic structure of Taxus baccata L. in the western Mediterranean Basin: past and present limits to gene movement over a broad geographic scale. Mol Phylog Evol 55:805–815
Grivet D, Robledo-Arnuncio JJ, Smouse PE, Sork VL (2009) Relative contribution of contemporary pollen and seed dispersal to the effective parental size of seedling population of California valley oak (Quercus lobata, Née). Mol Ecol 18:3967–3979
Guillot G, Santos F, Estoup A (2008) Analysing georeferenced population genetics data with Geneland: a new algorithm to deal with null alleles and a friendly graphical user interface. Bioinformatics 24:1406–1407
Hilfiker K, Holderegger R, Rotach P, Gugerli F (2004) Dynamics of genetic variation in Taxus baccata: local versus regional perspectives. Can J Bot 82:219–227
Hoff PD (2009) A first course in Bayesian statistical methods. Springer, New York
Holsinger KE (1999) Analysis of genetic diversity in geographically structured populations: a Bayesian perspective. Hereditas 130:245–255
Huang CC, Chiang TY, Hsu TW (2007) Isolation and characterization of microsatellite loci in Taxus sumatrana (Taxaceae) using PCR-based isolation of microsatellite arrays (PIMA). Conserv Genet. doi:10.1007/s10592-0079341-z
Hulme PE (1996) Natural regeneration of yew (Taxus baccata L.): microsite, seed or herbivore limitation? J Ecol 84:853–861
Iszkuło G, Boratyński A (2006) Analysis of the relationship between photosynthetic photon flux density and natural Taxus baccata seedlings occurrence. Acta Oecol 29:78–84
Iszkuło G, Jasińska AK, Giertych MJ, Boratyński A (2009) Do secondary sexual dimorphism and female intolerance to drought influence the sex ratio and extinction risk of Taxus baccata? Plant Ecol 200:229–240
Kimura M, Ohta T (1975) Distribution of allelic frequencies in a finite population under stepwise production of neutral alleles. Proc Natl Acad Sci USA 72:2761–2764
Lewandowski A, Burczyk J, Mejnartowicz L (1995) Genetic structure of English yew (Taxus baccata L.) in the Wierzchlas reserve: implications for genetic conservation. For Ecol Manag 73:221–227
Lindgren D, Mullin TJ (1998) Relatedness and status number in seed orchard crops. Can J For Res 28:276–283
Mistretta O (1994) Genetics of species re-introductions: applications of genetic analysis. Biodivers Conserv 3:184–190
Mohapatra KP, Sehgal RN, Sharma RK, Mohapatra T (2009) Genetic analysis and conservation of endangered medicinal tree species Taxus wallichiana in the Himalayan region. New For 37:109–121
Montgomery ME, Ballou JD, Nurthen RK, England PR, Briscoe DA, Frankham R (1997) Minimizing kinship in captive breeding programs. Zoo Biol 16:377–389
Myking T, Vakkari P, Skrøppa T (2009) Genetic variation in northern marginal Taxus baccata L. populations. Implications for conservation. Forestry 82:529–539
Mysterud A, Østbye E (2004) Roe deer (Capreolus capreolus) browsing pressure affects yew (Taxus baccata) recruitment within nature reserves in Norway. Biol Conserv 120:545–548
Noryśkiewicz M (2006) Historia cisa w okolicy Wierzchlasu w świetle analizy pyłkowej. Wydawnictwo Uniwersytetu Mikołaja Kopernika w Toruniu, Toruń (in Polish)
Petit RJ, El Mousadik A, Pons O (1998) Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12:844–855
Phillipsen IC, Funk WC, Hoffman EA, Monsen KJ, Blouin MS (2011) Comparative analyses of effective population size within and among species: ranid frog as a case study. Evolution 65:2927–2945
Pierson SAM, Keiffer CH, McCarthy BC, Rogstad SH (2007) Limited reintroduction does not always lead to rapid loss of genetic diversity: an example from the American chestnut (Castanea dentata; Fagaceae). Restor Ecol 15:420–429
Reynolds J, Weir BS, Cockerham CC (1983) Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics 105:767–779
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Res 8:103–106
Saura M, Pérez-Figueroa A, Fernández J, Toro M, Caballero A (2008) Preserving population allele frequencies in ex situ conservation programs. Conserv Biol 22:1277–1287
Senneville S, Beaulieu J, Daoust G, Deslauriers M, Bousquet J (2001) Evidence for low genetic diversity and metapopulation structure in Canada yew (Taxus canadensis): considerations for conservation. Can J For Res 31:110–116
Sheskin DJ (2000) Handbook of parametric and nonparametric statistical procedures. Chapman and Hall/CRC, Boca Raton
Sved JA (1971) Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor Pop Biol 2:125–141
Svenning JC, Magård E (1999) Population ecology and conservation status of the last natural population of English yew Taxus baccata in Denmark. Biol Conserv 88:173–182
Thomas PA, Polwart A (2003) Taxus baccata L. J Ecol 91:489–524
Waples RS, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Res 8:753–756
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Pop Biol 7:256–276
Willi Y, Van Buskirk J, Hoffmann AA (2006) Limits to the adaptive potential of small populations. Annu Rev Ecol Evol Syst 37:433–458
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Zarek M (2009) RAPD analysis of genetic structure in four natural populations of Taxus baccata from Southern Poland. Acta Biol Cracov S Bot 51:67–75
Acknowledgments
This study was supported by the research grant from the Polish Ministry of Science and The Higher Education (NN304 4216 33) to IJC. The authors would like to thank Maria Jennifer Sjölund as well as two anonymous reviewers for making valuable suggestions that improved the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Here we explain details of the Metropolis–Hastings algorithm used to estimate coancestry coefficients. In general, the algorithm subsequently proposes an update for each parameter, that is conditioned on remaining parameters of the model and the data. As a result a Markov Chain is built-up that provides an approximation of the joint posterior distribution, from which parameter estimates can be easily extracted (Hoff 2009).
Let us recall that the estimation procedure assumed that allelic counts within subpopulations follow the multinomial-Dirichlet distribution (Eq. 1). Because allele frequencies in the i-th subpopulations are integrated out, only frequencies in the ancestral population are to be estimated along with coancestry coefficients. We denote a set of subpopulation coancestry coefficients by {θ i } (where i = 1…N, N—number of subpopulations) and an array of ancestral allele frequencies by {p jk } (where j = 1…L and k = 1…A j , L—number of loci, A j —number of alleles at the j-th locus). We also assumed the truncated exponential distribution (X ∈ 0…1 with a mean μ) as a prior for θ i and a uniform distribution as a prior for {p jk }. The algorithm started with initial values of {θ i } and {p jk } equal to means of their prior distributions. Then, update for each θ i was obtained as follows. A new value of θ * i was drawn from a flat distribution centered on the current θ i with a range d (d was adjusted to reach an acceptance rate within 25–45 %). θ * i was accepted with the probability.
where L(…) is the likelihood function given by Eq. 1. Ancestral allele frequencies were updated similarly as in Falush et al. (2003). For the j-th locus, two integers m and n were drawn such that m ≠ n (m,n ∈ 1…A j ). A new value of p * jm was drawn from a flat distribution centered on the current p jm with a range e (e was adjusted to reach an acceptance rate within 25–45 %). Then, a new value of p * jn was computed as p * jn = p jn + (p jm −p * jm ). If either p * jm and/or p * jm felt outside (0…1) interval, the proposed values were rejected. Otherwise, they were accepted with the probability.
where L(…) is again the likelihood function given by Eq. 1.
The joint posterior distribution was approximated with 50,000 iterations, saving each 50th update. To estimate marginal distributions for {θ i } and {p jk }, first 10,000 updates were disregarded to avoid a dependence of final estimates on the initial values.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chybicki, I.J., Oleksa, A. & Kowalkowska, K. Variable rates of random genetic drift in protected populations of English yew: implications for gene pool conservation. Conserv Genet 13, 899–911 (2012). https://doi.org/10.1007/s10592-012-0339-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-012-0339-9