
Abstract
Chloride and pseudohalide (N3 −, NCS−) hydride-carbonyl ruthenium(II) complexes with 4-pyrrolidinopyridine as co-ligand were synthesized and characterized by IR, 1H, and 31P NMR, electronic absorption and emission spectroscopy and X-ray crystallography. The electronic structures of the complexes were calculated by density functional theory (DFT) on their crystal structures. The spin-allowed singlet–singlet electronic transitions of the complexes were calculated by time-dependent DFT, and the UV–Vis spectra have been discussed on these basis. The emission properties of the complexes were also studied.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pyridine ligands have energetically low-lying π-antibonding orbitals, which can accept electrons from the occupied d orbitals of metal atoms. Metal complexes with pyridine ligands can exhibit charge transfer bands with interesting spectroscopic properties in the visible region [1]. Hence, ligands containing pyridine rings have been widely studied, and their σ-donor and π-acceptor properties are often interesting. Their combination with other donor atoms should in principle afford complexes with tunable spectroscopic properties [2]. 4-Pyrrolidinopyridine (py-4P) is a stronger electron donor N-heteroaromatic ligand compared with pyridine (py-4P pKa = 18.33; pyridine pKa = 12.53) [3]. Hence, 4-pyrrolidinopyridine should be interesting as a ligand, but reports on this topic are rather scarce. The py-4-P ligand has been found to stabilize Zn–Zn bonded complexes [4, 5], five-coordinate zirconium(IV) and titanium(IV) complexes [6, 7] and iridium, hafnium, neodymium, and iron complexes with 4-pyrrolidinopyridine [8–11] have also been reported. Moreover, ruthenium and osmium catalysts containing 4-pyrrolidinopyridine have been claimed in several patents [12–14].
On the other hand, it is known that thiocyanate ligands tune the t 2g ruthenium orbitals by distributing the 4d Ru energy levels over a wide energy range, due to mixing with orbitals centered on the NCS ligand (2p N, 2p C and 3p S) [15]. The calculated density of states showed that both inter- and intramolecular interactions are important and can significantly influence the orbital composition in the frontier electronic structure. The N3 − ligand, which is similar in properties to thiocyanate, should exhibit comparable characteristics. Thus, studies of the electronic structures of these complexes are an important area of chemistry.
The complexes reported in this paper combine our interest in ruthenium coordination compounds and complexes containing pyridine derivative ligands [16–20]. We describe an experimental and quantum chemical study of ruthenium hydride-carbonyl chloride, isothiocyanate, and azide complexes with 4-pyrrolidinopyridine as co-ligand. As well as the syntheses and spectroscopic (1H, 31P NMR, IR) characterizations, the X-ray crystal structures and photophysical properties of the complexes are presented. The quantum chemical study included a characterization of the molecular and electronic structures of the complexes by analysis of the optimized molecular geometries and electronic populations using the natural bond orbitals scheme. The latter was also used to identify the nature of the interactions between the ligands and the metal. Finally, time-dependent density functional theory (TD-DFT) was used to calculate and interpret the electronic absorption spectra.
Experimental
All reagents used for the syntheses of the complexes were commercially obtained and were used without further purification. The starting complex [RuHCl(CO)(PPh3)3] was synthesized according to the literature method [21].
Synthesis of [RuHX(CO)(py-4-P)(PPh3)2] (X = Cl; N3; NCS)
The complexes were synthesized by reaction of [RuHCl(CO)(PPh3)3] (0.1 g, 1 × 10–4 mol), 4-pyrrolidinopyridine (0.015 g, 1 × 10–4 mol; py-4-P) (1), and sodium azide (0.007 g, 1 × 10–4 mol) (2) or ammonium thiocyanate (0.008 g, 1 × 10–4 mol) (3) in methanol solution (100 cm3). In each case, the mixture was refluxed in methanol for 4 h, then cooled and filtered. Crystals suitable for X-ray crystal analysis were obtained by slow evaporation of the filtrates.
Complex (1) ([RuHCl(CO)(py-4-P)(PPh3)2]): Yield 63 %. IR (KBr; cm−1): 2,058 (w) ν(Ru–H); 1,915 (s) ν(CO); 1,611, 1,528 (m) ν(C=N; C=C). UV–Vis (methanol; log ε; nm): 341 (1.58), 276 (3.12), 240 (3.74), 208 (4.89). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.32 (d, J = 7.2 Hz, py), 8.16 (d, J = 7.1 Hz, py), 7.95–7.15 (m, PPh), 6.25 (d, J = 6.9 Hz, py), 5.92 (d, J = 6.3 Hz, py), 5.74 (d, J = 7.2 Hz, py), 3.28 (s, pyrrolidine), 3.22 (dd, J = 32.4, 5.6 Hz, pyrrolidine), 2.19 (s, pyrrolidine), 1.62 (s, pyrrolidine), −4.45 (t, J = 19.4 Hz, 1H). 31P NMR (162 MHz, CDCl3): δ (ppm) 39.12 (s, PPh3).
Complex (2) ([RuH(N3)(CO)(py-4-P)(PPh3)2]): Yield 65 %. IR (KBr; cm−1): 2,052 (s) νN3; 1,939, 1,919 (w, s) ν(Ru–H)/ν(CO); 1,617, 1,572 (m) ν(C=N; C=C); 701 (s) δN3. UV–Vis (methanol; log ε; nm): 339 (1.92), 274 (3.47), 208 (4.96). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.83–6.85 (m, PPh3, py), 3.11 (d, J = 37.4 Hz, pyrrolidine), 2.19 (s, pyrrolidine), 1.62 (s, pyrrolidine), 1.34 (s, pyrrolidine), −7.17 (dt, J = 104.4, 24.8 Hz, HRu). 31P NMR (162 MHz, CDCl3) δ (ppm) 40.10 (d, J = 15.7 Hz. PPh3).
Complex (3) ([RuH(NCS)(CO)(py-4-P)(PPh3)2].CH3OH): Yield 68 %. IR (KBr; cm−1): 2,104 (s) νNCS; 2,004, 1,944 (w, m) ν(Ru–H)/ν(CO); 1,615, 1,585 (m) ν(C=N; C=C); 742, 694 (m) ν(SC from SCN); 519 (m) δ(NCS). UV–Vis (methanol; log ε; nm): 329 (1.99), 2,589 (3.62), 211 (4.92). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.84–7.00 (m, py, PPh3), 6.96 (t, J = 8.4 Hz, py), 3.04 (d, J = 6.7 Hz, pyrrolidine), 2.01 (d, J = 13.3 Hz, pyrrolidine), 1.63 (d, J = 12.1 Hz, pyrrolidine), 1.26 (d, J = 11.1 Hz, pyrrolidine), −7.18 (dt, J = 100.0, 24.4 Hz, HRu). 31P NMR (162 MHz, CDCl3) δ (ppm) 39.40 (d, J = 15.4 Hz).
Physical measurements
Infrared spectra were recorded on a Perkin–Elmer spectrophotometer in the range 4,000–450 cm−1 using KBr pellets. Electronic spectra were measured on a Lab Alliance UV–VIS 8500 spectrophotometer in the range of 600–180 nm in methanol solution. The 1H and 31P NMR spectra were obtained at room temperature in CDCl3 using a Bruker 400 MHz spectrometer. Luminescence measurements were taken in methanol solutions on an F-2500 FL spectrophotometer at room temperature.
Computational methods
The calculations were made using the Gaussian 09 [22] program. Molecular geometries of the singlet ground state of complexes (1), (2), and (3) were fully optimized in the gas phase at the B3LYP level of theory. [23, 24] For each complex, a frequency calculation was made, verifying that the optimized molecular structure corresponded to an energy minimum; thus, only positive frequencies were found. The DZVP basis set [25] with f functions with exponents 1.94722036 and 0.748930908 was used to describe the ruthenium atom, and the basis set used for the lighter atoms (C, N, O, P, H) was 6-31G with a set of d and p polarization functions. The TD-DFT method [26] was employed to calculate the electronic absorption spectra of the complexes using the solvent polarizable continuum model (PCM). In this work, 100 singlet excited states were calculated as vertical transitions for each complex. A natural bond orbital (NBO) analysis was also made for each of the complexes, using the NBO 5.0 package [27] included in Gaussian 09. Natural bond orbitals are orbitals localized on one or two atomic centers that describe molecular bonding in a manner similar to a Lewis electron pair structure, and they correspond to an orthonormal set of localized orbitals of maximum occupancy. NBO analysis provides the contribution of the atomic orbitals (s, p, d) to the NBO σ and π hybrid orbitals for bonded atom pairs. In this scheme, three NBO hybrid orbitals are defined, namely bonding orbital (BD), lone pair (LP), and core (CR), which were analyzed for the atoms directly bonded to, or presenting some kind of interaction with, the ruthenium atom. The contribution of a group (ligands, metal center) to a molecular orbital was calculated using Mulliken population analysis. GaussSum 2.2 [28] was used to calculate group contributions to the molecular orbitals and to prepare the partial density of states (DOS) spectra. The DOS spectra were created by convoluting the molecular orbital information with Gaussian curves of unit height and FWHM (full width at half maximum) of 0.3 eV.
Crystal structure determination and refinement
The crystals of [RuHCl(CO)(py-4-P)(PPh3)2] (1), [RuH(N3)(CO)(py-4-P)(PPh3)2] (2), and [RuH(NCS)(CO)(py-4-P)(PPh3)2] (3) were mounted in turn on an Xcalibur, Atlas, Gemini Ultra Oxford Diffraction automatic diffractometer equipped with a CCD detector, and used for data collection. X-ray intensity data were collected with graphite monochromated MoK α radiation (λ = 0.71073 Å) at a temperature of 295.0(2) K, with ω scan mode. Ewald sphere reflections were collected up to 2θ 50.10. The unit cell parameters were determined from least-squares refinement of the setting angles of 6,947, 15,306, and 10,064 strongest reflections for complexes (1), (2), and (3), respectively. Details concerning crystal data and refinement are gathered in Table 1. Lorentz, polarization, and empirical absorption corrections using spherical harmonics implemented in the SCALE3 ABSPACK scaling algorithm [29] were applied. The structures were solved by the Patterson method and subsequently completed by difference Fourier recycling. All the non-hydrogen atoms were refined anisotropically using full-matrix, least-squares techniques. Bearing in mind the limits of Fourier synthesis and the problems in recognizing artifacts in the immediate neighborhood of heavy atoms, it is doubtful if a reliable position for the hydrogen atom bound to the Ru atom can be found in the difference Fourier map while avoiding the danger of mistaking the effects of the series termination errors for a true atomic position. In the studied complexes, the Ru–H bond length of 1.50 Å is normal. The Olex2 [30] and SHELXS97, SHELXL97 [31] programs were used for all the calculations. Atomic scattering factors were incorporated in the computer programs.
Results and discussion
Spectroscopic characterization
In the 1H NMR spectra of the complexes, as well as signals corresponding to the PPh3, and 4-pyrrolidinopyridine ligands, there are signals at high field indicating the presence of the hydride ligands. The chemical shifts of these signals are due to the shielding effect of the metal and to the partial charge of the hydrogen atom. The Ru–H signals are observed as a triplet in (1) and doublet of triplets in (2) and (3) with J HP ~100 and 20 Hz. Even if (2) and (3) were asymmetric with inequivalent phosphines, a doublet of doublets would have been expected, but the asymmetry could be partially removed on the NMR timescale. The signals are observed at −4.45, −7.17, and −7.18 ppm for complexes (1), (2), and (3), respectively, and the differences are connected with the increasing π-acceptor properties in the chloride, nitride, and isothiocyanate ligands. The 31P NMR spectra of all three complexes show signals close to 40 ppm. The signals are doublets in the case of complexes (2) and (3) which suggests two triphenylphosphine groups, not in perfect trans positions. In the 31P spectrum of complex (1), the observed singlet may be caused by electronic interactions (π–π stacking) between PPh3 phenyl and pyridine rings.
The IR spectra of the complexes show strong bands at 1,934–2,004 and 1,915–1,944 cm−1, assigned to the Ru–H and C≡O stretching bands (see the selected IR frequencies given in Experimental section). The differences in the maxima of these bands are connected with the different (pseudo)halides present in the coordination sphere. The electron–donor hydride ligand delivers electron density via backbonding to the antibonding orbitals of the CO, resulting in a decrease in the frequency of the CO vibration. However, the acceptor properties of the (pseudo)halide ligands vary as Cl− < N3 − < NCS− and the positions of the νRu–H and νCO bands in the azide complex (2) suggest that this pseudohalide anion exerts a much weaker effect than isothiocyanate in these complexes. This is supported by the theoretically determined charge values which indicate charges on the Ru(II) centers of −0.905, −0.844, and −0.855 in (1), (2) and (3), respectively. The charges on the hydride ligands are close to zero, being 0.074, 0.036 and 0.021 in complexes (1), (2), and (3), respectively. The charges on the CO ligands, calculated by summing the individual charges on the carbon and oxygen atoms, are 0.213 (1), 0.198 (2), and 0.229 (3). Hence, in the complex (2), the charges on ruthenium and the carbonyl ligand are the smallest, and this is in accordance with the largest decrease in CO vibration frequency for this complex. The natural charges on the chloride, azide, and isothiocyanate ligands of −0.553, −0.602, and −0.663, respectively, are in accordance with their acceptor properties.
The stretching vibrations for N3 − and S=C=N− are observed at 2,052 and 2,104 cm−1, respectively. The coordination mode of thiocyanate in complex (3) cannot be determined from the IR spectrum. For N-bonded complexes, generally the C–N stretching band is in a lower region, around 2,050 cm–1, compared with 2,100 cm–1 for S-bonded complexes. However, the frequencies of the bands are sensitive to other factors such as co-ligands; hence, the structures of these complexes were determined by X-ray analysis. While the M–S–C angles of S-bonded thiocyanato ligands in such complexes are bent at around 110°, the M–N–C angles of N-bonded isothiocyanato ligands are close to linear. The Ru(1)–N(1)–C(1) angle in complex (3) is 174.7(2)°, indicating the isothiocyanato ligand.
Molecular structures
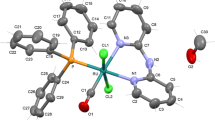
Crystals of the complexes suitable for single crystal X-ray analyses were obtained by slow evaporation of the reaction mixtures. Complexes (1) and (3) crystallize in monoclinic P21/c and P21/n space groups and (2) in triclinic P-1. The azide complex (2) has two independent molecules in the asymmetric unit, while complex (3) crystallizes as a solvate with one methanol molecule. The molecular structures of the complexes are displayed as ORTEP representations in Fig. 1, and selected bond distances and angles are collected in Table 2. The Ru(1)–N(1) bond lengths in the complexes are normal and comparable with other ruthenium hydride complexes with pyridine derivative ligands [17, 18].

ORTEP drawings of [RuHCl(CO)(py-4-P)(PPh3)2] (1), [RuH(N3)(CO)(py-4-P)(PPh3)2] (2) and [RuH(NCS)(CO)(py-4-P)(PPh3)2].CH3OH, (3) and complexes with 30 % probability displacement ellipsoids. Hydrogen atoms (except Ru–H) and solvent are omitted for clarity
The structures of all three complexes can be considered as distorted octahedral, with the largest deviation from the expected 90° bond angles for N(1)–Ru(1)–H(1), equal to 82.0(9)° in (1) and 84.5(10)° (average value) in (2), and P(1)–Ru(1)–H(1) (83.7(9)°) in complex (3). The angles between carbonyl C(1) and the chloride or pseudohalide (N3 −, NCS−) ligands differ by about 7° from a right angle. The P–Ru–P angles are lower than 180°, being in the 168.23(2)°–171.16(2)° range. As shown in Fig. 2, the CO groups are trans to the 4-pyrrolidinopyridine ligands, and the halide and hydride ligands are mutually trans disposed. In the parent complexes with general formula [RuHX(CO)(PPh3)3] where X = Cl−, N3 −, NCS−, the halide ligands are trans to the carbonyl, and the hydride and one PPh3 ligand are also mutually trans disposed [32, 33]. In the complexes with 4-pyrrolidinopyridine, the trans position to the X ligand is occupied by hydride, whilst the carbonyl is located opposite to the py-4-P ligand. The Ru–X bonds lengths in the complexes are longer by about 0.04, 0.097, 0.08 Å, and the Ru–CO bonds shorter by 0.015, 0.031, 0.014 Å in complexes (1), (2), and (3), respectively, compared with the [RuHX(CO)(PPh3)3] complexes. Moreover, the C≡O distances in the carbonyl ligands are longer than in the corresponding [RuHX(CO)(PPh3)3] complexes; in the chloride complex (1), the C≡O bond length is longer by 0.015 Å, in (2) by 0.07 Å and in (3) by 0.06 Å. Hence, the CO bond length is minimally elongated in the case of chloride complex (1), which is connected with the weak acceptor property of Cl− compared with pseudohalide ligands. The ruthenium–py-4-P bond distances are similar in the chloride and azide complexes (1) and (2), being close to 2.18 Å and in isothiocyanate complex (3) the distance is shorter by about 0.02 Å. Similarly, the Ru–N(3) distance in complex (3) is shorter by 0.04 Å than in complex (2). The Ru(1)–C(1) distances are similar, and the differences are more visible in the C≡O distances. The shortest carbonyl bond length is in the azide complex (2) (Table 2). In the molecular structures of the complexes several inter- and intra-molecular hydrogen bonds [34] exist and are collected in Table 3. Additionally, some π–π stacking between the PPh3 phenyl and pyridine rings is also visible. The plane-to-plane distances between the phosphine phenyl centroids, determined by C(11)–C(16) in (1), C(41)–C(46) and C(75)–C(80) in (2), and C(41)–C(46) in (3), and the pyridine rings are 3.595, 3.62 Å (average value) and 3.764 Å, indicating weak π–π stacking interactions. Moreover, in the structure of complex (2), a T-shaped C–H···π stacking interaction is visible between the two molecules in the asymmetric unit, involving the C(17)–C(22) and C(81)–C(86) phenyl rings with a distance of 2.829 Å.

The density-of-states (DOS) diagram for the complex (1) with partial density-of-states of azide and isothiocyanate ligands in frontier HOMO
Quantum calculations
The ground states geometries of the complexes were optimized in singlet states using the DFT method with the B3LYP functional. The calculations were made for gas phase molecules [without the solvent molecule of complex (3)], and in general, the predicted bond lengths and angles are over-estimated by about 0.1 Å and 5°, respectively. Nevertheless, the general trends observed in the experimental data are reproduced in the calculations, as can be seen from the data collected in Table 2. The calculated IR frequencies of the complexes show good agreement with the experimental spectra; the differences can be explained by the neglect of intermolecular interactions for the gas phase. From the data collected in Table 2, the major differences between the experimental and calculated geometries are found in the Ru(1)–N(1) and Ru(1)–P(1) distances (0.084 and 0.094 Å, respectively) in complex (3). In the case of chloride complex (1), the experimental and calculated Ru(1)–H(1) distances differ by about 0.12 Å. Based on the optimized geometries of the complexes, NBO analyses were performed in order to reveal the nature of the coordination between ruthenium and the donor atoms of the ligands. These analyses showed that the bonding between the py-4-P ligand and ruthenium is largely non-covalent; the Coulomb-type interaction between the ruthenium center and py-4-P ligand is clearly visible in the calculated Wiberg bond index, which is considerably lower than one and close to 0.4 (similar in all these complexes). The Ru–P bond orders are also smaller than 1 (0.7).
For the carbonyl ligands, three natural bond orbitals were detected for the C–O bond, and one for the Ru–C bond. The Ru–C bond orbitals are polarized toward the carbon atom, and the C–O bond orbitals are polarized toward oxygen. The oxygen atom of the carbonyl ligand has one lone pair (LP) orbital. The occupancies and hybridization of the Ru–H, Ru–C, and CO bonds are gathered in Table 4 (antibonding NBOs are given in round brackets). The Wiberg indexes of the CO bonds in the complexes are reduced (by about 0.2) with respect to free CO (W CO = 2.23). The maximum reduction of Wiberg index is calculated for complex (2) which is consistent with the lowest charge on the carbonyl ligand as well as the relatively short C≡O bond in the azide complex.
Analysis of the frontier molecular orbitals is useful for understanding the spectroscopic properties such as electronic absorption and emission spectra. The electronic structures of the complexes are similar because of their similar compositions. The density of states (DOS) in terms of Mulliken population analysis were calculated using the GaussSum program, and Fig. 2 presents the composition of the fragment orbitals contributing to the molecular orbitals for the complexes along with the N3 − and NCS− participations in the HOMOs. The HOMO of complex (1) is localized mainly on the ruthenium atom (54 %), with a contribution from the chloride ligand (36 % Cl). In complexes (2) and (3), the HOMOs are shifted to higher energy and composed of pseudohalide π orbitals (~80 %) with antibonding participation of ruthenium d orbitals (~17 %). This change in ordering of the molecular orbitals influences the luminescent properties of the complexes. The LUMOs are composed of π* orbitals on the PPh3 ligands (~80 %) with a contribution from the ruthenium d 2z orbital (~16 %). The py-4-P ligand plays a role in higher virtual orbitals (LUMO+3/+4 ~70 %). The HOMO-2 and HOMO-4 show antibonding interactions involving the π orbitals of 4-pyrrolidinopyridine and carbonyl ligands with ruthenium d orbitals.
Experimental and theoretical electronic spectra
The UV–Vis spectra of the complexes are similar and present two bands with maxima in the range 300–250 nm. A third high-energy band close to 210 nm may result from transitions in the PPh3 ligands and/or from π → π* excitations in the py-4-P type ligands. The electronic spectra of the complexes were calculated with the TD-DFT method with methanol as solvent in the polarizable continuum model (PCM). Table 5 shows the calculated electronic transitions for the complexes; only transitions to 300 nm are included so the character of the first band is presented. The lowest energy bands in the UV–Vis spectra have Metal–Ligand Charge Transfer (MLCT) with admixture of ligand field character. The frontier orbitals HOMO, HOMO-1, and HOMO-2 plus LUMO, LUMO+1 to LUMO+3 are engaged in transitions. These molecular orbitals are constructed from d ruthenium and π halide and py-4-P orbitals. The next bands in the vicinity of 260–270 nm have mixed MLCT and LMCT character with admixture of Ligand-to-Ligand Charge Transfer (LLCT) transitions.
The emission characteristics of the complexes have been examined in methanol solutions (concentration of 1 × 10–3 mol dm−3) at room temperature, as shown in Fig. 3. The solutions of the complexes excited at 327, 333, and 322 nm for complexes (1), (2), and (3), respectively, gave emissions with maxima at 381, 376, and 441 nm, respectively. The solution of isothiocyanate complex (3) when excited at 361 nm also results in emission at 441 nm. The red shifts of the emissions maxima are typical of ruthenium(II) complexes, and the emissions originating from the MLCT states are derived from excitation involving dπ → π*ligand transitions. The assignments are supported by the analysis of the frontier orbitals of the corresponding complexes, which reveal the contributions of the ligands. In Table 5, the transitions near the excitation wavelengths are marked in italics. The fluorescence of complex (1) is connected with dRu/πCl/πpy-4-P → dRu/π*PPh3 transitions that show considerable participation of the triphenylphosphine ligand. For this reason, the fluorescence of this complex has the lowest intensity. In the pseudohalide complexes (2) and (3), the 4-pyrrolidinopyridine ligands play a role in the emission processes which is possible due to the effect of the N3 − and NCS− ligands on the electronic structure (energies and compositions) of the frontier molecular orbitals.

The fluorescence spectra of the complexes in the methanol solutions (c = 1 × 10−3 mol dm−3)
Conclusion
In summary, three new ruthenium(II) complexes with 4-pyrrolidinopyridine ligands were synthesized and characterized by spectroscopy and X-ray crystallography. The crystal structures of the complexes reveal noncovalent interactions between the aromatic rings. The theoretical results obtained from NBO and analysis of the interactions between ruthenium and the pyridine derivative, and carbonyl and hydride ligands were used to explain the differences in bond lengths as well as the differences in the IR band positions of the complexes. Additionally, comparison of the carbonyl band position in the spectrum of complex (1) with similar ruthenium(II) hydride-carbonyl complexes with 4-phenylpyridimide or pyridine ligands [18, 35] confirms the strong σ-donor property of 4-pyrrolidinopyridine. Electronic structures of the complexes characterized in particular by density of states diagrams have been correlated with their fluorescence properties.
Supplementary data
CCDC 879949, CCDC 879950 and CCDC 879951 contain the supplementary crystallographic data for [RuHCl(CO)(py-4-P)(PPh3)2] (1), [RuH(N3)(CO)(py-4-P)(PPh3)2] (2) and [RuH(NCS)(CO)(py-4-P)(PPh3)2].CH3OH (3) complexes respectively. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Chandra M, Sahay AN, Pandey DS, Puerta MC, Valerga P (2002) J Organomet Chem 648:39
Singh AK, Kumar P, Yadav M, Pandey DS (2010) J Organomet Chem 695:567
Kaljurand I, Ktt A, Soovli L, Rodima T, Memets V, Leito I, Koppel IA (2005) J Org Chem 70:1019
Carrasco M, Peloso R, Rodríguez A, Álvarez E, Maya C, Carmona E (2010) Chem Eur J 16:9754
Carrasco M, Peloso R, Resa I, Rodríguez A, Sánchez L, Álvarez E, Maya C, Andreu R, Calvente JJ, Galindo A, Carmona E (2011) Inorg Chem 50:6361
Profilet RD, Zambrano CH, Fanwick PE, Nash JJ, Rothwell IP (1999) Inorg Chem 29:4362
Hill JE, Fanwick PE, Rothwell IP (1989) Inorg Chem 28:3602
Ionkin AS, Marshall WJ (2004) Organometallics 23:6031
Zambrano CH, Profilet RD, Hill JE, Fanwick PE, Rothwell IP (1993) Polyhedron 12:689
Evans WJ, Davis BL, Nyce GW, Perotti JM, Ziller JW (2003) J Organomet Chem 677:89
Friedle S, Kodanko JJ, Fornace KL, Lippard SJ (2008) J Mol Struct 890:317
Grubbs RH, Sanford MS, Moore JL, Love JA, Trnka TM (2003) Hexacoordinated ruthenium or osmium metal carbene metathesis catalysts, U.S. Pat. Appl. Publ., US 20030069374 A1 20030410
Grubbs RH, Sanford MS, Moore JL, Love JA, Trnka TM (2003) Hexacoordinated ruthenium or osmium metal carbene metathesis catalysts, their preparation and use in polymerization of olefins, PCT Int. Appl., WO 2003011455 A1 20030213
van Der Schaaf AP, Hafner A, Muhlebach A (1998) Hexacoordinated ruthenium and osmium carbene complexes, their preparation and use as metathesis polymerization catalysts, PCT Int. Appl., WO 9839346 A1 19980911
Johansson EMJ, Odelius M, Gorgoi M, Karis O, Ovsyannikov R, Schäfers F, Svensson S, Siegbahn H, Rensmo H (2008) Chem Phys Lett 464:192
Małecki JG, Maroń A (2012) Polyhedron 31:44
Małecki JG, Maroń A (2011) Polyhedron 30:1225
Małecki JG (2011) Polyhedron 30:79
Małecki JG (2010) Polyhedron 29:1973
Małecki JG, Maroń A (2012) Polyhedron 44:221
Ahmad N, Levinson JJ, Robinson SD, Uttely MF (1974) Inorg Synth 15:48
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision A.1. Gaussian, Inc., Wallingford CT
Becke AD (1993) J Chem Phys 98:5648
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785
Eichkorn K, Weigend F, Treutler O, Ahlrichs R (1997) Theor Chem Acc 97:119
Casida ME (1996) In: Seminario JM (ed) Recent developments and applications of modern density functional theory, theoretical and computational chemistry, vol 4. Elsevier, Amsterdam, p 391
Glendening ED, Reed AE, Carpenter JE, Weinhold F NBO (version 3.1)
O’Boyle NM, Tenderholt AL, Langner KM (2008) J Comput Chem 29:839
CrysAlis RED, Oxford Diffraction Ltd., Version 1.171.29.2
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) J Appl Cryst 42:339
Sheldrick GM (2008) Acta Cryst A64:112
Seetharaman SK, Chung M-C, Englich U, Ruhlandt-Senge K, Sponsler MB (2007) Inorg Chem 46:561
Małecki JG (2010) Polyhedron 29:2489
Desiraju GR, Steiner T (1999) The weak hydrogen bond in structural chemistry and biology. Oxford University Press, Oxford
Małecki JG, Maroń A (2012) Transition Met Chem 37:727
Acknowledgments
Calculations have been made in Wroclaw Centre for Networking and Supercomputing (http://www.wcss.wroc.pl).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Małecki, J.G., Maroń, A. Chloride and pseudohalide hydride-carbonyl ruthenium(II) complexes with 4-pyrrolidinopyridine as co-ligand. Transition Met Chem 38, 133–142 (2013). https://doi.org/10.1007/s11243-012-9670-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-012-9670-8