
Abstract
The reaction of [RuHCl(CO)(PPh3)3] with 4-phenylpyrimidine gave a new ruthenium(II) complex, namely [RuHCl(CO)(PPh3)2(pyrim-4-Ph)]. The complex has been studied by IR and UV–vis spectroscopy and by X-ray crystallography. The molecular orbitals of the complex have been calculated by density functional theory. The spin-allowed singlet–singlet electronic transitions of the complex have been calculated by time-dependent DFT, and the UV–vis spectrum of the compound has been discussed on this basis. The emission properties of the complex were also studied.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ruthenium hydride complexes of carbonyl and triarylphosphine ligands are interesting due to their reactivity and efficiency as catalysts in a wide variety of reactions [1–6]. The synthesis and characterization of ruthenium complexes containing nitrogen heteroaromatic ligands have also received considerable recent attention, owing to their interesting photophysical and photochemical properties. Pyrimidine and its derivatives as well as other 1,4-diazines, including compounds with partial pyrazine structures (e.g., quinoxaline, phenazine, pteridine, flavin and their derivatives), demonstrate distinctive physico-chemical properties that are caused by a low lying unoccupied π-molecular orbital and by their ability to act as bridging ligands.
Azine ligands generally have energetically low lying π-antibonding orbitals, which can accept electrons from filled metal d orbitals. In consequence, they can exhibit charge transfer bands with interesting spectroscopic properties in the visible region [7]. Ligands containing pyrimidine rings are widely studied and their π-donor properties are interesting. Their combination with other donor atoms should in principle afford complexes with tunable spectroscopic properties [8–10]. The hydride ligand as a powerful σ-donor is very efficient at compensating the electron deficiency at the metal center in hydride complexes. The “trans effect” of hydride and the interactions between carbonyl and donor ligands in mutually trans positions are factors, which explain the stability of these complexes [11].
Here, we report an experimental and quantum chemical study of a ruthenium hydridocarbonyl complex with a pyrimidine derivative as co-ligand. The quantum chemical study included a characterization of the molecular and electronic structures of the complex by analysis of the optimized molecular geometry and the electronic population by using the natural bond orbitals scheme. The latter was used to identify the nature of the interactions between the ligands and the central metal. Finally, time-dependent density functional theory (TD-DFT) was used to calculate the electronic absorption spectrum. These results allowed for the interpretation of the experimental UV–vis spectrum. The complex reported in this paper combines our interests in ruthenium hydride carbonyl coordination compounds and complexes containing nitrogen heterocyclic ligands [12–20].
Experimental
All reagents used to the synthesis of the complex are commercially available and were used without further purification.
Synthesis of [RuHCl(CO)(PPh3)2(pyrim-4-Ph)]
A suspension of [RuHCl(CO)(PPh3)3] (0.2 g, 0.2 mmol) and 4-phenylpyrimidine (0.03 g, 0.2 mmol) in methanol was refluxed until the solid dissolved and then for an additional 2 h, then cooled and filtered. Yellow crystals suitable for X-ray crystal analysis were obtained by slow evaporation the reaction mixture. Yield (0.17 g) 86 %.
Anal. calc. for C47H39ClN2OP2Ru: C 66.70 %, H 4.64 %, Cl 4.19 %, N 3.31 %, O 1.89 %, P 7.32 %, Ru 11.94 %. Found: C 66.62 %, H 4.67 %, N 3.28 %.
IR (KBr) (cm−1): 3,047 (m) νCH–pyrimidine; 2,039 (w) νRu–H; 1,943 (s) νCO; 1,602 (m) νCN; 1,566 (m) νC=C; 1,474 (m) δ(C–CH in the plane); 1,434 (m) νPh(P–Ph); 1,092 (m) δ(C–CH in the plane); 997 (w) δ(C–C out of the plane); 742 (m) δ(C–C out of the plane); 697 (s) δ(C–C in the plane); 520 (m) νRu–P/Ru–N 1H NMR (400 MHz, CDCl3) δ 9.34 (s, H(2) pyrimidine), 8.81 (d, J = 6.3 Hz, H(5) pyrimidine), 8.05–7.05 (m, PPh3; pyrimidine; phenyl), −4.44 (t, J = 19.2 Hz, Ru–H(1)). 31P NMR (162 MHz, CDCl3) δ 31.22 (s, PPh3).
UV–vis (methanol) (nm) (logε): 370 (1.01), 328 (1.93), 268 (2.68), 232 (3.06), 212 (4.27).
Physical measurements
Infrared spectra were recorded on a Perkin-Elmer spectrophotometer in the spectral range 4,000–400 cm−1 using KBr pellets. 1H and 31P NMR spectra were obtained at room temperature in CDCl3 using a Bruker 400 spectrometer. Electronic spectra were measured on a spectrophotometer Lab Alliance UV–VIS 8500 in the range 800–200 nm in methanol solution. Luminescence measurements were made in methanolic solution on an F-2500 FL spectrophotometer at room temperature. Elemental analyses (C, H, N) were obtained on a Perkin-Elmer CHN-2400 analyser.
Computational methods
The calculations were carried out using the Gaussian09 [21] program. The molecular geometry of the singlet ground state of the complex was fully optimized in the gas phase using the B3LYP functional [22, 23]. For the complex, a frequency calculation was carried out, verifying that the optimized molecular structure corresponds to energy minimum; thus, only positive frequencies were expected. The DZVP basis set [24] with f functions with exponents 1.94722036 and 0.748930908 was used to describe the ruthenium atom, and the basis set used for the lighter atoms (C, N, O, Cl, P, H) was 6-31G with a set of “d” and “p” polarization functions. The TD-DFT method [25] was employed to calculate the electronic absorption spectrum of the complex using the solvent PCM (Polarizable Continuum Model) model. In this work, 100 singlet excited states were calculated as vertical transitions for the complex. A natural bond orbital (NBO) analysis was also made for the complex using the NBO 5.0 package [26] included in Gaussian09. Natural bond orbitals are orbitals localized on one or two atomic centers that describe molecular bonding in a manner similar to a Lewis electron pair structure, and they correspond to an orthonormal set of localized orbitals of maximum occupancy. NBO analysis provides the contribution of atomic orbitals (s, p, d) to the NBO σ and π hybrid orbitals for bonded atom pairs. In this scheme, three NBO hybrid orbitals are defined, namely bonding orbital (BD), lone pair (LP) and core (CR), which were analyzed on the atoms directly bonded to or presenting some kind of interaction with the ruthenium atom. The contribution of a group (ligands, metal atom) to a molecular orbital was calculated using Mulliken population analysis. GaussSum 2.2 [27] was used to calculate group contributions to the molecular orbitals and to prepare the partial density of states (DOS) spectra. The DOS spectra were created by convoluting the molecular orbital information with Gaussian curves of unit height and full width at half maximum of 0.3 eV.
Crystal structure determination and refinement
A pale yellow plate shaped crystal of the complex was mounted in turn on an Xcalibur, Atlas, Gemini Ultra Oxford Diffraction automatic diffractometer equipped with a CCD detector and used for data collection. X-ray intensity data were collected with graphite monochromated MoK α radiation (λ = 0.71073 Å) at temperature 295.0(2) K, with ω scan mode. Ewald sphere reflections were collected up to 2θ = 50.10. The unit cell parameters were determined from least-squares refinement of the setting angles of 10,270 strongest reflections. Details concerning crystal data and refinement are gathered in Table 1. Lorentz, polarization and empirical absorption correction using spherical harmonics implemented in the SCALE3 ABSPACK scaling algorithm [28] were applied. The structure was solved by Patterson methods and subsequently completed by difference Fourier recycling. All the non-hydrogen atoms were refined anisotropically using full-matrix, least-squares techniques. Bearing in mind the limitations of Fourier synthesis and the problems in recognizing artifacts in the immediate neighborhood of heavy atoms, it is doubtful if a reliable position for the hydrogen atom bound to the Ru atom can be found in the difference Fourier map, avoiding the danger of mistaking the effects of the series termination errors for a true atomic position. In this complex, the Ru–H bond length of 1.86(4) Å is normal. The Olex2 [29] and SHELXS97, SHELXL97 [30] programs were used for all the calculations. Atomic scattering factors were used as incorporated in the programs.
Results and discussion
Spectroscopic characterization
The reaction of [RuHCl(CO)(PPh3)3] with 4-phenylpyrimidine gave the complex [RuHCl(CO)(PPh3)2(pyrim-4-Ph)] as a pale yellow crystalline solid. In the 1H NMR spectrum, signals at 9.34 and 8.81 ppm are assigned to the pyrimidine protons. In the range of 8.05–7.05 ppm, the PPh3 phenyl and 4-pyrimidinepheny protons are observed but the various signals in this region are difficult to resolve. A signal at high field (−4.44 ppm) indicates the presence of the hydride ligand. The Ru–H signal is a triplet, due to coupling with the two trans phosphorus atoms (J HP ~ 19 Hz). The 31P NMR spectrum of the complex shows a singlet at 31.22 ppm, which suggests that the two triphenylphosphine groups are in equivalent trans positions. The molecular structure of the complex as determined by X-ray crystallography shows that the angle between triphenylphosphine ligands is not linear but this may be due to the crystal packing interactions.
The IR spectrum displays Ru–H and C≡O stretching bands at 2,039 and 1,943 cm−1, respectively. The νRu–H and νCO stretching bands in the parent [RuHCl(CO)(PPh3)3] complex are at 2,020 and 1,922 cm−1, respectively, hence a decrease in the carbonyl stretch is clearly seen for the studied complex. The inclusion of electron-acceptor ligands such as chloride or 4-pyrimidinephenyl in the coordination sphere should decrease the electron density on the metal, therefore increasing the bond order of the CO bond and increasing the vibration frequency. However, the electron-donor hydride ligand delivers electron density via backbonding to the antibonding orbitals of the CO, which will decrease the vibration frequency of the CO bond. Overall, the changing of the positions of νRu–H and νRu–CO indicates a decrease in the metal—carbonyl carbon interaction and the Ru–H bond order. Stretching vibrations for the C=C and C=N bonds of the 4-pyrrolidinephenyl ligand are observed in the 1,566–1,602 cm−1 range.
Crystal structure
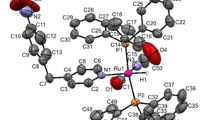
This complex crystallizes in the triclinic P-1 group. The molecular structure of the complex is displayed as an ORTEP representation in Fig. 1, and selected bond distances and angles are collected in Table 2. The Ru(1)–N(1) bond length in the complex is normal and comparable with other ruthenium hydride complexes with pyrimidine derivative ligands [16]. The structure of the complex can be considered as distorted octahedral, with the largest deviation from the expected 90° bond angles for P(1)–Ru(1)–H(1) at 85.1(12)° and N(1)–Ru(1)–H(1) at 83.8(12)°. The angles between the carbonyl and pyrimidine C(1)–Ru(1)–N(1) ligands and chloride and hydide Cl(1)–Ru(1)–H(1) differ by about 9° from a right angle. The P–Ru–P angle is slightly lower than 180° with a value of 177.67(3)°.

The ORTEP plot of the [RuHCl(CO)(pyrim-4-Ph)(PPh3)2] complex. The displacement ellipsoids are drawn with 30 % probability. The hydrogen atoms, except Ru(1)–H(1) are omitted for clarity
As shown in Fig. 1, the CO group is trans to the 4-phenylpyrimidine ligand and the chloride and hydride ligands are mutually trans. In the parent complex with formula [RuHCl(CO)(PPh3)3], the chloride ligand is trans to the carbonyl, and the hydride and one PPh3 ligand are also mutually trans [31]. In spite of the changes in the coordination sphere in the studied complex, the Ru–Cl and Ru–CO bond lengths are almost unchanged compared with the parent complex. Moreover, the C≡O bond lengths are also similar in both complexes (1.151(5) Å in [RuHCl(CO)(PPh3)2(pyrim-4-Ph)] and 1.141 Å in [RuHCl(CO)(PPh3)3]). Based on the data and taking into account the IR shifts discussed above, the 4-phenylpyrimidine can be considered as a rather strong σ-donor.
In the molecular structure of the complex, several inter- and intra- molecular hydrogen bonds [32] are observed and are collected in Table 3. Additionally, some electronic interactions (π–π stacking) between the PPh3 phenyl and pyrimidine rings are visible, as shown in Fig. 2. The plane-to-plane distances between the phosphine phenyl centroids, determined by C(12) to C(17) and C(30) to C(35) carbons, and the pyrimidine ring are 3.708 and 3.660 Å, respectively, indicating weak π–π stacking interactions.

π-stacking interactions in the molecule of [RuHCl(CO)(pyrim-4-Ph)(PPh3)2] complex
Quantum calculations
The ground state geometry of the complex was optimized in the singlet state, using the B3LYP functional. The calculation was carried out for the gas phase molecule, and in general, the predicted bond lengths and angles are over-estimated by about 0.1 Å and 3o, respectively. Nevertheless, the general trends observed in the experimental data are reproduced in the calculations, as can be seen from the Table 2. The calculated IR frequencies of the complex show good agreement with the experimental spectrum; the differences can be explained by the neglect of intermolecular interactions for the gas phase. From the data collected in Table 2, one may see that the major differences between the experimental and calculated geometries are found in the Ru(1)–H(1) and Ru(1)–Cl(1) distances (0.25 and 0.1 Å, respectively). Based on the optimized geometry of the complex, a NBO analysis was performed in order to reveal the nature of the coordination between ruthenium and the donor atoms of the ligands. This analysis showed that the pir-4-P ligand does not show covalent bonding with ruthenium; the Coulomb-type interaction between the ruthenium center and the 4-phenylpyrimidine ligand is clearly visible in the calculated Wiberg bond index, which is considerably lower than one and close to 0.38. The Ru–P bond orders are also smaller than 1 (0.7).
For the carbonyl ligand, three natural bond orbitals were detected for the C–O bond, and one for the Ru–C bond. The Ru–C bond orbitals are polarized toward the carbon atom, while the C–O bond orbitals are polarized toward the oxygen. The carbonyl oxygen has one lone pair (LP) orbital. The Wiberg index of the CO bond in the complex is reduced (by 0.21) with respect to free CO (W CO = 2.23). The charge on the carbonyl group calculated simply by summing the individual charges on the carbon and oxygen atoms is +0.205. The charges on the hydride and chloride ligands are +0.05 and −0.53, respectively. The Wiberg indices of the Ru–H, Ru–C(O) and Ru–Cl bonds are equal to 0.79, 1.32 and 0.57, respectively. The nitrogen atoms in the pyrimidine ring have similar natural charges, equal to −0.44 for N(1) and −0.49 for N(2). The natural charge on the ruthenium is −0.92, which confirms the strong donor property of 4-phenylpyrimidine and also provides supporting evidence for ligand to d Ru electron transfer. The data suggest that donation from the ligands to the d Ru orbitals plays a role in the electronic structure of the complex, and in order to determine the extent of donation, the stabilization energies were calculated. ΔE ij (kcal/mol) associated with delocalization is estimated by second-order perturbation as: ΔE ij = q i (F(i, j)2)/(ε j − ε i ) where q i is the donor orbital occupancy, ε i , ε j are diagonal elements (orbital energies), and F(i, j) is the off-diagonal NBO Fock or Kohn–Sham matrix element.
The stabilization energies show that the pyrimidine ligand in this complex donates charge to ruthenium, with a stabilization energy (ΔE ij ) of 53.5 kcal/mol. The back donation to 4-phenylpyrimidine provides 24.5 kcal/mol, and the data highlight the higher σ-donor than π-acceptor properties of the ligand. For comparison, the stabilization energy for donation from chloride to ruthenium is 44.6 kcal/mol, and the back donation Ru → Cl only 6.0 kcal/mol.
Analysis of the frontier molecular orbitals is useful for understanding the spectroscopic properties such as electronic absorption and emission spectra. The density of states (DOS) in terms of Mulliken population analysis was calculated using the GaussSum program [27], and Fig. 3 presents the composition of the fragment orbitals contributing to the molecular orbitals of the complex. The HOMO is localized on the ruthenium atom (49 %) with a significant contribution from the chloride ligand (43 % Cl), while the LUMO is composed of π* orbitals of 4-phenylpyrimidine (97 %). The ruthenium d orbitals contribute to the HOMO-1 and HOMO-2 (42 and 44 %) while in the HOMO-1 the carbonyl (10 %) and chloride (45 %) ligands are also engaged, and the HOMO-2 has a contribution from the PPh3 ligands (43 %). The d 2 z (~16 %) ruthenium orbital is a component of the LUMO+2, along with the π* triphenylphosphine orbitals, and the d 2 x 2−y orbital plays a role in the higher virtual orbital LUMO + 15 along with the π* carbonyl and PPh3 orbitals.

DOS diagram for [RuHCl(CO)(pyrim-4-Ph)(PPh3)2] complex
Experimental and theoretical electronic spectra
The UV–vis spectrum of the complex shows maxima at 370, 328, 268, 232 and 212 nm. The electronic spectrum was calculated with the TD-DFT method with methanol as solvent in the polarizable continuum model (PCM). Computation of 100 excited states of the complex allowed the interpretation of the experimental spectrum down to ~230 nm. The assignment of the calculated orbital excitations to the experimental bands was based on an overview of the composition and relative energies of the HOMO and LUMO. As one can see from the data collected in Table 4, the Charge Transfer transitions have Metal-to-Ligand Charge Transfer character. These transitions mainly involve the HOMO and LUMO, LUMO+1/+2. The bands with maxima at 268 and 232 nm have Ligand-to-Ligand Charge Transfer character. The highest experimental band close to 212 nm may result from transitions based on the PPh3 ligands and from π → π* excitations in the pyrimidine ligand.
The emission characteristics of the complex were examined in methanol solution (concentration of 1 × 10−3 mol/dm3) at room temperature. Excitation at 303 nm gave fluorescence with a maximum at 425 nm, as shown Fig. 4. The red shift of the emission maximum is typical of ruthenium(II) complexes, and the emission originates from excitation involving a \( d_{\pi } \to \pi_{\text{ligand}}^{*} \) MLCT transition. This assignment is supported by the analysis of the frontier orbitals of the complex. Moreover, the transition between the HOMO-1 and LUMO+1 was calculated to lie in this region. The LUMO+1 is localized on the 4-phenylpyrimidine ligand, while the HOMO-1 has a significant contribution from the carbonyl ligand, whose presence is associated with the fluorescence of the complex.

Fluorescence spectrum of [RuHCl(CO)(pyrim-4-Ph)(PPh3)2] complex solution in methanol
Conclusion
A new ruthenium(II) hydride carbonyl complex with 4-phenylpyrimidine as co-ligand has been synthesized and characterized. In the crystal structure of the complex, some non-covalent interactions between the aromatic rings have been found. The electronic structure of the complex, characterized in particular by the density of states diagram, has been correlated with its fluorescence properties.
References
Salvi L, Salvini A, Micoli F, Bianchini C, Oberhauser W (2007) J Organomet Chem 692:1442
Po Lau C, Man Ng S, Jia G, Lin Z (2007) Coord Chem Rev 251:2223
Jun Yue C, Liu Y, He R (2006) J Mol Catal A: Chem 259:17
Nipa Haque F, Lough AJ, Morris RH (2008) Inorg Chim Acta 361:3149
Buskens P, Giunta D, Leitner W (2004) Inorg Chim Acta 357:1969
Bravo J, Castro J, Garcia-Fontana S, Rodriguez-Martinez MC, Albertin G, Antoniutti S, Manera A (2007) J Organomet Chem 692:5481
Chandra M, Sahay AN, Pandey DS, Puerta MC, Valerga P (2002) J Organomet Chem 648:39
Małecki JG, Maroń A (2011) Trans Met Chem 36:297
Małecki JG (2010) Trans Met Chem 35:801
Michalik S, Machura B, Kruszynski R, Kusz J, Krompiec M (2010) Polyhedron 29:2629
Coe BJ, Glenwright SJ (2000) Coord Chem Rev 203:5
Małecki JG, Maroń A (2011) Polyhedron 30:1225
Małecki JG (2011) Polyhedron 30:79
Małecki JG (2010) Polyhedron 29:2489
Małecki JG, Kruszyński R, Mazurak Z (2009) Polyhedron 28:3891
Małecki JG, Kruszyński R, Mazurak Z (2008) J Coord Chem 61:2186
Małecki JG, Kruszyński R, Tabak D, Kusz J (2007) Polyhedron 26:5120
Małecki JG, Kruszynski R, Mazurak Z (2007) Polyhedron 26:4201
Małecki JG, Kruszyński R (2007) Polyhedron 26:2686
Małecki JG, Kruszyński R (2007) J Coord Chem 60:2085
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb,MA Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, revision A.1. Gaussian Inc., Wallingford
Becke D (1993) J Chem Phys 98:5648
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785
Eichkorn K, Weigend F, Treutler O, Ahlrichs R (1997) Theor Chim Acc 97:119
Casida ME (1996) In: Seminario JM (ed) Recent developments and applications of modern density functional theory, theoretical and computational chemistry, vol 4. Elsevier, Amsterdam, p 391
Glendening ED, Reed AE, Carpenter JE, Weinhold F, NBO (version 3.1)
O’Boyle NM, Tenderholt AL, Langner KM (2008) J Comp Chem 29:839
CrysAlis RED, Oxford Diffraction Ltd., Version 1.171.29.2
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) J Appl Cryst 42:339
Sheldrick GM (2008) Acta Cryst A64:112
Seetharaman SK, Chung M-C, Englich U, Ruhlandt-Senge K, Sponsler MB (2007) Inorg Chem 46:561
Desiraju GR, Steiner T (1999) The weak hydrogen bond in structural chemistry and biology. Oxford University Press, Oxford
Acknowledgments
Calculations have been carried out in Wroclaw Centre for Networking and Supercomputing (http://www.wcss.wroc.pl).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Appendix: supplementary data
Appendix: supplementary data
CCDC 891199 contains the supplementary crystallographic data for [RuHCl(CO)(pyrim-4-Ph)(PPh3)2] complex. This data can be obtained free of charge from http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44-1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Małecki, J.G., Maroń, A. A ruthenium(II) hydride carbonyl complex with 4-phenylpyrimidine as co-ligand. Transition Met Chem 37, 727–734 (2012). https://doi.org/10.1007/s11243-012-9644-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-012-9644-x