
Abstract
The mechanistic aspects of the hetero Diels–Alder reactions between strongly electrophilic (E)-2-aryl-1-cyano-1-nitroethenes and ethyl vinyl ether have been analyzed on the basis of kinetic experimental results as well as quantum chemical simulations of reaction paths. These reactions may theoretically proceed via one step or stepwise, zwitterionic mechanism. It was found that the conversion of addends into adducts carried out via polar, but not zwitterionic mechanism. This is confirmed by kinetic substituent and solvent effects. Subsequently, a detailed analysis of internal reaction coordinate trajectories shows that the title reactions should be considered as examples of “two-stage one-step” cycloadditions according to Domingo terminology.
Similar content being viewed by others
Introduction
Nitronates are very useful precursors in organic synthesis [1–5]. The general, most universal method of the preparation of six-membered cyclic nitronates is a hetero Diels–Alder (HDA) reaction between conjugated nitroalkenes (CNA) as heterodienes and ethylenic dienophiles [3–6]. Most of these reactions follow the one-step [4 + 2] cycloaddition mechanism. However, in the case of reactions between strongly electrophilic CNA and π-electron-rich ethylenes as dienophiles, the one-step mechanism may compete with the two-step, zwitterionic mechanism [7]. For example, HDA reactions between (E)-3,3,3-trifluor-1-nitroprop-1-ene and enamines take place via zwitterionic intermediates [8, 9]. A similar mechanism (1a-f + 2 → 5/6a-f → 3/4a-f) is very likely in the case of HDA reaction of (E)-2-aryl-1-cyano-1-nitroethenes (global electrophilicity ω > 3 eV [10]) with ethyl vinyl ether Scheme (1)

Theoretically possible mechanisms of HDA reactions between (E)-2-aryl-1-cyano-1-nitroethenes 1 and ethyl vinyl ether 2
These reactions were examined experimentally by Fringuelli et al. [11]. Unfortunately, the authors analyzed only the reaction products. In the light of this experimental support, it is very difficult to determine a reaction mechanism. In this work, we decided to resolve this problem on the basis of an experimental kinetic study as well as quantum chemical considerations. In particular, we performed: (i) analysis of the nature of addends interactions with respect to the recently strongly developed [12–15] application of the reactivity indices defined within the conceptual DFT, (ii) study about kinetic substituent effects, (iii) study about kinetic solvent effects, (iv) quantum chemical simulations of reaction paths. In our study, we used a series of (E)-2-aryl-1-cyano-1-nitroethenes nitroalkenes (1a-f) containing substituents with different electronic properties (Scheme 2). We hope that this comprehensive research will resolve the above mentioned mechanistic problem, which is important both from theoretical and practical point of view.

(E)-2-aryl-1-cyano-1-nitroethenes 1 which were tested in HDA reactions with ethyl vinyl ether 2
Experimental procedure
Reagents and equipment
All solvents employed for the kinetic measurements were purified by standard methods [16]. Commercially available (Aldrich) ethyl vinyl ether was used. Just before use, it was distilled under atmospheric pressure, using 25 cm Vigreux column. (E)-2-aryl-1-cyano-1-nitroethenes 1a-f was obtained by the condensation of appropriate aromatic aldehydes with nitroacetonitrile [17], according to a reported procedure [18]. The purity was confirmed by HPLC and 1H NMR analyses.
Kinetic experiments were carried out in a glass reactor supplied with a thermostatically controlled jacket, magnetic stirrer, thermometer, reflux condenser and sampling device. Liquid chromatography (HPLC) was done using a Knauer apparatus, equipped with a UV–vis detector (λ = 205 nm). For monitoring of the reaction progress, a LiChrospher 100RP 3 μm column (4 × 240 mm) was used, and, 75 % methanol at flow rate 0.6 mL min−1 was applied as the eluent.
Kinetic procedure
The overall rate constants (ktotal) were determined by monitoring the decrease of the HPLC peak area (A) corresponding to ethyl vinyl ether. The kinetic experiments were carried out at 25 °C up to 70–80 % completion, using in each case the equimolar quantities of substrates. At determined time intervals, 250 μl samples were taken out of the reactor with a microsyringe and analyzed immediately by HPLC. In this way, 11 series of measurements were completed. The regression analysis was carried out using MATCAD 07. It showed excellent linear relationships (r > 0.99) between 1/A and time t for all kinetic runs, using the second-order kinetic equation of the form:
The second-order rate constants (ktotal) obtained by this method, are summarized in Table 1.
Theoretical exploration of reaction paths
The quantum chemical calculations were performed on a SGI-Altix-3700 computer in the Cracow Computing Center “CYFRONET”. The hybrid B3LYP functional and the 6-31G(d) basis set included within the Gaussian 09 software [19] were applied. Reports published recently show that the same theoretical level was used e.g. for the exploration of reaction pathways of several DA reactions involving CNA [10, 20–22]. In particular, the B3LYP/6-31G(d) calculations illustrate well the structure of transition states (TS) in cycloadditions between cyclopentadiene and CNA. This has been confirmed previously by the compilation of deuterium secondary kinetic isotope effects (SKIE) and results of quantum chemical calculations for polar Diels–Alder (P-DA [23] ) reaction between cyclopentadiene and (E)-2-phenylnitroethene [24]. The critical structures on the reaction paths were localized as in the case of the previously analysed reaction of (E)-2-phenyl-1-cyano-1-nitroethene with cyclopentadiene [22]. For the simulations of dielectric medium presence (methanol ε = 32.63; diethyl ether ε = 4.34), the PCM algorithm of Tomasi et al. [25] was used. The same solvation model was used recently successfully in exploring DA reactions paths between (E)-2-phenylnitroethene and cyclopentadiene [21, 26], 3-nitro-1-(p-toluenesulfonyl)-indole and cyclopentadiene [27] as well as in many other similar cases [28–30].
All calculations were performed for T = 298 K and p = 1 atm. The results are shown in Tables 2 and 3. The letters LM and TS denote the pre-reaction complex, and transition state, respectively, and a subscript is added to denote the reaction path.
Results and discussion
Electronic interactions of addents
To investigate reactions between (E)-2-aryl-1-cyano-1-nitroethenes 1a-f and ethyl vinyl ether 2 before beginning experimental study, we analyzed the electronic interactions of addents using the theory of DFT reactivity indices [13, 14, 32] (Table 4). Indices of global electrophilicity (ω) [33], global nucleophilicity (N) [34] as well as local descriptors ωk, Nk required for this purpose were calculated by using the Parr functions [32] and the equations recommended by Domingo et al. [13, 15]. In particular:
Here, μ is the electronic chemical potential, and η is the chemical hardness of the substrates
Here, P + k , and, P − k , are electrophilic and nucleophilic Parr functions based on the spin density distribution at the radical anion and at the radical cation of a neutral molecule.
So, the global electrophilicity of the nitroalkenes confirms its strongly electrophilic nature [13]. In particular, (E)-2-(4-methoxyphenyl)-1-cyano-1-nitroethene (1a, ω = 3.14 eV [10] ) is the weakest electrophile in the series of nitroalkenes studied, while (E)-2-(4-carbomethoxyphenyl)-1-cyano-1-nitroethene (1f, ω = 3.80 eV [10] ) is the strongest one. On the other hand, ethyl vinyl ether 2, which is a weak electrophile (ω = 0.41 eV), can be considered as a strong nucleophile (N = 3.24 eV). The large difference in the electrophilicity (Δω > 2.7 eV) of the substrates indicates that reactions 1a-f + 2 should be considered as P-DA reactions on the basis of Domingo terminology [23].
We also performed an analysis of local interactions. It was found that the coruse of the 1a-f + 2 reaction should be determined by a nucleophilic attack from the terminal, one position of ethyl vinyl ether 2 (NC1 = 2.27 eV) to the β-position of nitroalkene (ωβ = 1.29–1.58 eV [10] ). Such attack favors reaction channels leading to nitronates three and four, which—as confirmed experimentally [11]—actually formed during the reaction analyzed.
Kinetic substituent effects
First, we have investigated the influence of the nature of the substituent on the reaction kinetics. It was found [35] that in the case of two-step cycloadditions EWG/EDG, substituents influence the intermediate stability, and, in consequence the values of rate constants.
Our study about kinetic substituent effects (Table 1) showed that the rate constants of P-DA reactions 1a-f + 2 increase with the CNA electrophilicity. In particular, the rate constant for the reactions involving the weakest electrophilic (E)-2-(4-methoxyphenyl)-1-cyano-1-nitroethene 1a is equal to 1.34 × 10−2 L mol−1 s−1, whereas a similar reaction involving most electrophilic (E)-2-(4-carbomethoxyphenyl)-1-cyano-1-nitroethene 1f—features the value 9.34 × 10−2 L mol−1 s−1. To obtain a quantitative description of the substituent effect on the reaction rate, we studied correlations between substrate reactivity (log k) and CNA electrophilicity (ω). It was found that there is a good linear correlation (R = 0.953) between log k and ω values (Fig. 1).
A similar correlation (R = 0.915) was obtained for the relations between log k and Hammett constants of substituents in CNA phenyl rings (Fig. 2).

Plot of log k versus global electrophilicity index ω for DA reactions between (E)-2-aryl-1-cyano-1-nitroethenes 1a-f and ethyl vinyl ether 2

Plot of log k versus Hammett constants σp for for DA reactions between (E)-2-aryl-1-cyano-1-nitroethenes 1a-f and ethyl vinyl ether 2
This proves that the substituent effect is transferred into reaction sites via both resonance and induction effects. The absolute value of the ρ constant confirmed that the reactions tested here are of polar nature [36]. However, the ρ values are outside the range typical for zwitterionic reactions [35].
Kinetic solvent effects
The nature of kinetic substituent effect suggests polar, but one-step mechanism for the reactions between 1a-f and 2. In order to explain the problem of existence of the hypothetical zwitterionic intermediate, we studied the kinetics of DA reaction involving most electrophilic CNA 1f in several solvents with different polarity. It was found that the rate constants for the reactions in weakly polar diethyl ether (ε = 4.34, ΕΤ(30) = 34.7) is equal to 0.7 × 10−2 L mol−1 s−1, whereas in the case of the same reaction in strongly polar methanol, the value is 9.34 × 10−2 L mol−1 s−1.
We examined the kinetic solvent effect on the reaction quantitatively using classical correlation analysis, seeking the best match between log k and the values of constants which characterized solvents polarity. The best linear correlations (R = 0.939) were obtained with the Reichardt–Dimroth constants ΕΤ(30) [36] (Fig. 3).
These data prove that the sensitivity constants in Eq. 3 did exceed 0.1 and are beyond the typical range observed for ionic reactions [35].

Plot of log k versus Dimroth ET(30) constants for for DA reactions between (E)-2-(4-carbomethoxyphenyl)-1-cyano-1-nitroethene 1f and ethyl vinyl ether 2
Quantum chemical exploration of reaction paths
The experimental results exclude a stepwise mechanism of P-DA reactions between ether 2 and CNA 1a-f. Unfortunately, they do not provide precise information about nature of critical structures, which exist on the reaction profile. Therefore, in the next step of our investigations, we decided to realize quantum chemical simulations of the reaction channels leading to adducts three and four. In this study, we have performed for P-DA reactions involving more electrophilic (1f) and less electrophilic (1a) CNA. The simulations included the presence of a more polar (methanol) as well as a less polar (diethyl ether) solvent, which were tested during the experimental study.
Reaction profiles
B3LYP/6-31g(d) calculations prove that the energy profiles of 1a + 2 and 1f + 2 reactions in a weakly polar medium are similar (Fig. 4). Only one TS, preceded by a shallow trough related to the pre-reaction complex (LM), was located between the minima corresponding to individual addents and products in all cases. All attempts to find pathways leading from addents to products through hypothetical intermediates 5 and 6 were unsuccessful.

Enthalpy profiles for DA reactions between (E)-2-aryl-1-cyano-1-nitroethenes 1a,f and ethyl vinyl ether 2 in diethyl ether according to B3LYP/6-31G(d) (PCM) calculations (298 K)
Interactions between the addents in the first stage lead to LM complexes. This involves the enthalpy of the reaction system reduced by 0.6–1.5 kcal mol−1. The energy minima related to the LMs are purely enthalpic, because ΔG > 0. Therefore, they may not exist as stable intermediates.
Further movement of the reaction system leads to the TS. For reactions involving much less electrophilic CNA 1a, this involves an increase in activation enthalpy by more than 25 kcal mol−1, while for reactions with more electrophilic CNA 1f, the increase is lower (approximately 22 kcal mol−1). Therefore, B3LYP/6-31g(d) calculations suggest an identical nature of the substituent effect as experimental kinetic studies performed earlier (see “Kinetic procedure”). This generally means that the reaction kinetics is determined by the strength of polar electrophile–nucleophile interactions (see “Reagents and equipment”).
It is noted by analyzing the activation parameters barriers of competing reaction pathways leading to adducts three and four that the former cycloaddition is always favored irrespective of the substituent. This is perfectly consistent with the stereoselectivity observed experimentally [11].
When diethyl ether is replaced by a more polar methanol, the reaction energy profiles do not change; it is only the quantitative description of the respective critical points that changes. In particular, for the 1a + 2 and 1f + 2 reactions, the activation barriers are considerably reduced. This is perfectly consistent with the results of our experimental kinetic studies (see “Theoretical exploration of reaction paths”).
It should be noted at this point that in all cases, profiles of relationship between intrinsic reaction coordinate and reaction force constant is different as in the case one-stage one-step reaction, and similar to that in the case of two-stage one-step reaction [37].
Geometries and electronic properties of critical structures
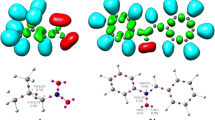
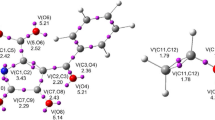
Distances between the reaction centers within the LMs are still higher than the range typical for the TS σ-bonds (Fig. 5). At this stage, the reaction centers have yet to adopt orientations found later in TSs. Therefore, they are not orientation complexes (OC). They are not charge transfer complexes (CT), either, because electron transfer within them does not occur (see t indexes in Table 3). Medium polarity does not generally affect the character of LMs.

Views of transitions states for HDA reactions between (E)-2-(4-carbomethoxy)-1-cyano-1-nitroethenes 1f and ethyl vinyl ether 2 in methanol according to B3LYP/6-31G(d) (PCM) calculations (298 K)
The natures of all the located TS are similar. They all have biplanar structures, typical of TS in HDA reactions (Fig. 5). Within the TS’s two new σ bonds always form. Their degree of advancement, however, is different. That is, while the formation of the C4–C5 bond is more advanced, the formation of C6–O1 bond is more delayed. This is justified by the nature of local nucleophile-electrophile interactions. TS asymmetry may be to some extent increased by increasing the polarity of the reaction medium. The extent, however, is insufficient to force the change of a two-stage one-step mechanism [23] to a stepwise mechanism.
It is noted that all attempts to optimize the hypothetical zwitterions five and six as stable structures were unsuccessful.
Conclusions
The compilation of quantum chemical computations and experimental kinetic measurements proves it beyond any doubt that the mechanism of (E)-2-aryl-1-cyano-1-nitroethenes reactions with ethyl vinyl ether is polar. Both their regioselectivity and the character of the substituent effect can be interpreted quite accurately according to the theory of electrophilicity and nucleophilicity indexes.
The reactions in question proceed through polar, asymmetric transition complexes. Their asymmetry, however, is insufficient to encourage a zwitterionic mechanism. Detailed analysis of IRC trajectories of the TS proved that according to Domingo’s terminology [23], the reactions may be considered “two-stage one-step cycloadditions”.
Refereneces
Namboothiri INN, Tastogi N (2008) Synthesis of heterocycles via cycloadditions. Springer, Heidelberg
Barański A, Kelarev VI (1990) Chem Heterocycl Compd 26:371–386
Ioffe SL (2007) In: Feuer H (ed) Nitrile oxides, nitrones and nitronates in organic synthesis. Wiley-Interscience, New Yersey
Denmark SE, Cottell JJ (2002) In: Padwa A, Pearson WH (eds) Wiley, Chem Heterocycl Compd vol 59. New York
Ono N (2001) The nitro group in organic synthesis. Wiley-VCH, Weinheim
Tietze LF, Kettschau G (1997) Top Curr Chem 1(189):70–74
Jasiński R, Kwiatkowska M, Barański A (2007) Wiad Chem 61:485–514
Korotayev VY, Barkov AY, Slepukhin PA, Kodess MI, Sosnovskikh VYa (2011) Mendeleev Commun 21:112–114
Korotayev VY, Sosnovskikh VYa, Barabanov MA, Barkov AY, Kodess MI (2010) Mendeleev Commun 20:17–19
Jasiński R, Barański A (2010) J Mol Struct (Theochem) 949:8–13
Fringuelli F, Matteucci M, Piermatti O, Pizzo F, Burla MC (2001) J Org Chem 66:4661–4666
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Tetrahedron 58:4417–4423
Pérez P, Domingo LR, Aizman A, Contreras R (2007) In: Toro-Labbé A (ed) Theoretical aspects of chemical reactivity, vol 19. Elsevier, Amsterdam
Chattaraj PK, Giri G, Duley S (2011) Chem Rev 111:43–75
Domingo LR, Perez P, Saez JA (2013) RSC Adv 3:1486–1494
Leniewski A (1993) In: Wróbel A (ed) Preparatyka i elementy syntezy organicznej. PWN, Warszawa
Jasiński R (2013) Preparatyka alifatycznych nitrozwiązków. RTN, Radom
Jasiński R, Kwiatkowska M, Barański A (2006) Czasopismo Techn (Chemia) 2006:41–46
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09. Gaussian Inc, Wallingford
Jasiński R, Kwiatkowska M, Sharnin V, Barański A (2013) Monatsh Chem 144:327–335
Jasiński R, Kwiatkowska M, Barański A (2011) J Phys Org Chem 24:843–853
Jasiński R, Kwiatkowska M, Barański A (2009) J Mol Struct (Theochem) 910:80–87
Domingo LR, Sáez JA (2009) Org Biomol Chem 7:3576–3583
Kwiatkowska M, Jasiński R, Mikulska M, Barański A (2010) Monatsh Chem 141:545–548
Tomasi J, Mennucci B, Cancés E (1999) J Mol Struct (Theochem) 464:211–226
Jasiński R, Koifman O, Barański A (2011) Central Eur J Chem 9:1008–1018
Gómez AV, Aranda AI, Moreno A, Cossio FP, de Cózar A, Diaz-Ortiz Á, de la Hoz A, Prieto P (2009) Tetrahedron 65:5328–5336
Arroyo P, Picher MT, Domingo LR, Terrier F (2005) Tetrahedron 61:7359–7365
Arroyo P, Picher MT, Domingo LR (2004) J Mol Struct (Theochem) 709:45–52
Domingo LR, Aurell MJ, Kneeteman MN, Mancini PM (2008) J Mol Struct (Theochem) 853:68–76
Leroy G, Sana M, Burke LA, Nguyen MT (1980) Quantum Theory Chem React 1:91–144
Parr RG, Yang W (1989) Density functional theory of atoms and molecules. Oxford University, New York
Parr RG, von Szentpaly L, Liu S (1999) J Am Chem Soc 121:1922–1924
Domingo LR, Chamorro E, Perez P (2008) J Org Chem 73:4615–4624
Schwetlick K (1971) Kinetische Metoden zur Untersuchung von Reaktionsmechanismen. VEB, Berlin
Reichardt Ch, Welton T (2011) Solvents and solvent effects in organic chemistry. Wiley-VCH, Weinheim
Yepes D, Donoso-Tauda O, Perez P, Murray JS, Politzerb P, Jaque P (2013) Phys Chem Chem Phys 15:7311–7320
Acknowledgments
The generous allocation of computing time by the regional computer center “Cyfronet” in Cracow (Grant Number MNiSW/Zeus_lokalnie/PK/009/2013) and financial support from the Polish Ministry of Science and Higher Education (Grant Number C-2/63/2013/DS) are gratefully acknowledged. Ewa Dresler is a recipient of a Ph.D. scholarship under a project funded by the European Social Fund.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Jasiński, R., Kubik, M., Łapczuk-Krygier, A. et al. An experimental and theoretical study of the hetero Diels–Alder reactions between (E)-2-aryl-1-cyano-1-nitroethenes and ethyl vinyl ether: one-step or zwitterionic, two-step mechanism?. Reac Kinet Mech Cat 113, 333–345 (2014). https://doi.org/10.1007/s11144-014-0753-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-014-0753-8