
Abstract
Reaction paths for [3 + 2] cycloaddition (32CA) between 2-methyl-1-nitroprop-1-ene and (Z)-C-aryl-N-phenylnitrones were explored in detail at the B3LYP/6-31G(d) level of theory. All of the 32CA processes considered were found to be initiated by the attack of the most nucleophilic oxygen atom in the nitrone molecule on the most electrophilic carbon atom (Cβ) in the nitroethylene moiety. This type of interaction favors the formation of 4-nitro-substituted cycloadducts. Additionally, based on a molecular electron density theory (MEDT) study, the 32CA processes of interest should be considered polar processes with asynchronous transition states (TSs). However, the asynchronicity of the localized TSs is unexpectedly low and clearly insufficient to enforce a stepwise zwitterionic mechanism.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Conjugated nitroalkenes are valuable materials for organic synthesis. This is a consequence of the wide range of potential transformations of the NO2 group; for example, nitro compounds can be converted into nitronates [1,2,3,4], hydroxylamines [1, 5,6,7], amines [1, 5, 6, 8], oximes [1, 7, 9, 10], carbonyl compounds [1, 11, 12], and many other types of compounds. Considering their biological and pharmacological activities, nitro compounds have a variety of important applications [13,14,15,16,17]. Additionally, conjugating the NO2 group with a vinyl system can activate the alkene for cycloaddition reactions with nucleophilic three-atom components (TACs [18]) or conjugated dienes, which yield various five-membered [19] and six-membered [20] carbo- and heterocycles that are difficult or impossible to regio- and stereoselectively synthesize through an alternative approach.
There are many known [3 + 2] cycloaddition (32CA) reactions of the parent nitroethene molecule with nitrones [21,22,23,24,25]. These reactions have been also explored in detail using various theoretical approaches [21, 22, 26]. Additionally, similar cycloadditions of 1-substituted nitroethenes (such as 2-nitroprop-1-ene [27], 1-chloronitroethene [28, 29], and 1-bromonitroethene [27]) and 2-substituted nitroethenes (e.g., (E)-2-arylnitroethenes [30, 31], (E)-1-nitroprop-1-ene [32], (E)-3,3,3-trichloro-1-nitroprop-1-ene [27, 33, 34], (E)-3,3,3-trifluor-1-nitroprop-1-ene [35], and (E)-3-nitroacrylate [27]) analogs have been explored. Recently, preliminary studies of 32CA processes between 1,2-disubstituted nitroethenes and (Z)-(3,4,5-trimethoxyphenyl)-N-methylnitrone have also been published [28, 36]. However, no examples of the 32CA of 2,2-disubstituted nitroethene analogs and nitrones have been described in the literature so far. Thus, the influence of bis substitution at the β position of the nitroethylene moiety on the course of the 32CA is completely unknown. It should also be noted that the participation of 2,2-disubstituted nitroethene analogs in other types of cycloadditions has only rarely attracted the attention of chemists. Therefore, we decided to initiate comprehensive studies in this area. To this end, we analyzed the theoretically possible reaction paths (Scheme 1) of 32CA reactions involving 2-methyl-1-nitroprop-1-ene (1) as a model electrophilic component and a homogeneous group of nitrones (2a–g) as model TACs, and we report the results of that study in the present paper.

Theoretically possible paths of cycloadditions between 1a-g and 2
In particular, we decided to (i) analyze the nature of the intermolecular interactions involved in the elementary reaction steps in the framework of molecular electron density theory (MEDT) [37], (ii) predict regio- and stereoselectivity, and (iii) explore reaction profiles and examine all critical structures. It should be underlined at this point that the mechanistic aspects of these types of reactions require detailed studies. Due to the high global electrophilicity of the nitroethylene moiety, a “classical” one-step mechanism may compete with a stepwise zwitterionic mechanism. Such a stepwise mechanism has recently been assigned to several 32CA processes involving nitroethene as well as those of its 1- or 2-substituted analogs. For example, cycloadditions of (E)-3,3,3-trichloro-1-nitroprop-1-ene to (Z)-C-anthryl-C-phenylnitrone [33], 1,1-dinitroethene to (Z)-C,N-diphenylnitrone [38], nitroethene to 2,2,4,4-tetramethyl-3-thiocyclobutanone S-methylide, and some other cycloadditions [39, 40] all proceed via zwitterionic intermediates.
Computational details
The quantum-chemical calculations reported in this paper were performed using the B3LYP functional along with the 6-31G(d) basis set included in the GAUSSIAN 09 package [41]. All calculations were carried out at the same level of theory that was used to study the 32CA reactions of different type of nitrones and 2-substituted nitroethenes. It is important to note that good correlations between the calculated parameters of the critical structures [27, 32, 42], the results of a comprehensive kinetic study (Eyring parameters) [34, 42], the nature of solvent effects [34, 42], and a quantitative description of secondary isotope effects [43] were obtained when this level of theory was applied. This suggests that the B3LYP/6-31G(d) level can accurately illustrate the nature of the critical structures in the cycloaddition process of interest in the present work. However, in order to prove that this rather low level of theory was adequate for our needs, we also performed additional calculations for the model process at higher levels of theory (that also take, for example, the dispersion correction into account [44]).
Optimizations of the critical structures were performed with the Berny algorithm, whereas the transition states (TSs) were calculated using the QST2 procedure. TSs along the considered reaction paths were localized through an alternative methodology which involved gradually changing the distance between the reaction centers (with optimization performed after each step). This approach yielded TSs identical to those obtained previously.
Localized critical points were successfully verified via frequency calculations. All reactants and products were found to be characterized by positive Hessian matrices. All TSs showed only one negative eigenvalue in their diagonalized Hessian matrices, and their associated eigenvectors were confirmed to correspond to the motion along the reaction coordinate under consideration. To further verify the TSs, IRC calculations were performed. The effect of the solvent on the reaction paths was included using the polarizable continuum model (PCM) [45]. Global electron density transfer between substructures (GEDT) [46] was calculated according to the equation
where qA is the net charge, and the sum is performed over all the atoms of the nitroalkene.
New σ-bond development (l) was expressed based on the following equation involving the distance between the reaction centers in the transition structure (rTSX–Y) and the corresponding distance in the product (rPX–Y) [29]:
ELF studies were performed with the TopMod [47] program, using the corresponding gas-phase B3LYP/6-31G(d) monodeterminantal wavefuctions. ELF localization domains were obtained for an ELF value of 0.75.
Electronic properties of the reactants were estimated via the following previously reported recommended relations [37, 48, 49]:
Global nucleophilicities (N) [50] were calculated using the equation
The local electrophilicity (ωk) of atom k was calculated using the index ω and the respective Parr function P+k [51]:
The local nucleophilicity (Νk) of atom k was calculated using the index N and the respective Parr function P−k [51]:
Results and discussion
Investigating the intermolecular interactions through a MEDT study
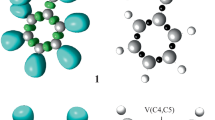
We first analyzed the nature of the interactions between the reactants by probing the electronic properties of the reactants. A topological analysis of the ELF of 2-methyl-1-nitroprop-1-ene 1 and C,N-diphenylnitrone 2d was performed in order to characterize electronic structure. The ELF attractors, together with the basin populations and the ELF localization domains, are shown in Fig. 1. ELF topological analysis of 2-methyl-1-nitroprop-1-ene 1 shows the presence of two disynaptic basins within the C4–C5 bonding region, V(C4–C5) and V′(C4–C5), involving a total population of 3.64 e. This indicates that the C4–C5 bond possesses strong double-bond character (Fig. 1). ELF topological analysis of C,N-diphenylnitrone 2d shows the presence of two monosynaptic basins, V(O1) and V′(O1), involving a total population of 6.02 e; one single-bond disynaptic basin V(O1,N2) involving 1.29 e; and one disynaptic basin between the N2 and C3 centers, V(N2,C3). Thus, based on a series of studies carried out by Domingo and coworkers [26, 52, 53] and the topological analysis of the ELF, the electronic structure of C,N-diphenylnitrone 2d (just like that of C,N-dimethylnitrone) indicates that it is zwitterionic, enabling its participation in zw-type 32CA reactions.

B3LYP/6-31G(d) ELF localization domains of 2-methyl-1-nitroprop-1-ene 1 and C,N-diphenylnitrone 2d at an isosurface value of ELF = 0.75, ELF basin attractor positions, and the most representative valence basin populations
The respective global and local indices were then estimated using equations defined on the basis of conceptual density functional theory [37, 48, 49]. A similar approach was recently successfully used to explain the courses of a number of different biomolecular processes involving unsaturated nitrocompounds (see, for example, [32, 54,55,56]). In light of the results of the MEDT study, 2-methyl-1-nitroprop-1-ene 1 was classified as a strong electrophile (ω > 2 eV) (Table 1). For comparison, the value of N for 1 is only 1.53 eV. Thus, 2-methyl-1-nitroprop-1-ene 1 should be considered an electrophilic component in the 32CA processes of interest here.
On the other hand, the electronic properties of the C-aryl-N-phenylnitrones 2a–g vary to a considerable extent (Table 1). In particular, the global electrophilicity of C,N-diphenylnitrone 2d is 1.67 eV, which classifies it as a rather strong electrophile. However, gradually increasing the electron-donating properties of the substituent at the 4-position in the phenyl ring conjugated with the carbon atom of the NO moiety causes a change in electrophilicity. For example, for the dimethylamino-substituted nitrone 2a, ω drops below 1.3 eV, indicating that it will exhibit only moderate electrophilic properties. Replacing the dimethylamino group with a NO2 group again modifies the properties of the nitrone. For instance, the nitro-substituted nitrone 2g is characterized by very strong electrophilicity (ω > 2.8 eV). That said, the values of the global N indices show, without any doubt, that all of the considered nitrones are nucleophilic. Therefore, the cycloadditions considered here should be interpreted as polar processes [37]. This is a consequence of the highly nucleophilic nature of the TACs and the notably electrophilic nature of ethylene derivative 1. The courses of the reactions of interest here are influenced by the nucleophilic attack of the oxygen atom of the CNO moiety of nitrone (NO > 1.2 eV; NC < 0.6 eV) on the most electrophilic (ωβ = 0.94 eV) carbon atom (Cβ) of the nitroalkene. If we assume that this governs the course of the reaction, then the products of the cycloaddition should be the stereoisomeric 4-nitroisoxazolidines 3a–g and/or 4a–g.
Energy profiles
In this part of the study, we first carried out DFT simulations of theoretically possible reaction channels for the 32CA of 2-methyl-1-nitroprop-1-ene 1 with the parent C,N-diphenylnitrone 2d. Next, in a similar manner, we analyzed the 32CA processes involving the most nucleophilic nitrone 2a and the least nucleophilic nitrone 2g.
Quantum-chemical calculations showed that the reaction 1 + 2d in toluene solution leads to molecular complexes (MCs) at the initial stage in all four of the reaction channels considered (A–D; see Scheme 2, Figs. 2 and 3). The MCs are formed without having to surmount an activation barrier. The decrease in enthalpy caused by the occurrence of MCs along the reaction path is not significant (it does not exceed 2 kcal mol−1). It should be noted that the MCs are enthalpic at room temperature (Tables 2 and 3). Due to the value of the entropic factor (ΤΔS), ΔG is > 0, which excludes the existence of MC structures that may be considered stable from a thermodynamic point of view. A similar type of intermediate was also localized in the analogous 32CAs involving nitrones 2a and 2g.

Reaction pathways

Energy profiles for paths A and B of the 32CA of 2-methyl-1-nitroprop-1-ene 1 with (Z)-C,N-diphenylnitrone 2d in toluene solution, according to B3LYP/6-31G(d) (PCM) calculations

Energy profiles for paths C and D of the 32CA of 2-methyl-1-nitroprop-1-ene 1 with (Z)-C,N-diphenylnitrone 2d in toluene solution, according to B3LYP/6-31G(d) (PCM) calculations
Further conversion of the 1 + 2d intermolecular system along the reaction path, regardless of the 32CA channel, leads to a transition complex (TS). The existence of a TS is confirmed by the presence of one imaginary eigenvalue in the Hessian. Based on the corresponding activation enthalpies, the order of preference for the reaction channels is B > A > D ≈ C. The ΔH values are relatively low (19.1 and 17.9 kcal mol−1 for channels A and B, respectively) for the TSs of the reaction channels that yield isoxazolidines in which the nitro group is at the C4 position in the heterocyclic ring. Significantly higher values (> 25 kcal mol−1) are observed in the reaction channels (C and D) that yield isoxazolidines with the nitro group at the C5 position. It should be noted that the order of preference for the channels based on the kinetics (as gauged through the Gibbs free energy of activation, ΔG) is similar to the order of preference based on the ΔH values. It should therefore be assumed that the reaction channels leading to 5-nitro-substituted isoxazolidines are kinetically forbidden. This suggests that only the 4-nitroisoxazolidines 3a and 4a are formed during the reaction. This observation correlates well with the analysis of two-center interactions discussed above. It should be noted that all attempts to identify alternative TSs leading to adducts 3–6 were unsuccessful.
When the structure of 2a is modified by introducing substituents onto the phenyl rings of nitrone 2d, the profiles of all the considered reaction channels do not change qualitatively. However, some transformations do change from a quantitative perspective (Tables 2 and 3). In particular, the presence of the electron-donating (EDG) dimethylamino group results in a lower activation barrier in channels A and B. At the same time, the presence of the electron-withdrawing (EWG) nitro group has the opposite effect. This conclusion correlates well with the previous analysis of the global nucleophilicities of the nitrones. It is interesting that the influence of the nature of the substituent on the kinetics of reaction channels C and D is marginal. For example, the ΔG value for the activation of the reaction involving the parent nitrone 2d is only 0.1–0.3 kcal mol−1 higher than that for the same transformation involving nitrone 2a.
When more polar solvents (Table 2) were included as dielectric media in DFT calculations, the reaction profiles did not change qualitatively, but they did change quantitatively, albeit to a minor degree. In particular, the MCs in all the profiles were shallower (Table 2), and all activation barriers were slightly higher. However, even in the strongly polar solvent nitromethane, the order of reaction channel preference based on the kinetics was the same as that obtained in toluene solution. Additionally, in all of the considered solvents, reaction channels C and D appeared to be kinetically forbidden.
Critical structures
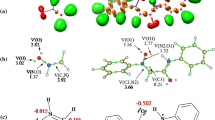
As noted in previous paragraphs, the first stage in each of the 32CA processes considered is always the formation of a molecular complex (MC). Analysis of the structural aspects of each MC showed that the lengths of the O1–N2, N2–C3, and C4–C5 bonds were practically identical to their lengths in the individual reactants. However, in the transition state (TS) structures, the interatomic distances C3–C4 and C5–O1 (Tables 4 and 5) are outside of the typical bond length ranges for carbon–carbon and carbon–oxygen bonds. Also, in the MCs, the orientations of the reaction centers with respect to each other are not the same as they are in the final products (Fig. 4). Additionally, based on the GEDT [46] values (0.0 e), none of the MCs are charge-transfer complexes. Similar molecular complexes were observed in the 32CA reactions between allenyl-type TACs and nitroacetylene [57], benzonitrile N-oxides and nitroethene [55], as well as between diazocompounds and hexafluoroacetone [58].

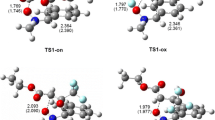
Critical structures in the 32CA of 2-methyl-1-nitroprop-1-ene 1 with (Z)-C,N-diphenylnitrone 2d in toluene solution, according to B3LYP/6-31G(d) (PCM) calculations
Conversion of the MC into the respective adduct was always found to occur via a single TS (Figs. 2 and 3, Tables 2 and 3). The nature of the TS depends to some extent on the relative orientations of its substructures. In particular, in the TSs obtained in the channels that ultimately yield 4-nitroisoxazolidines (A and B), the C5–O1 sigma bond is formed earlier than the sigma bond between C3 and C4. However, in the TSC and TSD structures, the C3–C4 sigma bond is formed faster than the C5–O1 sigma bond. In all cases, the synchronicity of the localized TS is controlled by the formation of the new bond at the reaction center associated with the β carbon atom from the nitroethylene moiety. This conclusion correlates well with the earlier discussion of the local reactivities of the reactants. Regardless of the synchronicity of the new sigma bond, all of the considered TSs were observed to exhibit a significantly polar nature. This was confirmed by the results of the GEDT analysis (Tables 4 and 5).
Our study shows that the synchronicity of the TS may be controlled to some degree by the particular substituents present in the nitrone molecule or/and the polarity of the reaction environment. This substituent effect is readily apparent in the kinetically favored reaction channels A and B. In particular, for the TSs TSA and TSB in the 32CA involving the EDG-substituted nitrone 2a, the difference between the values of the new bond development index for the two bonds C3–C4 and C5–O1 (see the Δl values in Table 5) is 0.29 and 0.23, respectively. The analogous index-value differences for the 32CA involving the EWG-substituted nitrone 2g are 0.15 and 0.11, respectively.
On the other hand, the solvent polarity was observed to influence the synchronicity of the TS in all of the considered reaction channels. For example, for the 32CA of 1 + 2d, the asynchronicity of the TS is higher in nitromethane than in toluene by 44%, 64%, 31%, and 25% for channels A, B, C, and D, respectively (see the Δl values in Table 4). It is important to note, however, that not even this asynchronicity of the TS is sufficient to enforce a stepwise zwitterionic mechanism.
Lastly, we performed full calculations of the reaction paths for the 32CA of 1 + 2d at more advanced levels of theory (that accounted for dispersion functions). The results obtained (i.e., the order of preference of the reaction channels based on the kinetics, as well as the key geometric parameters of the TSs) were very close to those given by the B3LYP/6-31G(d) computational study (Tables 6 and 7). In other words, there was no need to apply a high level of theory to obtain accurate results for the systems of interest. It should also be noted that a B97D study underestimated the enthalpies of activation. According to those calculations, the ΔH≠ values for the preferred reaction paths were about 5–6 kcal mol−1, whereas the experimentally estimated values for the preferred channels of the 32CA between 1-nitroprop-1-ene 1 (which is not very sterically crowded) and the same nitrone 2d are 9.5 and 11.3 kcal mol−1, respectively [43]. These values imply that including dispersion functions [44] in a DFT study of the cycloadditions of interest here is not a good approach.
Conclusions
Our B3LYP/6-31G(d) computational study utilizing molecular electron density theory (MEDT) undoubtedly showed that 32CA processes between 2-methyl-1-nitropro-1-ene and (Z)-C-aryl-N-phenylnitrones should be treated as polar reactions. This was confirmed by our analysis of global electron density transfer for the localized TSs. However, in contrast to the analogous process involving 1,1-dinitroethene, the cycloadditions of interest here involved only one TS. Each of the localized TSs was found to be asynchronous, but this asynchronicity was not sufficient to enforce a stepwise zwitterionic mechanism. Every attempt to find zwitterionic intermediates during the reaction paths considered here was unsuccessful.
A detailed analysis of the reaction paths indicated that all of the considered 32CA processes are initiated by two-center interactions between the most nucleophilic oxygen atom in the nitrone molecule and the most electrophilic carbon atom (the βC) in the nitroethylene moiety. This type of interaction favors the formation of 4-nitro-substituted cycloadducts, in good agreement with the results of the detailed exploration of theoretically possible reaction channels. It appears that competitive channels leading to 5-nitroisoxazolidines are kinetically forbidden.
References
Kabalka GW, Varma RS (1987) Syntheses and selected reductions of conjugated nitroalkenes—a review. Org Prep Proc Int 19:283–328
Jasiński R, Kubik M, Łapczuk-Krygier A, Kącka A, Dresler E, Boguszewska-Czubara A (2014) An experimental and theoretical study of the hetero Diels–Alder reactions between (E)-2-aryl-1-cyano-1-nitroethenes and ethyl vinyl ether: one-step or zwitterionic, two-step mechanism? React Kinet Mech Cat 113:333–345
Mikhaylov AA, Zhmurov PA, Naumova AS, Khoroshutina YA, Sukhorukov AY, Ioffe SL (2015) Stereoselective synthesis of spirocyclic nitronates by SnCl4-promoted reaction of nitroalkenes with C-2 substituted 4-methylidene-1,3-dioxolane. Mendeleev Commun 25:449–451
Tabolin AA, Sukhorukov AY, Ioffe SL, Dilman AD (2017) Recent advances in the synthesis and chemistry of nitronates. Synthesis 49:3255–3268
Laso NM, Quiclet-Sire B, Zarda SZ (1996) A new selective reduction of nitroalkenes into enamides. Tetrahedron Lett 37:1605–1608
Guy M, Freeman S, Alder JF, Brandt SD (2008) The Henry reaction: spectroscopic studies of nitrile and hydroxylamine by-products, formed during synthesis of psychoactive phenylalkylamines. Cent Eur J Chem 6:526–534
Rappaport Z (2009) The chemistry of functional groups. In: Rappaport Z, Liebman JF (ed) The chemistry of hydroxylamines, oximes and hydroxamic acids, part 1. Wiley, Chichester
Leroux M, Gall TL, Mioskowski C (2001) Enantioselective synthesis of α,α-disubstituted amines from nitroalkenes. Tetrahedron Asymmetry 12:1817–1823
Lee SH, Park YJ, Yoon CM (2003) Catalytic transfer hydrogenation of conjugated nitroalkenes using decaborane: synthesis of oximes. Org Biomol Chem 1:1099–1100
Cai S, Zhang S, Zhao Y, Wang DZ (2013) New approach to oximes through reduction of nitro compounds enabled by visible light photoredox catalysis. Org Lett 15:2660–2663
Halimehjani AZ, Namboothiri I NN, Hooshmand SE (2014) Nitroalkenes in the synthesis of carbocyclic compounds. RSC Adv 4:31261–31299
Ballini R, Petrini M (2015) The nitro to carbonyl conversion: new perspectives for a classical transformation. Adv Synth Catal 357:2371–2402
Boguszewska-Czubara A, Lapczuk-Krygier A, Rykala K, Biernasiuk A, Wnorowski A, Popiolek L, Maziarka A, Hordyjewska A, Jasiński R (2016) Novel synthesis scheme and in vitro antimicrobial evaluation of a panel of (E)-2-aryl-1-cyano-1-nitroethenes. J Enzyme Inhib Med Chem 31:900–907
Olender D, Żwawiak J, Zaprutko L (2018) Multidirectional efficacy of biologically active nitro compounds included in medicines. Pharmaceuticals 11:1–29
Herrera C, Vallejos GA, Loaiza R, Zeledón R, Urbina A, Sepúlveda-Boza S (2009) In vitro activity of thienyl-2-nitropropene compounds against Trypanosoma cruzi. Mem Inst Oswaldo Cruz 104:980–985
Shenvi S, Diwakar L, Reddy GC (2014) Nitro derivatives of naturally occurring β-asarone and their anticancer activity. J Med Chem 2014:1–5
Salman HH, Majeed NN (2013) Synthesis, characterization and study of biological activity of some new nitrone and isoxazolidine compounds. J Basrah Res (Sci) 39:99–111
Domingo LR, Emamian SR (2014) Understanding the mechanisms of [3+2] cycloaddition reactions. The pseudoradical versus the zwitterionic mechanism. Tetrahedron 70:1267–1273
Jasiński R (2015) 1,3-Dipolar cycloaddition reactions: mechanistic aspects and applications in organic synthesis. RTN, Radom
Ono N (2001) The nitro group in organic synthesis. Wiley, Weinheim, pp 159–181
Kula K, Łapczuk-Krygier A (2018) A DFT computational study on the [3+2] cycloaddition between parent thionitrone and nitroethene. Curr Chem Lett 7:27-34
Jasiński R (2009) Regio- and stereoselectivity of [2+3]cycloaddition of nitroethene to (Z)-N-aryl-C-phenylnitrones. Collect Czech Chem Commun 74:1341–1349
Jasiński R (2009) The question of the regiodirection of the [2+3] cycloaddition reaction of triphenylnitrone to nitroethene. Chem Heterocycl Compd 45:748–749
Burdisso M, Gandolfi R, Grünanger P (1989) Control of regiochemistry in nitrone cycloadditions. Regioselectivity of the reactions of trisubstituted nitrones with electron-deficient and conjugated dipolarophiles. Tetrahedron 45:5579–5594
Marzilli LG, Epps RCSLA, Allen JB (1973) Reversal of nitrone cycloaddition regioselectivity with electron-deficient dipolarophiles. J Am Chem Soc 95:5798–5800
Domingo LR, Ríos-Gutiérrez M, Pérez P (2018) Molecular electron density theory study of the reactivity and selectivities in [3+2] cycloaddition reactions of C,N-dialkyl nitrones with ethylene derivatives. J Org Chem 83:2182–2197
Jasiński R, Mróz K, Kącka A (2016) Experimental and theoretical DFT study on synthesis of sterically crowded 2,3,3,(4)5-tetrasubstituted-4-nitroisoxazolidines via 1,3-dipolar cycloaddition reactions between ketonitrones and conjugated nitroalkenes. Heterocyclic Chem 53:1424–1429
Jasiński R, Dresler E, Mikulska M, Polewski D (2016) [3+2] Cycloadditions of 1-halo-1-nitroethenes with (Z)-C-(3,4,5-trimethoxyphenyl)-N-methyl-nitrone as regio- and stereocontrolled source of novel bioactive compounds: preliminary studies. Curr Chem Lett 5:123–128
Jasiński R (2015) A stepwise, zwitterionic mechanism for the 1,3-dipolar cycloaddition between (Z)-C-4-methoxyphenyl-N-phenylnitrone and gem-chloronitroethene catalysed by 1-butyl-3-methylimidazolium ionic liquid cations. Tetrahedron Lett 56:532–535
Banerji A, Gupta M, Biswas PK, Prangé T, Neuman A (2007) 1,3-Dipolar cycloadditions. Part XII—Selective cycloaddition route to 4-nitroisoxazolidine ring systems. J Heterocycl Chem 44:1045–1049
Ramamoorthy V, Ramasubbu A, Muthusubramanian S, Sivasubramanian S (1999) Pillared buserite as a new catalytic material for the 1,3-dipolar cycloaddition of α-phenyl-N-(p-methyphenyl) nitrone with electron deficient olefins. Synth Commun 29:21–26
Jasiński R, Ziółkowska M, Demchuk OM, Maziarka A (2014) Regio- and stereoselectivity of polar [2+3] cycloaddition reactions between (Z)-C-(3,4,5-trimethoxyphenyl)-N-methylnitrone and selected (E)-2-substituted nitroethenes. Cent Eur J Chem 12:586–593
Jasiński R, Żmigrodzka M, Dresler E, Kula K (2017) A full regioselective and stereoselective synthesis of 4-nitroisoxazolidines via stepwise [3+ 2] cycloaddition reactions between (Z)-C-(9-anthryl)-N-arylnitrones and (E)-3,3,3-trichloro-1-nitroprop-1-ene: comprehensive experimental and theoretical study. J Heterocycl Chem 54:3314–3320
Jasiński R, Mróz K (2015) Kinetic aspects of [3+2] cycloaddition reactions between (E)-3,3,3-trichloro-1-nitroprop-1-ene and ketonitrones. React Kinet Mech Cat 116:35–41
Bigotti S, Malpezzi L, Molteni M, Mele A, Zanda M (2009) Functionalized fluoroalkyl heterocycles by 1,3-dipolar cycloadditions with γ-fluoro-α-nitroalkenes. Tetrahedron Lett 50:2540–2542
Jasiński R (2018) Competition between one-step and two-step mechanism in polar [3+2] cycloadditions of (Z)-C-(3,4,5-trimethoxyphenyl)-N-methyl-nitrone with (Z)-2-EWG-1-bromo-1-nitroethenes. Comput Theor Chem 1125:77–85
Domingo LR, Ríos-Gutiérrez M Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21:748
Jasiński R (2013) Competition between the one-step and two-step, zwitterionic mechanisms in the [2+3] cycloaddition of gem-dinitroethene with (Z)-C,N-diphenylnitrone: a DFT computational study. Tetrahedron 69:927–932
Jasiński R (2015) In the searching for zwitterionic intermediates on reaction paths of [3+2] cycloaddition reactions between 2,2,4,4-tetramethyl-3-thiocyclobutanone S-methylide and polymerizable olefins. RSC Adv 5:101045–101048
Huisgen R, Penelle J, Mloston G, Buyle Padias A, Hall Jr HK (1992) Can polymerization trap intermediates in 1,3-dipolar cycloadditions? J Am Chem Soc 114:266–274
Frisch MJ, Trucks GW, Schlegel HB et al (2009) Gaussian 09, rev A.1. Gaussian, Inc., Wallingford
Szczepanek A, Jasińska E, Kącka A, Jasiński R (2015) An experimental and quantumchemical study of [2+3] cycloaddition between (Z)-C-(m,m,p-trimethoxyphenyl)-N-(p-methyphenyl)-nitrone and (E)-3,3,3-trichloro-1-nitroprop-1-ene: mechanistic aspects. Curr Chem Lett 4:33–44
Jasiński R (2004) PhD thesis. Cracov University of Technology, Cracov
Grimme S (2006) Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J Comp Chem 27:1787–1799
Cossi M, Rega N, Scalmani G, Barone V (2003) Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J Comp Chem 24:669–681
Domingo LR (2014) A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4:32415–32428
Noury S, Krokidis K, Fuster F, Silvi B (1999) Computational tools for the electron localization function topological analysis. Comput Chem 23:597–604
Perez P, Domingo LR, Aurell MJ, Contreras R (2003) Quantitative characterization of the global electrophilicity pattern of some reagents involved in 1,3-dipolar cycloaddition reactions. Tetrahedron 59:3117–3125
Parr RG, Szentpaly L, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922–1924
Domingo LR, Chamorro E, Perez P (2008) Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions: a theoretical study. J Org Chem 73:4615–4624
Domingo LR, Perez P, Saez JA (2013) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3:1486–1494
Domingo LR, Ríos-Gutiérrez M (2017) A molecular electron density theory study of the reactivity of azomethine imine in [3+2] cycloaddition reactions. Molecules 22:750–770
Domingo LR, Ríos-Gutiérrez M, Adjieufack AI, Zdassa IM, Nouhou CN, Mbadcam JK (2018) Molecular electron density theory study of fused regioselectivity in the intramolecular [3+2] cycloaddition reaction of cyclic nitrones. Chem Select 3:5412–5420
Jasiński R (2018) β-Trifluoromethylated nitroethenes in Diels–Alder reaction with cyclopentadiene: a DFT computational study. J Fluor Chem 206:1–7
Jasiński R, Jasińska E, Dresler E (2017) A DFT computational study of the molecular mechanism of [3+2] cycloaddition reactions between nitroethene and benzonitrile N-oxides. J Mol Model 23:1–13
Jasiński R (2017) One-step versus two-step mechanism of Diels–Alder reaction of 1-chloro-1-nitroethene with cyclopentadiene and furan. J Mol Graph Model 75:55–61
Jasiński R (2015) Nitroacetylene as dipolarophile in [2+3] cycloaddition reactions with allenyl-type three-atom components: DFT computational study. Monatsh Chem 146:591–599
Jasiński R (2015) On the question of zwitterionic intermediates in 1,3-dipolar cycloadditions between hexafluoroacetone and sterically crowded diazocompounds. J Fluor Chem 176:35–39
Acknowledgements
The authors thank the regional computer center “Cyfronet” in Cracow (grant no. MNiSW/Zeus_lokalnie/PK/009/2013) for allocating them computing time.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dresler, E., Kącka-Zych, A., Kwiatkowska, M. et al. Regioselectivity, stereoselectivity, and molecular mechanism of [3 + 2] cycloaddition reactions between 2-methyl-1-nitroprop-1-ene and (Z)-C-aryl-N-phenylnitrones: a DFT computational study. J Mol Model 24, 329 (2018). https://doi.org/10.1007/s00894-018-3861-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-018-3861-y