
Abstract
Background and aims
To assess the impacts of soil microbes and plant genotype on the composition of maize associated bacterial communities.
Methods
Two genotypes of Brazilian maize were planted indoors on sterile sand, a deep underground subsoil, and a nutrient-rich topsoil from the Amazon jungle (terra preta). DNA was extracted from rhizospheres, phyllospheres, and surface sterilized roots for 16S rDNA fingerprinting and next generation sequencing.
Results
Neither plant genotype nor soil type appeared to influence bacterial diversity in phyllospheres or endospheres. Rhizospheres showed strikingly similar 16S rDNA ordination of both fingerprinting and sequencing data, with soil type driving grouping patterns and genotype having a significant impact only on sterile sand. Rhizospheres grown in non-sterile soils contained greater bacterial diversity than sterile-sand grown ones, however the dominant OTUs (species of Proteobacteria and Bacteroidetes) were found in all rhizospheres suggesting seeds as a common source of inoculum. Rhizospheres of the commercial hybrid appeared to contain less bacterial diversity than the landrace.
Conclusions
Maize rhizospheres receive diverse bacteria from soil, are influenced by the genotype or treatment of the seed, and are dominated by species of Proteobacteria, Actinobacteria, Bacteroidetes, and Firmicutes. As many dominant 16S rDNA sequences were observed in rhizospheres grown in both sterile and non-sterile substrate, we conclude that the most common bacterial cells in juvenile maize rhizospheres are seed transmitted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Maize (corn, Zea mays ssp. mays L.) is one of the most productive and important plants in modern agriculture. Research into the maize microbiome aims to further improve its productivity and resistance to stress, by understanding the ecology of populations of microbes that live within and on the plants. Microbes living inside plants are known as endophytes and may have a direct effect on plant growth and health, given the intimate contact between the two (Johnston-Monje and Raizada 2011b). Microbes living on plant surfaces are known as epiphytes; those living in phyllospheres, or leaf surfaces inhabit the largest biological surface area on Earth (Whipps et al. 2008), but bacteria living on root surfaces and the attached soil (rhizospheres) exist at the site of critical uptake of nutrients and water. Common examples of rhizosphere bacteria that aid in plant growth, health and nutrition include Pseudomonas species which secrete antibiotics into the rhizosphere protecting wheat against take-all pathogens in the soil (Raaijmakers and Weller 1998) and species of Azospirrillum which secrete phytohormones into rhizospheres, promoting root growth, development and function (Okon and Itzigsohn 1995). Plants influence the microbial composition of their rhizospheres, by secreting a variety of compounds through their roots into the surrounding soil to feed and manipulate the microbes that live there. These secretions represent a drain of plant energy and resources, totalling up to 40 % of the carbon fixed by the plant (Badri and Vivanco 2009; Keister et al. 1991). Plants appear to favour the colonization of their rhizospheres and endospheres by Proteobacteria above all other bacterial phyla, although Bacteroidetes, Actinomycetes and Firmicutes are also commonly observed in both niches, while Acidobacteria and Planctomycetes are only common in rhizospheres (Bulgarelli et al. 2013; Hawkes et al. 2007; Philippot et al. 2013).
While the origin of phyllosphere bacteria remains poorly defined (Bulgarelli et al. 2013), the origins of both endophytic bacteria (Bulgarelli et al. 2012; Edwards et al. 2015; Long et al. 2008; Lundberg et al. 2012) and rhizosphere bacteria (Berg and Smalla 2009; Normander and Prosser 2000; Philippot et al. 2013; Singh et al. 2007) are traditionally thought to be from the soil. Contrary to a soil origin, there is evidence that bacterial endophytes in and/or on maize seeds contribute to the majority of the root endosphere bacterial population (Johnston-Monje et al. 2014; Johnston-Monje and Raizada 2011a), and that at least some of these bacteria are able to travel within the plant, exit the roots and colonize the rhizosphere (Johnston-Monje and Raizada 2011a). Similar evidence exists that seed endophytes are found in the endosphere and rhizosphere microbiota of rice plants (Hardoim et al. 2012), while studies on the cardon cactus which grows on bare desert rock, have shown that seedlings depend on seed transmitted bacteria to colonize the rhizosphere in order to help dissolve the rock substrate to help the plant absorb nutrients (Puente et al. 2009).
The advent of sensitive molecular fingerprinting and affordable next generation sequencing technologies has sparked a renaissance in rhizosphere research, with many new studies concerning the bacterial diversity present in maize rhizospheres (Bakker et al. 2015; Bouffaud et al. 2012; Castellanos et al. 2009; Dalmastri et al. 1999; Peiffer and Ley 2013; Peiffer et al. 2013), Arabidopsis rhizospheres (Bulgarelli et al. 2012; Lundberg et al. 2012; Micallef et al. 2009) and the rhizospheres of other important plant species (Costa et al. 2006; Edwards et al. 2015; Garbeva et al. 2008; Germida and Siciliano 2001; Inceoglu et al. 2010; van Overbeek and van Elsas 2008; Weinert et al. 2011).
To better tease apart the origins of, and influences on bacterial populations in phyllospheres, endopheres and rhizospheres of maize plants, in this study we acquired two different genotypes of maize and grew them in three very distinct substrates. The Lenha landrace is a chemically untreated seed of an ancient, open pollinated variety from Brazil with thick cobs which are so named because indigenous peoples are believed to have used them as firewood (Paterniani and Goodman 1977). For genotypic contrast, the EMBRAPA bred hybrid BRS 1030 (referred to as EMBRAPA in this paper) was grown from pesticide and fungicide coated seed, and represents an elite variety bred for stress tolerance and disease resistance in industrial agriculture, although this particular variety is known to be responsive to inoculation with beneficial bacteria (Alves et al. 2010). These different genotypes were grown on three very microbially different substrates to investigate the ability of soil to act as a source of rhizosphere, endosphere or phyllosphere microbes. One of the substrates was included as a microbial negative control, by repeatedly heat sterilizing quartz sand in an autoclave. The second substrate was chosen to represent a soil with no previous exposure to plant life, and was expected to be devoid of plant associated microbes; this powdery subsoil was taken from 400 m underground in a Brazilian iron mine. Finally, to observe the effect of a nutrient and microbe rich topsoil on the maize microbiome, we acquired terra preta do indio (“black earth of the natives”), harvested from an archeological site within the Brazilian Amazon jungle near the city of Manaus. This biochar was created by humans more than 500 years ago, probably by burning mounds of garbage under low oxygen conditions (Marris 2006).
To characterize the influence of plant genotype and soil substrate on the maize microbiome, the two maize genotypes were grown in the three substrates, side by side in a greenhouse. By extracting and amplifying the bacterial DNA in plant associated microbial niches (spermosphere, phyllosphere, root endosphere and rhizosphere) all samples were analyzed for species diversity (alpha diversity) and compared for changes in bacterial population structure (beta diversity) using principle component analysis (PCA) of 16S rDNA data from the DNA fingerprinting technique, terminal fragment length polymorphism analysis (TRFLP). Rhizosphere DNA was additionally studied using Miseq analysis of 16S rDNA. Sequencing data allowed us to measure the abundance of different phylotypes (not only their diversity), giving us the ability to estimate the relative size of bacterial populations in maize rhizospheres. We expected to observe that both phyllosphere and root endosphere bacterial diversity were most influenced by plant genotype, while rhizospheres were composed mostly of soil transmitted bacteria. We discuss the results and what the data suggests about origin of these bacterial populations.
Methods
Sources of seed
Two varieties of Zea mays ssp. mays were obtained from EMBRAPA Agrobiologia for the experiment: the Brazilian landrace Lenha and a variety originally developed by EMBRAPA, BRS 1030 (referred to as “EMBRAPA”). Lenha seed was chemically untreated and was shared courtesy of EMBRAPA Milho e Sorgo. EMBRAPA seeds were purchased from Brasmilho and treated with K-obiol 25 CE (containing deltamethrin insecticide), Actellic 500 CE (containing Pirimifos-Metil insecticide) and Maxim XL (fungicide containing Fludioxonil and Metalazyl-M).
Sources of soil
Sterile sand
Quartz sand was provided by EMBRAPA Agrobiologia and prepared by washing, mixing 4:1 with vermiculite, then sterilized by autoclaving twice at 121 °C for 1 h.
Iron mine subsoil
The powdery, white subsoil used in this experiment was originally extracted from 400 m underground in an iron mine belonging to the mining company Vale in the municipality of Itabira, Minas Gerais. It was kindly provided for this experiment by Dr. Sergio de Faria of EMBRAPA Agrobiologia.
Terra preta do índio
A rich, organic, black anthrosol was excavated from an archeological site called Hatahara in the municipality of Iranduba near the present day city of Manuas, Brazil. Apparently ancient pottery shards were removed from the sample before use in the experiment. This soil was kindly provided by Siu Mui Tsai of the Brazil Center of Nuclear Energy in Agriculture.
The physico-chemical properties of the three soils were analyzed at EMBRAPA Agrobiologia (Table 1: Physiochemical Properties) using methods developed at EMBRAPA (1997).
Culturing bacteria from soil extracts
In order to visualize the culturable bacterial diversity in the three different soils, 1 g samples from each of the autoclaved sand, subsoil and terra preta were measured into 2 mL Eppendorf tubes and suspended with 1 mL of sterile Na2HPO4 buffer. This liquid was then diluted 100X and 100 μl of this dilution was spread on R2A agar, then incubated at 30 °C for 3 days.
Plant experimental design and growth conditions
For each seed/substrate combination, 5 sterile, 500 mL plastic cups were filled with 200 mL of sterile 4:1 sand/vermiculite, plus another 200 mL of sand/vermiculite, or 200 mL of subsoil or 200 mL of terra preta (a total of 30 cups). Substrates were mixed to uniformity after placement in the cup. Seeds were not sterilized or soaked before planting, and two seeds were planted per cup and later culled to leave one seedling per container. Cups were placed in a climate controlled, locked greenhouse at the EMBRAPA Agrobiologia main campus and watered daily with autoclaved tap water. Plants received an average of 50 μmol m−2 s−1 of light during the 11 h day and the average daily temperature was 23 °C.
Harvesting phyllospheres, rhizospheres and root endospheres
To study root and shoot surfaces, whole plants at the 5-leaf stage (21 days old) were carefully shaken free from any soil, cut at the root/shoot boundary, the rotten seed hulk removed, roots were further shaken to remove any visible excess soil (including rocks and clods), then roots and shoots placed into separate sterile 50 mL conical tubes. 40 ml of sterile 50 mM Na2HPO4 buffer with 0.1 % Tween 20 was added to each tube, shaken briefly by hand, and then sonicated in a model T7 sonicating water bath (Thornton, Brazil) at 22.5 kHz for 10 min to dislodge soil particles and microbes from plant surfaces. The rhizosphere and phyllosphere washes were then centrifuged at 15,000 g for 15 min at 4 °C, generating a pellet. The supernatant was removed, and the pellet was suspended in 4 mL of phosphate buffer, then frozen for later DNA extraction.
Root tissues were placed in clean conical tubes and further rinsed with sterile phosphate buffer until no further cloudiness was observed in the wash. The washed roots were then treated with 2.5 % sodium hypochlorite and sonication as before for 10 min. The bleach was drained, and the treatment repeated with 2.5 % sodium hypochlorite and sonication for 10 min. The samples were then drained and rinsed with autoclaved, distilled water, then washed in 70 % ethanol for 10 min. The ethanol was removed, and samples rinsed three times with autoclaved, distilled water. To check for surface sterility, one piece of tissue per treatment was transiently placed on sterile R2A agar plates which were incubated for 10 days at 25 °C. Surface sterilized roots were placed in an autoclaved mortar and ground by crushing with sterile ceramic pestles. Crushed roots were washed and blended with 5 mL of 50 mM Na2HPO4 buffer and this “root juice” was decanted into a fresh tube then frozen for later DNA extraction.
For collecting spermospheres, five seeds each of both Lenha and EMBRAPA genotypes were soaked in 5 mL of sterile 50 mM potassium phosphate buffer with 0.1 % Tween 20 for 24 h, then shaken briefly by hand, and then sonicated in a model T7 sonicating water bath at 22.5 kHz for 10 min to dislodge microbes from seed surfaces. The resulting seed wash was concentrated by centrifugation at 15,000 g for 15 min at 4 °C, the supernatant decanted off, and the pellet suspended in 500 μL of fresh phosphate buffer and then frozen for later DNA extraction.
DNA extraction and terminal restriction fragment length polymorphism (TRFLP) from plant tissues and soil
Total DNA was extracted from three samples of each soil, and five samples of each set of phyllosphere, rhizosphere, spermosphere and root endosphere using the PowerSoil® DNA Isolation Kit (MoBio, USA). DNA was quantified using a Nanodrop (Thermo Scientific, USA).
Low titre microbial samples, including root endospheres and phyllospheres, were amplified using a nested PCR approach to generate sufficient product for TRFLP. A PCR mastermix was made with the following components per 25 μl volume: 2.5 μl Standard Taq Buffer (New England Biolabs), 0.5 μl of 25 mM dNTP mix, 0.5 μl of 10 mM 27 F-Degen primer with sequence 5′-AGRRTTYGATYMTGGYTYAG-3′ (Frank et al. 2008), 0.5 μl of 10 mM 1492r primer with sequence 5′-GGTTACCTTGTTACGACTT-3′, 0.25 μl of 50 mM MgCl2, 0.25 μl of bovine serum albumin, 0.25 μl of Standard Taq (New England Biolabs), 20 ng of total DNA, and double distilled water up to 25 μl total. Amplification was for 35 cycles in a PTC200 DNA Thermal Cycler (MJ Scientific, USA) using the following program: 96 °C for 3 min, 35X (94 °C for 30 s, 48 °C for 30 s, 72 °C for 1:30 min), 72 °C for 7 min.
Using the same conditions as above, 1.0 μl of the above PCR product was used as a template in a nested, fluorescently labelled PCR reaction. For the nested PCR, primer 799f with sequence 5′-AACMGGATTAGATACCCKG-3′ (Chelius and Triplett 2001) was labelled with 6FAM, and 1389r primer with sequence 5′-ACGGGCGGTGTGTRC-3′ was labelled with Max550, both by Integrated DNA Technologies (USA). The forward primer 799 F was chosen as it is strongly biased against amplifying chloroplast 16S rDNA; the much larger mitochondrial 18S fragments were later removed in silico after amplification and restriction, but before statistical analysis was performed.
Rhizosphere, spermosphere, soil, and water samples were amplified without nesting by amplifying 10 ng of DNA 40 times with the primers 799f and 1389r. 1.5 μl of the labelled PCR product was then added to a 8.5 μl restriction mixture [1U DdeI (NEB), 1X Buffer 3 (NEB)] and incubated in darkness at 37 °C for 16 h before being analyzed by sequencing gel using a 3730 DNA Analyzer alongside GeneScan 1200 LIZ Size Standards (Applied Biosystems, USA). There were 5 biological replicates per genotype/treatment combination and 3 replicates per soil.
TRFLP analysis
TRFLP results were analyzed using Genemarker software (SoftGenetics, USA) using default TRFLP settings with a modified fragment peak height cut off of 30 fluorescence units and manual verification for correct peak identification. The 6FAM and Max550 fragment sizes and peak heights were exported to Microsoft Excel and transformed into counts of presence or absence (see “Binary Transformed TRFLP data” in the electronic supplementary material). Probable primer dimer fragments were removed in silico (peaks 1–30 bp) as were mitochondrial and chloroplast fragment sizes.
For PCA, both forward and reverse fragments were used. To reduce experimental noise inherent in TRFLP analysis as recommended by others (Culman et al. 2008), peak height data was transformed into presence/absence (binomial values 0 or 1), and PCA of covariance was performed using XLStat software (Addinsoft, France).
MiSeq analysis of rhizosphere samples
PCR amplification was performed using universal bacterial 16S rDNA primers 515 F and 806R following a previously published protocol (Lundberg et al. 2013), where each template molecule was given a unique sequence, or molecule tag, through a linear amplification step prior to exponential amplification via PCR. Anti-chloroplast (5′-GGCTCAACCCTGGACAG-3′) and anti-mitochondria (5′-GGCAAGTGTTCTTCGGA-3′) peptide nucleic acid (PNA) blockers were added to the PCR reactions to block amplification of chloroplast and mitochondria as previously described (Lundberg et al. 2013). The PCR program was denaturation at 95 °C for 45 s followed by 34 cycles of denaturation at 95 °C for 15 s, PNA annealing at 78 °C for 10 s, primer annealing at 60 °C for 30 s, extension at 72 °C for 30 s and then a cooldown to 4 °C. All samples were cleaned with Agencourt beads using 35 μL of beads to clean the 50 μL PCR (0.7:1). DNA was eluted in 50 μL water. Amplicons of hypervariable region 4 of the 16S rRNA gene were sequenced in a single 2 × 250 bp paired-end run on the Illumina MiSeq platform. Demultiplexing of sequence reads was performed using the CASAVA v1.8.2 software.
Sequence reads were processed using MT-Toolbox software (Yourstone et al. 2014). This software makes consensus sequences (conseq) from two or more sequences carrying the same molecule tag, correcting sequence errors and biases. Only conseq representing two or more raw reads were used for analysis. Conseq were then binned into operational taxonomic units (OTUs) at a 97 % sequence similarity threshold using USEARCH 6 (www.drive5.com), and OTUs were assigned a taxonomic identity with the RDP classifier (Wang et al. 2007) trained on the Los Alamos National Laboratory’s GreenGenes taxonomic reference (4 February 2011 release, gg_97_otus_4feb2011), using the scripts in the QIIME v1.5.0 package (Caporaso et al. 2010). OTU counts were exported to Excel (Microsoft, USA) for further analysis including that shown in Figs. 4, 5, and 6 and are included in the electronic supplementary material as “Raw Conseq Counts”. 1059 OTU sequences observed by Miseq are included as a FASTA file “OTU Sequences” in the electronic supplementary material.
Statistical analysis of Miseq data: Heatmaps
OTU count tables were rarified to 1000 consensus sequences per sample (note: only 20 of the 30 rhizosphere samples amplified and of these only 15 had more than 1000 conseq). The data were log2 transformed to improve visualization of relative differences in the heatmap. Heatmaps were created using the heatmap.2 function in R (library gplots). Samples and OTUs were clustered by Bray-Curtis dissimilarity.
Statistical analysis of Miseq data: OTU diversity and consensus sequence taxonomy
In order to view bacterial diversity in individual rhizospheres, Miseq data in the best samples were rarified to 1900 conseq (the highest number of consensus sequences shared by these samples) then tallied to estimate OTU diversity (Fig. 4a) and total conseq counts to estimate the proportion of conseq belonging to each phylum (Fig. 4b).
Results
Root substrate analysis and study design
The three root substrates used in this study were characterized: a heat sterilized sand (Fig. 1a), a powdery subsoil from 400 m underground which had never been exposed to growing plants (Fig. 1b), and terra preta do indio – a carbon and nutrient rich biochar created by humans in tropical rainforests (Fig. 1c). To visualize the bacterial diversity in these substrates, aqueous extracts were taken from each and plated on R2A media. No microbes from the sterile sand grew on R2A, a few diverse species of bacteria grew from the subsoil, and an abundance of colonies grew from the terra preta extract (Fig. 1d, e and f). The physio-chemical analysis of these soils (Table 1) showed that both the sand and subsoil were low in carbon and nutrients, while the terra preta was much higher in levels of carbon, nitrogen and phosphorus – the pH of all three was close to neutral, varying from 6.3 for the terra preta to 7.3 for the sand.

Substrates, cultured bacteria from each substrate, seeds, and a typical maize plant used in this study. a Autoclaved sand b Subsoil. c Terra preta do indio (ancient clay pottery shards found in this soil sample are not shown). d Culturable microbes in an extract of autoclaved sand. e Culturable microbes from an extract of subsoil. f Culturable microbes from an extract of terra preta. g Seeds of the Brazilian landrace Lenha on the left and the chemically treated EMBRAPA hybrid BRS 1030 on the right. Scale bar indicates 10 mm. h An EMBRAPA maize plant grown in autoclaved sand for 21 days and being processed to harvest samples of phyllosphere, rhizosphere, and root endosphere. Scale bar indicates 10 mm
Lenha and EMBRAPA seeds (Fig. 1g) were grown to seedlings and then harvested (Fig. 1h).
TRFLP community profiling
To characterize bacterial communities, DNA was extracted from the starting materials of the experiment including heat sterilized water and sand, subsoil, terra preta, and spermospheres (wash water retained after rinsing seeds). DNA was also extracted from rhizospheres (wash water retained after rinsing and sonicating roots), phyllospheres (wash water retained after rinsing and sonicating shoots) and root endospheres (surface sterilized root tissue).
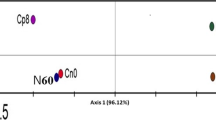
The bacterial communities in all DNA sample types were compared by conducting principle component analysis (PCA) using presence/absence counts from TRFLP profiles based on restriction of a 590 base pair section of the bacterial 16S rDNA (binary transformed TRFLP data supplied as electronic supplementary material). The starting materials showed clustering of autoclaved water, autoclaved sand, and subsoil, suggesting the three sample types did not contain sufficiently different bacterial DNA to distinguish each other (Fig. 2a). The terra preta soil group was significantly different from both subsoil and sterile sand, probably reflecting the diverse and abundant bacterial population evidenced in Fig. 1f.

PCA of bacterial 16S rDNA TRFLP profiles (both 6FAM and Max550 labelled fragments) after transformation to signal presence/absence data. Shown are the profiles of the starting materials used in this experiment as well as phyllospheres, root endospheres, and rhizospheres of two different genotypes of young maize plants grown in three different substrates. Ninety-five percent confidence ellipses were plotted around each group of samples based on chi-squared distribution showing treatment effects in (a) experimental materials at the start of the experiment, (b) maize phyllospheres (collected as shoot washings), (c) maize endospheres (from surface sterilized roots), and (d) rhizospheres (collected as root washings). 16S rDNA amplicons were generated using primers 799f/1389r and then were restricted using DdeI. Small fragments and those corresponding to 16S chloroplast rDNA or 18S rDNA were removed in silico before analysis
The two groups of spermosphere bacterial 16S TRFLP profiles were significantly different from those found in any of the soils or water, suggesting that seed surfaces harbour bacterial populations that are distinct from those in soil. Lenha and EMBRAPA spermospheres were not significantly distinct from each other suggesting similar populations of bacteria on both varieties of seed. PCA analysis also showed that 16S TRFLP profiles (95 % confidence ellipses) of neither phyllospheres (Fig. 2b), nor root endospheres (Fig. 2c) were distinct from each other. This result suggested that neither soil, seed treatment, nor plant genotype significantly influence bacterial diversity on leaf surfaces or inside roots of juvenile maize plants.
With respect to the rhizosphere, TRFLP data (Fig. 2d) showed significant differences in bacterial diversity between Lenha rhizospheres and EMBRAPA rhizospheres when plants were grown in sterile sand, suggesting a genotypic or seed treatment effect. Plants that were grown on either subsoil or terra preta had rhizospheres that were significantly different from those of sterile sand grown plants, suggesting that a diverse assortment of bacteria in soil do in fact colonize root surfaces. There was some overlap between 95 % confidence intervals drawn around terra preta and subsoil rhizospheres, suggesting that some of the same 16S TRFLP signals were added to root surfaces by both soil types (Fig. 2d). Of note was that the statistical separation observed between genotypes grown on sand was completely absent from rhizospheres of non-sterile soil grown plants.
MiSeq bacterial community profiling
Except for rhizospheres, PCR amplification and Miseq analysis of 16S rDNA worked (poorly) only for two Lenha spermospheres, one sand, and one endosphere sample (see “Raw ConSeq Counts” in the electronic supplementary material). All of the OTUs observed in Lenha spermospheres belonged to OTU 212, which was also observed in the EMBRAPA endosphere sample, but not in the sterile sand sample. For rhizospheres, we rarified conseq counts to 1000 per sample (only 15 of the 30 rhizosphere samples had more than 1000 consensus sequences), log2 transformed the counts, then used Bray-Curtis dissimilarity to cluster the data for display in heatmaps at the OTU (Fig. 3a) and phylum (Fig. 3b) levels. In these heatmaps, three distinct patterns of OTU diversity were observed in maize rhizospheres corresponding to the three substrates that the plants were grown on (Fig. 3a). The sterile sand clade with an average of 32 different OTU per sample had the lowest species diversity with an average Shannon index of 1.97, while the non-sterile soil clade contained two distinct sub-branches, one for subsoil with an average of 95 different OTU per sample and an average Shannon index of 2.58 and the other for terra preta rhizospheres with an average of 163 different OTU per sample and an average Shannon index of 3.17. These results were shown to be significant by Mann–Whitney statistical analysis which compared bacterial OTU diversity counts between soil treatments: sand to subsoil rhizosphere P = 0.006, sand to terra preta rhizosphere P = 0.006 and subsoil to terra preta rhizosphere P = 0.006. There were not enough samples to allow non-parametric statistics between genotypes, however EMBRAPA rhizospheres did segregate away from Lenha rhizospheres within each cluster of sterile sand, subsoil and terra preta (Fig. 3a).

Heatmaps of bacterial 16S rDNA OTUs derived from Miseq analysis of rhizospheres from maize plants grown in different substrates in a greenhouse. Consensus sequences were rarified to 1000 per sample and log2 transformed, then clustered by Bray-Curtis dissimilarity. Rhizosphere samples from sand grown plants are shaded in blue, samples from subsoil grown plants are shaded in green, and samples from terra preta grown plants are shaded in brown. a Bray-Curtiss dissimilarity clustered OTU profiles observed in rhizospheres of Lenha and EMBRAPA plants grown on sterile sand, subsoil or terra preta. Shannon diversity values are displayed next to each rhizosphere sample. b Bray-Curtis dissimilarity clustered conseq profiles grouped by phylum in rhizospheres of Lenha and EMBRAPA plants grown on sterile sand, subsoil or terra preta. The major branches grouping samples by substrate were found to be statistically significant by using Mann–Whitney, with P = 0.006 for all comparisons
Figure 3b shows 16S OTUs classified by bacterial phyla. Proteobacteria were an especially abundant group in maize rhizospheres, representing up to 957 of the 1000 randomly chosen consensus sequences per sample. Terra preta added the greatest microbial diversity to rhizospheres, including representatives from the SPAM and Chloroflexi phyla which were largely undetected in subsoil or sterile sand rhizospheres. Subsoil grown plants yielded rhizospheres which had an intermediate level of OTU diversity (more than sterile sand but less than terra preta) and had unique OTUs observed from the phyla ZB2 and Chlorobi. Non-sterile soils in this experiment added representatives from the bacterial phyla Gemmatimonadetes, Fibrobacteres, Armatimonadetes, Planctomyces, TM7, Verrumicrobia and Acidobacteria to maize rhizospheres, which were not observed in plants grown on sterile sand. Surprisingly, Firmicutes, Actinobacteria, Bacteroidetes, and Proteobacteria, which were the most common phyla of bacteria in subsoil and terra preta rhizospheres, were also detected in sterile sand rhizospheres, suggesting either that sterile sand contributed a very large amount of contaminating DNA to rhizospheres of plants grown in all three substrates, or (much more likely) that at least a subset of these rhizosphere colonizing microbes may be seed transmitted. The most ubiquitous and abundant phyla in all rhizosphere samples were Bacteroidetes and Proteobacteria, suggesting these bacteria are the best adapted to both seed transmission and rhizosphere colonization.
Detailed alpha diversity and phylum level analysis of rhizosphere OTUs
To better understand the bacterial diversity in the Miseq data, the most comprehensive rhizosphere samples of each genotype/soil combination were further analyzed. The conseq were rarified to the highest shared number (1900 conseq in EMBRAPA rhizosphere grown in terra preta sample #2), the number of distinct OTUs in each rarefied set was counted as a measure of alpha diversity (Fig. 4a) and the number of conseq that belong to each phylum was counted (Fig. 4b). Both the Lenha and EMBRAPA rhizosphere samples grown in terra preta contained higher bacterial diversity (192 and 183 different OTUs respectively) than did the subsoil rhizospheres (136 and 111 different OTUs respectively), which in turn showed higher diversity than the rhizospheres grown in sterile sand (50 and 24 different OTUs respectively) (Fig. 4a). Bacterial OTUs found in sterile sand grown rhizospheres were predominantly Proteobacteria (33 Lenha, 18 EMBRAPA), Bacteroidetes (9 Lenha, 2 EMBRAPA) and Firmicutes (2 Lenha, 2 EMBRAPA), while subsoil grown rhizospheres were rich in Proteobacteria (68 Lenha, 55 EMBRAPA), Bacteroidetes (18 Lenha, 12 EMBRAPA) and Actinobacteria (8 Lenha, 11 EMBRAPA), and terra preta grown rhizospheres were rich in Proteobacteria (83 Lenha, 75 EMBRAPA), Actinobacteria (22 Lenha, 36 EMBRAPA) and Bacteroidetes (27 Lenha, 12 EMBRAPA). Lenha rhizospheres grown in all three substrates had higher bacterial diversity than their corresponding EMBRAPA ones (Fig. 4a).

Alpha diversity and phylum level classification of OTUs of bacterial 16S rDNA isolated from rhizospheres of one sample each of the maize landrace Lenha and the EMBRAPA hybrid BRS 1030 grown in either sterile sand, subsoil or terra preta. Results from the rhizosphere sample with the greatest conseq count were chosen from each genotype/soil combination, rarified to the highest shared conseq count (1900), classified at the phylum level, and counted as contributing to OTU diversity (a) or summed to show conseq taxonomic abundance (b)
While both subsoil and terra preta contributed a large diversity of bacterial species to the rhizospheres, this diversity represented a small proportion of the actual bacterial population when the number of conseq was considered instead of alpha diversity (Fig. 4b). For example for Lenha rhizospheres grown on subsoil or terra preta, there were only 356 and 567 out of 1900 conseq per sample (19 and 30 % respectively) from novel OTUs that were not observed in sterile sand grown rhizospheres. A similar phenomenon was observed in the rhizospheres of EMBRAPA plants, where the subsoil and terra preta contributed only 25 and 27 % of the conseq that belonged to novel OTUs not observed in sterile sand rhizospheres. In all samples, Proteobacteria made up the bulk of sequences observed, ranging from 1514 out of 1900 conseq in Lenha grown on subsoil, to 1818 for EMBRAPA grown in sterile sand (Fig. 4b). Bacteroidetes conseq were present in all samples, ranging from 22 out of 1900 conseq in EMBRAPA grown on sterile sand, to 315 in Lenha grown on subsoil. Actinobacteria were the third most abundant phylum of conseq detected but appeared to be largely soil derived as none were detected in rhizospheres of Lenha grown in sterile sand, only seven in EMBRAPA grown on sterile sand, while as many as 104 were detected in rhizospheres grown on non-sterile soil (Fig. 4b). Note that a small proportion of conseq were in fact non-target amplicons coming from the mitochondria of fungi, oomycetes and protists which were not effectively inhibited by the PNA blockers targeting plant mitochondria and chloroplasts.
Conservation of rhizosphere OTUs between substrates
All rhizosphere Miseq data (rarefied to 1000) was overlapped to determine which of the 1059 OTUs observed were shared between genotypes grown on the three different substrates (Fig. 5). Lenha rhizospheres grown on sterile sand, subsoil and terra preta shared 35 OTUs which represented on average 90 % of the conseq from sand grown samples, 74 % of the conseq from subsoil grown samples and 64 % of the conseq in terra preta grown samples (Fig. 5a). EMBRAPA rhizospheres from plants grown on sterile sand, subsoil or terra preta shared 23 OTUs which represented on average 88 % of the conseq from sand grown samples, 74 % of the conseq from subsoil grown samples and 55 % of the conseq in terra preta grown samples (Fig. 5b). In both genotypes on all three substrates, more than 50 % of conseq came from a select group of OTUs, suggested that these dominant bacterial groups are transmitted by seed. Seventeen of these shared OTUs were found in both Lenha and EMBRAPA rhizospheres, suggesting these as the most conserved seed transmitted bacteria in maize (Fig. 5a and b). There was also a higher number of overlapping OTUs between plants grown on non-sterile soils (92 for Lenha and 44 for EMBRAPA), than between plants grown on sterile sand versus subsoil or terra preta (from 5 to 13), which suggests that some rhizosphere colonizers are cosmopolitan microbes that do not require special adaptations to life in the rhizosphere and may be found in ecologically disparate soils.

Venn diagrams and average proportions of shared conseq per sample in maize rhizospheres grown in sterile sand, subsoil or terra preta. a Shared OTUs, their ID # and average proportion of conseqs per sample between Lenha plants grown in sterile sand, subsoil and terra preta. b Shared OTUs, their ID # and average proportion of conseq per sample between EMBRAPA plants grown in sterile sand, subsoil and terra preta. 17 shared OTUs found in both maize genotypes are highlighted in brown
Most abundant OTUs in the rhizospheres
By averaging the proportion of each conseq type across samples, it was possible to rank OTUs by abundance. The average proportion of conseq in each the 25 most abundant OTUs observed in rhizospheres is shown (Fig. 6). Seven of the OTUs displayed (212, 216, 183, 162, 196, 96, 155) make up the majority of the conseq that are shared by plants of the same genotype growing in the 3 different substrates. These 7 OTUs show the following levels of abundance in the respective rhizosphere communities: Lenha in sand - 78.1 %; EMBRAPA in sand - 81.1 %; Lenha in subsoil - 65.2 %; EMBRAPA in subsoil - 64.4 %; Lenha in terra preta - 56.4; and EMBRAPA in terra preta - 49.5 %. These OTUs were predicted to represent genera of Proteobacteria including Burkholderia, Pantoea/Enterobacter, Stenotrophomonas/Pseudomonas, Massilia/Telluria, Sphingobium/Sphingomonas, Agrobacterium/Rhizobium, Pseudorhodoferax, Bradyrhizobium and Ochrobacterium, of which many are elsewhere described as endophytes. OTU 155 represents the mitochondria of a fungus (Penicillium) which may also be seed transmitted although it was a non-target amplicon in this experiment.

Average OTU proportion per sample of the 25 most commonly observed sequences in rhizospheres of Lenha and EMBRAPA maize grown in sterile sand, subsoil and terra preta. OTU sequences were BLASTed against the nucleotide collection at Genbank and annotated based on the family of the top hits
The single most common OTU observed in maize rhizospheres was 212, taxonomically of the family Burkholderiaceae, which was observed to be very abundant in Lenha rhizospheres grown on sand (33.6 % of conseq) but was surprisingly rare in EMBRAPA rhizospheres grown on sterile sand (0.4 % of conseq). Both Lenha and EMBRAPA genotypes when grown on soil were found to possess a large proportion of conseq OTU 212, perhaps suggesting that soils in addition to seeds can transmit species of Burkholderiaceae. The two Lenha spermosphere samples that yielded Miseq data had conseq counts that were 100 % OTU 212, while it was not detected at all in sterile sand (electronic supplementary material).
The second most common OTU was 216, which had a 100 % match with Pantoea stewartii and several other species of Pantoea and Enterobacter of the family Enterobacteriaceae, was observed to be very abundant in both Lenha and EMBRAPA rhizospheres grown on sand (25.9 and 46.7 % respectively) but this proportion went down when the plants were grown in subsoil (15.9 and 5.3 % respectively) and even lower when they were grown in terra preta (3.3 and 1.8 % respectively).
Discussion
The main purpose of this experiment was to characterize the importance of soil microbes in the make-up of plant-associated microbiomes, and to accomplish that we compared plants grown in two microbially different soils to plants grown on sterile sand. A secondary question was whether plant genotype might affect bacterial populations in the microbiome of juvenile plants, so we used two distinct maize varieties; an ancient landrace and a modern hybrid coated with pesticides (Fig. 1g). We were unable to observe differences in bacterial diversity between any phyllosphere or root endosphere, however we did find that soil has significant effect on rhizosphere bacterial diversity and that plant genotype/seed treatment has an effect on rhizosphere bacterial diversity, especially when grown on sterile sand where there is no colonization pressure from soil microbes. We were surprised to find evidence that the most abundant rhizosphere bacteria come from seeds: 35 OTUs in Lenha and 23 in EMBRAPA plants were found in rhizospheres from all three soil conditions and are thus likely seed transmitted endophytes or epiphytes (Fig. 5). The bacteria represented by these OTUs appear to be robust colonizers of the maize rhizosphere, making up as little as 55 % of the population when plants were grown in non-sterile terra preta, and as much as 90 % when the plants were grown in sterile sand.
As a tertiary objective, we wanted to contrast the ability of TRFLP to profile bacterial diversity in rhizospheres in comparison to next generation sequencing; TRFLP is a rapid and economical (albeit less sensitive and less informative) PCR based technique of DNA fingerprinting which can benefit scientists who mostly need to observe trends within microbial communities (Hamady and Knight 2009). We transformed TRFLP signals into presence or absence counts (binary data only reflects diversity) and after statistical analysis we observed that neither soil, nor plant genotype significantly shifted bacterial diversity within roots or on leaf surfaces (Fig. 2c). TRFLP also showed that soils do significantly change bacterial diversity in rhizospheres, while plant genotype/seed treatment was only observed to influence microbial diversity on root surfaces grown in sterile sand (Fig. 2d). MiSeq as we’ve used it here, is deep sequencing technology that yields detailed knowledge about the diversity and number of sequences in a PCR product. By using Bray-Curtis dissimilarity clustering of log2 transformed MiSeq data (log transformation reduces the statistical importance of abundant OTUs), we were able to observe the same ordination patterns as with PCA of binary TRFLP data, showing that TRFLP can be sensitive enough to observe community level differences in bacterial diversity. Our Miseq data should show representative patterns of bacterial diversity, as the universal primers we used are believed to be unbiased towards any bacterial phylum (Bergmann 2011). On the other hand, as TRFLP and Miseq are both PCR based techniques which have been shown to miss as much as 50 % of the sequence diversity in an environmental sample (Hong et al. 2009), our results cannot be assumed to show all the bacteria present in juvenile maize microbiomes.
The influence of soil on the maize microbiome
As the most microbially diverse habitat on Earth, soil would be expected to influence the microbiome of plants that are growing in it. Although it is not well known where phyllosphere microbial populations originate from, we found no clear evidence that soil is a dominant factor shaping leaf surface communities (Fig. 2b). In a previous study, we observed that soil did not appear to dramatically affect bacterial endophyte diversity inside maize roots (Johnston-Monje et al. 2014) and we appear to have replicated those results here, with no significant difference between root endospheres that were grown on sterile sand, subsoil or terra preta (Fig. 2c).
Soil did affect rhizosphere bacterial diversity, with average Shannon diversity of rhizosphere samples being lowest in sterile sand rhizospheres (1.97), somewhat higher in subsoil rhizospheres (2.58) and the highest in terra preta rhizospheres (3.17). Previous studies suggest salinity, calcium and soil organic carbon are the most important factors driving maize rhizosphere microbial diversity (Castellanos et al. 2009), however we found in statistical analysis that terra preta rhizospheres (high in calcium and carbon) clustered with subsoil rhizospheres and away from sterile sand rhizospheres (Figs. 2d and 3a), likely because the sand was microbe free while terra preta and subsoil contained taxonomically related bacteria. Terra preta is topsoil from a biotically rich jungle, while subsoil was mined from 400 m underground and had never before been exposed to plant life, suggesting that the bacteria deposited into rhizospheres by these soils are not specially adapted rhizosphere colonizers, but rather generalist soil microbes that were able to compete successfully to colonize the root. There were 101 such OTUs contributed by both subsoil and terra preta but not found in sand rhizospheres (92 OTU from Lenha + 9 more from EMBRAPA – Fig. 5), and these were mostly classified to the phyla Proteobacteria (52 %), Actinobacteria (13 %), and Bacteroidetes (11 %), while only 17 % belonged to oligotrophic phyla of soil bacteria such as Acidobacteria, Verrumicrobia, Gemmatimonadetes, Armatimonadetes and Planctomyces. Rhizospheres are nutrient rich niches, well suited for copiotrophs including many types of Proteobacteria, and less so for oligotrophic soil bacteria such as Acidobacteria and Verrumicrobia (Fierer et al. 2007). Few of the unique OTUs that were added by either subsoil or terra preta to rhizospheres were observed in great abundance (the greatest was 4 % of total conseq for OTU 151 in terra preta rhizospheres as compared to OTU 212 which was 40 %), implying that most soil transmitted microbes are poor colonizers of the rhizosphere, either being outcompeted by other microbes or growing poorly under suboptimal conditions.
The effect of plant genotype on the maize microbiome
A question of interest to plant geneticists and breeders is whether genotype is able to shape or affect the microbiome. Using TRFLP, it was possible to observe that seed surfaces had bacterial populations that were different from soil but there was no seed genotype or treatment effect observed before germination (Fig. 2a). Based on previous experiments using similar methods (Johnston-Monje et al. 2014), we had expected that bacterial diversity in root endospheres would be more influenced by plant genotype than by soil, however in this experiment we saw neither genotype nor soil effects, perhaps because the plants were not genetically different enough (i.e., they are both tropical varieties of domesticated Zea mays). We were likewise surprised to observe that leaf and stem surfaces weren’t influenced by plant genotype or soil either; although little is understood regarding the origin of phyllosphere microbes (Bulgarelli et al. 2013), genotype is thought to play a major role in shaping the phyllosphere (Horton et al. 2014; Whipps et al. 2008).
Plant genotypes have previously been shown to impact rhizosphere bacterial populations in field grown maize at flowering (Peiffer and Ley 2013; Peiffer et al. 2013), field grown strawberry (Costa et al. 2006), field grown potato (Weinert et al. 2011), field and greenhouse grown rice (Edwards et al. 2015) and lab grown Arabidopsis (Bulgarelli et al. 2012; Lundberg et al. 2012). Using TRFLP we did observe a clear and significant difference between bacterial diversity in Lenha and EMBRAPA rhizospheres, but only when grown in sterile sand (Fig. 2d) perhaps because the microbial diversity from non-sterile soils overshadows the diversity of seed transmitted bacteria. Looking at the more sensitive Miseq data, there was a consistently lower diversity of bacteria in rhizospheres of EMBRAPA plants compared to Lenha (Figs. 4a and 5a and b); future experiments will have to include greater replication to ensure statistical significance of these trends, and to discover plant phenotypic differences which might correlate with such differences in microbial populations.
Transmission of bacteria from seed to rhizosphere
Most research until now suggests that “soil is the main reservoir for rhizosphere microorganisms” (Berg and Smalla 2009), with experiments ignoring or actively trying to eliminate the possibility of vertical transmission of bacteria by sterilizing seeds and/or not including a microbe free substrate as a control. Contrary to a soil origin for rhizosphere bacteria, we found evidence that a majority of cells in the rhizosphere might be seed transmitted endophytes and epiphytes instead. To be clear, our data suggests that while rhizosphere bacterial diversity is most influenced by soil, the majority of conseq observed belonged to a limited group of OTUs which were present in both sterile sand and subsoil/terra preta grown rhizospheres; except for dust which may have been a small source of contamination, the only way for bacteria to be in plants grown on both sterile sand and non-sterile soil is if they were transmitted on or in the seed.
Seed transmitted OTUs in Lenha landrace rhizospheres accounted for as much as 89 % of the total conseq in sterile sand grown plants, to as little as 64 % from terra preta grown plants (Fig. 5a), while in EMBRAPA hybrid rhizospheres they accounted for as much as 88 % of the total conseq from sterile sand grown plants, to as little as 55 % from terra preta grown plants (Fig. 5b). In both plant varieties in all three substrates, a select group of seed transmitted bacteria appear to dominate the rhizosphere despite competition from soil microbes and must be well adapted to both transmission through the seed and life in the rhizosphere. These OTUs were mostly Bacteroidetes, and Proteobacteria; members of which have been previously isolated or observed as endophytes or epiphytes from a variety of plant seeds (Adams and Kloepper 2002; Ferreira et al. 2008; Kaga et al. 2009; Mano et al. 2006; Mundt and Hinkle 1976; Truyens et al. 2013), including maize (Johnston-Monje and Raizada 2011a; Liu et al. 2013a). The most abundant conseq observed in maize rhizospheres belonged to OTU 212, and this was also the only OTU observed in Lenha spermospheres, suggesting a connection between seed surfaces and root surfaces.
If these findings can be replicated, they suggest a system of bacterial inheritance similar to that of mammals, where the microbiome is transmitted through placental connections and surface contact with the mother during birth (Dominguez-Bello et al. 2010; Funkhouser and Bordenstein 2013; Romano-Keeler and Weitkamp 2014). Seed endophytes have been shown to colonize grass seedlings (Johnston-Monje et al. 2014; Hardoim et al. 2012; Rijavec et al. 2007), travel within the plant and even exit the roots and colonize the rhizosphere (Johnston-Monje and Raizada 2011a; Hardoim et al. 2012). Seed transmitted microbes colonizing the rhizosphere would be guaranteed first access to that habitat, perhaps creating a founder effect, blocking later colonization by less adapted soil microbes (Ait Barka et al. 2002; Bacilio-Jimenez et al. 2001; Raibaud et al. 1980). Seed epiphytes should have no barrier to getting on root surfaces, however endophytes might struggle to exit the plant in order to gain access to the rhizosphere; a newly discovered mechanism endophytes might use to do this involves root border cells containing intracellular populations of bacteria that are sloughed off and released into the surrounding substrate almost like little water balloons full of inoculant (Cope-Selby 2013). Seed endophytes which colonize the rhizosphere can play an important role in plant nutrition, for example in the cardon cactus, where they help to mineralize the surrounding rock for nutrient absorption by the roots (Puente et al. 2009) or in grasses where dying bacteria release organic nitrogen for absorption by the plant (White et al. 2015).
Although these are interesting results, it should be noted that the abundance of seed transmitted bacteria we have observed in these juvenile rhizospheres may be exaggerated because they have not yet had a chance to be more heavily colonized during passage through soil as would older and larger root systems (Inceoglu et al. 2011). As plants age, their rhizosecretions also change which can alter the microbial populations living around the root (Chaparro et al. 2014). Older and larger plants would also have more time to interact with other organisms such as fungi, nematodes and insects, which may vector bacteria into the rhizosphere and reduce the dominance of seed transmitted bacteria as they are eaten and killed or displaced.
Abundant and conserved OTUs in the rhizosphere
Despite observing 1059 different OTUs in this experiment (OTU sequences in the electronic supplementary material), just the top seven of the most commonly observed OTUs (ID# 212, 216, 183, 162, 196, 96, 155) comprised the majority of the shared conseq in each rhizosphere, ranging from 49.5 % of conseq in EMBRAPA grown in terra preta to 81 % for EMBRAPA grown in sterile sand (Fig. 6). The single most abundant OTU in rhizospheres was 212, representing one or more species of Burkholderia, which comprised 34 % of the conseq in Lenha rhizosphere from sterile sand, 28 % in Lenha rhizosphere from subsoil, and 47 % in Lenha rhizosphere from terra preta. The idea that OTU 212-like bacteria can be transmitted from seeds is supported by data from the two sequenced Lenha spermospheres which had 100 % of their conseqs identified as OTU 212 (Raw conseq counts in electronic supplemental material). Conversely, EMBRAPA rhizospheres may have acquired their OTU 212 from soils as only 0.4 % of the conseq in sterile sand rhizospheres belonged to that OTU, compared to 35 and 39 % for rhizospheres from subsoil and terra preta grown plants. The apparent dominance of Burkholderia species that we have observed in juvenile maize plants is not unprecedented: Burkholderia have been reported to be the most abundant genera of bacteria in rhizospheres of turf grasses (Vandenkoornhuyse et al. 2007), in rhizospheres of maize grown under field conditions (Bouffaud et al. 2012; Peiffer et al. 2013), in hybrid maize seeds in China (Liu et al. 2013b), in moss sporophytes and gametophytes (Bragina et al. 2013) and in mimosa roots (Elliott et al. 2009). More than one species of Burkholderia matches OTU 212, highlighting a general problem with relying only on 16S data to predict specific taxonomy, ecology or behaviour of bacteria. To identify specific strains of Burkholderia in maize rhizospheres, and more convincingly show that bacteria are seed derived under all conditions and not just when grown on sterile sand, future work to develop strain specific identification methods such as strain specific real time PCR or FISH will be necessary.
The second most abundant 16S rDNA OTU was #216, with 100 % identity to Pantoea stewartii; the most important bacterial pathogen of maize and also known to be transmitted endophytically inside seeds (Block et al. 1998). The proportion of this OTU seemed to be highest in sterile sand grown plants, suggesting that competition from soil microbes may reduce the dominance of these Enterobacteraceae in the rhizosphere. Again, because OTU 216 was binned at 97 % and BLASTing the sequence yields 100 % matches to many species of Pantoea and Enterobacter, it is difficult to predict the exact taxonomy or ecological role of these bacteria in the rhizosphere.
Conclusions
By comparing the populations of bacteria found in rhizospheres grown in sterile sand, to those of plants grown in a microbe poor soil from deep underground and to a microbe rich topsoil from the Amazon jungle, our data implies that the majority of bacterial cells found in this niche come from the seed, either as endophytes within, or as epiphytes on the surface. Evidence of differences in rhizosphere bacterial diversity between the two varieties of maize may be explained as either a genotype effect or as a seed treatment effect (EMBRAPA was coated with pesticides and fungicides). Soils of course do add to the diversity of maize rhizosphere inhabitants, but contribute small numbers of many diverse bacterial genera rather than installing dominant groups into that niche. Neither soil, nor genotype appeared to affect phyllosphere or root endosphere bacterial populations, although experiments with older or more genetically different plants could show different results. Our data suggests that scientists and farmers attempting to alter and optimize the maize rhizosphere to aid in disease and stress resistance, nutrient acquisition and root development, may be more successful if they focus efforts to perturb the seed associated microbiome rather than the soil microbiome.
References
Adams PD, Kloepper JW (2002) Effect of host genotype on indigenous bacterial endophytes of cotton (Gossypium hirsutum L.). Plant and Soil 240:181–189
Ait Barka E, Gognies S, Nowak J, Audran JC, Belarbi A (2002) Inhibitory effect of endophyte bacteria on Botrytis cinerea and its influence to promote the grapevine growth. Biol Control 24:135–142
Alves GC, Rodrigues dos Santos CL, Urquiaga S, Reis VM (2010) Milho inoculado com a estirpe BR 11417 em experimentos de campo nas safras e safrinhas de 3 anos consecutivos. In Agronomia/Ciência do Solo. UFRRJ, Seropedica
Bacilio-Jimenez M, Aguilar-Flores S, del Valle MV, Perez A, Zepeda A, Zenteno E (2001) Endophytic bacteria in rice seeds inhibit early colonization of roots by Azospirillum brasilense. Soil Biol Biochem 33:167–172
Badri DV, Vivanco JM (2009) Regulation and function of root exudates. Plant Cell Environ 32:666–681
Bakker M, Chaparro J, Manter D, Vivanco J (2015) Impacts of bulk soil microbial community structure on rhizosphere microbiomes of Zea mays. Plant Soil 1–12
Berg G, Smalla K (2009) Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol Ecol 68:1–13
Bergmann GT, Bates ST, Eilers KG, Lauber CL, Caporaso JG, Walters WA, Knight R, Fierer N. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol Biochem 43:1450–1455
Block CC, Hill JH, McGee DC (1998) Seed transmission of Pantoea stewartii in field and sweet corn. Plant Dis 82:775–780
Bouffaud M-L, Kyselková M, Gouesnard B, Grundmann G, Muller D, Moënne-Loccoz Y (2012) Is diversification history of maize influencing selection of soil bacteria by roots? Mol Ecol 21:195–206
Bragina A, Cardinale M, Berg C, Berg G (2013) Vertical transmission explains the specific Burkholderia pattern in Sphagnum mosses at multi-geographic scale. Front Microbiol 4:394
Bulgarelli D, Rott M, Schlaeppi K, Ver Loren van Themaat E, Ahmadinejad N, Assenza F, Rauf P, Huettel B, Reinhardt R, Schmelzer E, Peplies J, Gloeckner FO, Amann R, Eickhorst T, Schulze-Lefert P (2012) Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488:91–95
Bulgarelli D, Schlaeppi K, Spaepen S, van Themaat EVL, Schulze-Lefert P (2013) Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol 64:807–838
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Castellanos T, Dohrmann AB, Imfeld G, Baumgarte S, Tebbe CC (2009) Search of environmental descriptors to explain the variability of the bacterial diversity from maize rhizospheres across a regional scale. Eur J Soil Biol 45:383–393
Chaparro JM, Badri DV, Vivanco JM (2014) Rhizosphere microbiome assemblage is affected by plant development. ISME J 1;8(4):790–803
Chelius MK, Triplett EW (2001) The diversity of archaea and bacteria in association with the roots of Zea mays L. Microb Ecol 41:252–263
Cope-Selby NL (2013) Diversity and characterization of bacteral endophytes in the C4 energy crop Miscanthus (PhD thesis). Aberystwyth University, Aberystwyth
Costa R, Gotz M, Mrotzek N, Lottmann J, Berg G, Smalla K (2006) Effects of site and plant species on rhizosphere community structure as revealed by molecular analysis of microbial guilds. FEMS Microbiol Ecol 56:236–249
Culman SW, Gauch HG, Blackwood CB, Thies JE (2008) Analysis of T-RFLP data using analysis of variance and ordination methods: a comparative study. J Microbiol Methods 75:55–63
Dalmastri C, Chiarini L, Cantale C, Bevivino A, Tabacchioni S (1999) Soil type and maize cultivar affect the genetic diversity of maize root–associated Burkholderia cepacia populations. Microb Ecol 38:273–284
Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R (2010) Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 107:11971–11975
Edwards J, Johnson C, Santos-Medellín C, Lurie E, Podishetty NK, Bhatnagar S, Eisen JA, Sundaresan V (2015) Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci 112:E911–E920
Elliott GN, Chou J-H, Chen W-M, Bloemberg GV, Bontemps C, Martínez-Romero E, Velázquez E, Young JPW, Sprent JI, James EK (2009) Burkholderia spp. are the most competitive symbionts of Mimosa, particularly under N-limited conditions. Environ Microbiol 11:762–778
EMBRAPA (1997) Manual de métodos e análises de solo. Centro Nacional de Pesquisa de Solos, Rio de Janeiro
Ferreira A, Quecine MC, Lacava PT, Oda S, Azevedo JL, Araújo WL (2008) Diversity of endophytic bacteria from Eucalyptus species seeds and colonization of seedlings by Pantoea agglomerans. FEMS Microbiol Lett 287:8–14
Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology 88:1354–1364
Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ (2008) Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl Environ Microbiol 74:2461–2470
Funkhouser LJ, Bordenstein SR (2013) Mom knows best: the universality of maternal microbial transmission. PLoS Biol 11:e1001631
Garbeva P, Van Elsas JD, Van Veen JA (2008) Rhizosphere microbial community and its response to plant species and soil history. Plant Soil 302:19–32
Germida J, Siciliano S (2001) Taxonomic diversity of bacteria associated with the roots of modern, recent and ancient wheat cultivars. Biol Fertil Soils 33:410–415
Hamady M, Knight R (2009) Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res 19:1141–1152
Hardoim PR, Hardoim CC, van Overbeek LS, van Elsas JD (2012) Dynamics of seed-borne rice endophytes on early plant growth stages. PLoS One 7:e30438
Hawkes CV, DeAngelis KM, Firestone MK (2007) Root interactions with soil microbial communities and processes. In The rhizosphere: an ecological perspective. ZG Cardon, JL Whitbeck (eds.) pp 1–29. Elsevier Academic Press
Hong SH, Bunge J, Leslin C, Jeon S, Epstein S (2009) Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J 3:1365–1373
Horton MW, Bodenhausen N, Beilsmith K, Meng D, Muegge BD, Subramanian S, Vetter MM, Vilhjálmsson BJ, Nordborg M, Gordon JI, Bergelson J (2014) Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nat Commun 5
Inceoglu O, Salles JF, van Overbeek L, van Elsas JD (2010) Effects of plant genotype and growth stage on the betaproteobacterial communities associated with different potato cultivars in two fields. Appl Environ Microbiol 76:3675–3684
Inceoglu O, Al-Soud WA, Salles JF, Semenov AV, van Elsas JD (2011) Comparative analysis of bacterial communities in a potato field as determined by pyrosequencing. PLoS One 6:e23321
Johnston-Monje D, Raizada MN (2011a) Conservation and diversity of seed associated endophytes in Zea across boundaries of evolution, ethnography and ecology. PLoS One 6:20396–20396
Johnston-Monje D, Raizada MN (2011b) Plant and endophyte relationships: Nutrient management. In: Moo-Young M (ed) Comprehensive Biotechnology, 2nd edn. Elsevier, Oxford, pp 713–727
Johnston-Monje D, Mousa WK, Lazarovits G, Raizada MN (2014) Impact of swapping soils on the endophytic bacterial communities of pre-domesticated, ancient and modern maize. BMC Plant Biol 14:233
Kaga H, Mano H, Tanaka F, Watanabe A, Kaneko S, Morisaki H (2009) Rice seeds as sources of endophytic bacteria. Microbes Environ 24:154–162
Keister D, Cregan P, Lynch JM, Whipps JM (1991) Substrate flow in the rhizosphere. In The Rhizosphere and Plant Growth. Springer, Netherlands, pp 15–24
Liu Y, Zuo S, Xu L, Zou Y, Song W (2013a) Study on diversity of endophytic bacterial communities in seeds of hybrid maize and their parental lines. Arch Microbiol 194:1001–1012
Liu Y, Zuo S, Zou Y, Wang J, Song W (2013b) Investigation on diversity and population succession dynamics of endophytic bacteria from seeds of maize (Zea mays L., Nongda108) at different growth stages. Ann Microbiol 63:71–79
Long HH, Schmidt DD, Baldwin IT (2008) Native bacterial endophytes promote host growth in a species-specific manner; Phytohormone manipulations do not result in common growth responses. PLoS One 3:e2702
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, Rio TGD, Edgar RC, Eickhorst T, Ley RE, Hugenholtz P, Tringe SG, Dangl JL (2012) Defining the core Arabidopsis thaliana root microbiome. Nature 488:86–90
Lundberg DS, Yourstone S, Mieczkowski P, Jones CD, Dangl JL (2013) Practical innovations for high-throughput amplicon sequencing. Nat Meth 10:999–1002
Mano H, Tanaka F, Watanabe A, Kaga H, Okunishi S, Morisaki H (2006) Culturable surface and endophytic bacterial flora of the maturing seeds of rice plants (Oryza sativa) cultivated in a paddy field. Microbes Environ 21:86–100
Marris E (2006) Putting the carbon back: black is the new green. Nature 442:624–626
Micallef SA, Shiaris MP, Colón-Carmona A (2009) Influence of Arabidopsis thaliana accessions on rhizobacterial communities and natural variation in root exudates. J Exp Bot 60:1729–1742
Mundt JO, Hinkle NF (1976) Bacteria within ovules and seeds. Appl Environ Microbiol 32:694–698
Normander B, Prosser JI (2000) Bacterial origin and community composition in the barley phytosphere as a function of habitat and presowing conditions. Appl Environ Microbiol 66:4372–4377
Okon Y, Itzigsohn R (1995) The development of Azospirillum as a commercial inoculant for improving crop yields. Biotechnol Adv 13:415–424
Paterniani E, Goodman MM (1977) Races of maize in Brazil and adjacent areas. CIMMYT, Mexico City
Peiffer JA, Ley RE (2013) Exploring the maize rhizosphere microbiome in the field: A glimpse into a highly complex system. Commun Integr Biol 6:e25177
Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler ES, Ley RE (2013) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci 110:6548–6553
Philippot L, Raaijmakers JM, Lemanceau P, van der Putten WH (2013) Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Microbiol 11:789–799
Puente ME, Li CY, Bashan Y (2009) Endophytic bacteria in cacti seeds can improve the development of cactus seedlings. Environ Exp Bot 66:402–408
Raaijmakers JM, Weller DM (1998) Natural plant protection by 2,4-diacetylphloroglucinol-producing pseudomonas spp. in take-all decline soils. Mol Plant Microbe Interact 11:144–152
Raibaud P, Ducluzeau R, Dubos F, Hudault S, Bewa H, Muller MC (1980) Implantation of bacteria from the digestive tract of man and various animals into gnotobiotic mice. Am J Clin Nutr 33:2440–2447
Rijavec T, Lapanje A, Dermastia M, Rupnik M (2007) Isolation of bacterial endophytes from germinated maize kernels. Can J Microbiol 53:802–808
Romano-Keeler J, Weitkamp J-H (2014) Maternal influences on fetal microbial colonization and immune development. Pediatr Res 77:189–195
Singh BK, Munro S, Potts JM, Millard P (2007) Influence of grass species and soil type on rhizosphere microbial community structure in grassland soils. Appl Soil Ecol 36:147–155
Truyens S, Weyens N, Cuypers A, Vangronsveld J (2013) Changes in the population of seed bacteria of transgenerationally Cd-exposed Arabidopsis thaliana. Plant Biol 15:971–981
van Overbeek L, van Elsas JD (2008) Effects of plant genotype and growth stage on the structure of bacterial communities associated with potato (Solanum tuberosum L.). FEMS Microbiol Ecol 64:283–296
Vandenkoornhuyse P, Mahé SP, Ineson P, Staddon P, Ostle N, Cliquet J-B, Francez A-J, Fitter AH, Young JPW (2007) Active root-inhabiting microbes identified by rapid incorporation of plant-derived carbon into RNA. Proc Natl Acad Sci 104:16970–16975
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Weinert N, Piceno Y, Ding G-C, Meincke R, Heuer H, Berg G, Schloter M, Andersen G, Smalla K (2011) PhyloChip hybridization uncovered an enormous bacterial diversity in the rhizosphere of different potato cultivars: many common and few cultivar-dependent taxa. pp. 497–506.
Whipps JM, Hand P, Pink D, Bending GD (2008) Phyllosphere microbiology with special reference to diversity and plant genotype. J Appl Microbiol 105:1744–1755
White JF, Chen Q, Torres MS, Mattera R, Irizarry I, Tadych M, Bergen M (2015) Collaboration between grass seedlings and rhizobacteria to scavenge organic nitrogen in soils. AoB plants 7, plu093
Yourstone SM, Lundberg DS, Dangl JL, Jones CD (2014) MT-Toolbox: improved amplicon sequencing using molecule tags. BMC Bioinforma 15:284
Acknowledgments
Open access funding provided by the Max Planck Society. We thank Dr. Jeff Dangl (University of North Carolina at Chapel Hill) who graciously sponsored the next-generation sequencing of bacterial communities in the rhizosphere samples in this study. We would like to thank Dr. Siu Mui Tsai (Centro de Energia Nuclear na Agricultura), Dr. Amanda Lima and Dr. Weceslau Teixeira (EMBRAPA Solos, Brazil) who helped us procure the terra preta used in this study. We are grateful to Dr. Sergio de Faria of EMBRAPA Agrobiologia who helped us acquire the iron mine subsoil. Thank you to Dr. David Weisman (Symbiota, USA) for his advice on statistical analysis. This work was made possible through funding by the Canadian office of the Inter-American Institute for Cooperation on Agriculture which allowed DJM to travel to and conduct experiments in the lab of VMR in Seropédica, Brazil.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
David Johnston-Monje conceived of and executed this project as a grad student at the University of Guelph, but at the time of publishing is employed by the plant microbiome company Symbiota. George Lazarovits is an employee of the agricultural microbiology detection and diagnosis company A&L Biologicals.
Additional information
Responsible Editor: Andrea Campisano.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Johnston-Monje, D., Lundberg, D.S., Lazarovits, G. et al. Bacterial populations in juvenile maize rhizospheres originate from both seed and soil. Plant Soil 405, 337–355 (2016). https://doi.org/10.1007/s11104-016-2826-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-016-2826-0