
Abstract
Purpose
Farming practices on farmlands aim to improve nutrients in the fields or crops, soil quality and functions, as well as boost and sustain crop yield; however, the effect of loss of ecological diversity and degradation have impacted ecosystem functions. The beneficial rhizosphere-microorganism network and crop rotation may enhance a stable ecosystem. The use of next-generation sequencing technique will help characterize the entire bacterial species in the sunflower rhizosphere compared with the nearby bulk soils. We investigated the potential of the bacterial community structure of sunflower rhizosphere and bulk soils cultivated under different agricultural practices at two geographical locations in the North West Province of South Africa.
Methods
DNA was extracted from rhizosphere and bulk soils associated with sunflower plants from the crop rotation (rhizosphere soils from Lichtenburg (LTR) and bulk soils from Lichtenburg (LTB) and mono-cropping (rhizosphere soils from Krayburg (KRPR) and bulk soils from Krayburg (KRPB) sites, and sequenced employing 16S amplicon sequencing. Bioinformatics tools were used to analyse the sequenced dataset.
Results
Proteobacteria and Planctomycetes dominated the rhizosphere, while Firmicutes and Actinobacteria were predominant in bulk soils. Significant differences in bacterial structure at phyla and family levels and predicted functional categories between soils (P < 0.05) across the sites were revealed. The effect of physicochemical parameters was observed to influence bacterial dispersal across the sites.
Conclusion
This study provides information on the predominant bacterial community structure in sunflower soils and their predictive functional attributes at the growing stage, which suggests their future study for imminent crop production and management for enhanced agricultural yields.
Similar content being viewed by others


Introduction
Comprehending the rhizosphere's geographical distribution of microbial communities has opened up several possibilities for exploiting their agricultural potential. Various microbial communities inhabit the rhizosphere, each with the ability to induce maximal adaptive responses in the plant via specific metabolic pathways. The rhizosphere is the area near the plant's roots where exudates containing various metabolites are discharged, as well as a variety of microorganisms (Agomoh et al. 2020; Ai et al. 2012). Roots are engaged in the release of exudates of various chemical components into the rhizosphere, in addition to providing nutrients and anchoring the entire plant. Through the secreted root exudates, the rhizosphere plays an important role in the modification of its microbiome component.
Plant-microbial interactions are complicated and can enhance plant growth and development (Igiehon and Babalola 2018; Berlanas et al. 2019). Bacteria are the predominant microorganisms in the rhizosphere and are indicators of soil quality, health and fertility due to their responses to biotic and abiotic pressures (Igiehon et al. 2019). The actions of bacteria are dynamic because they accelerate most biogeochemical processes, thus inducing mineral nutrient availability in soil (Nwachukwu and Babalola 2021). Bacterial communities in the rhizosphere can resist pathogens and stimulate tolerance to abiotic stressors, hence promoting plant growth, health and yield (Li et al. 2020; Meena et al. 2014). Bacterial communities that colonize the rhizosphere could be valuable, however, most do not affect plant health (Nwachukwu et al. 2021; Maquia et al. 2020).
Although, various researchers have explored the microbiome of oil food crops such as sunflower root microbiome, studies on the impact of plants on microorganisms are still ongoing; thus, necessitating this study. The sunflower (Helianthus annuus), a major oilseed crop in modern agriculture, is used for various food and industrial purposes (Majeed et al. 2018). Owing to its growing agricultural importance, some continents, such as South America, Europe, and Africa especially, South Africa, have exploited the potential for its usage (Pandey et al. 2013; Majeed et al. 2018).
Reports on the plant growth-promoting bacteria associated with sunflower plants for improved productivity in South Africa are limited, perhaps due to the inadequate studies on sunflower plants using the next-generation sequencing techniques. Hence, it is imperative to determine bacterial community structures that are resident in the sunflower rhizosphere soils using the 16S rRNA gene and their associated predictive functions (Yadav et al. 2017; Lu et al. 2020). Given this, to distinguish the effects of plants, we evaluated bacterial communities in the rhizopheric soil of sunflower and the corresponding bulk soils. Furthermore, we explored the dissimilarities in the associated predicted functional compositions of the soils.
We postulated that the soil properties and agricultural practices, such as the use of chemical fertilizer, cropping type (mono-cropping and mixed cropping) and organic manure would influence the structure and metabolic potential of sunflower rhizosphere bacterial communities compared to the bulk soil. A good knowledge of the predicted metabolic pathways of bacterial communities in the rhizosphere region is essential since functional heterogeneity is a delicate signal of the quality elements of the soil management. It also speeds up the amplification of bacterial community functions as a comprehension of biochemical and molecular components in the rhizosphere zone and controls particular bacterial enhancement.
Materials and methods
Site location, sampling, and climatic conditions
In March 2020, the rhizosphere and bulk replicate soil samples from the two commercial sunflower fields (at the growing stage) of different cultivars, PAN 7160 CLP and PAN 7011 Pannar, were collected from Lichtenburg (LT) (S26°4′31.266′′ E25°58′44.442) and Krayburg/Kraaipan (KRP) (S26°17′24.186′′ E25°13′33.258), North West Province, South Africa. A total of 12 samples each for the rhizosphere and bulk soil were collected from 4 points of sunflower plant and 15–20 cm depth from the two farms and pooled into labelled zip lock bags and were homogenized to get a composite sample as described by (Oberholster et al. 2018). The soils were immediately transported to the Microbial Biotechnology Research Laboratory, North-West University, South Africa. The soils were placed separately, sieved, and stored in zip lock bags in the dark at -80 °C for DNA extraction and high throughput sequencing.
Usually, North West Province has a summer temperature ranging from 17 °C to 31 °C and a winter temperature ranging from 3 °C to 21 °C. The annual rainfall ranges between 300 and 600 mm. According to the farm owner, the farmland in Lichtenburg has been cultivated for over 40 years. Sunflower has been rotationally cultivated with other agricultural crops, such as soybean and maize. Water supply is mainly by rainfall during the summer while irrigation during winter. The main farm activities are clearing, tilling, plowing, and ridging. Also, the application of chemical fertilizers (NPK 15:8:4), pre-emergence and post-emergence herbicides (Metagon Gold and Judo 50EC) the soil before and after planting. Foliar insecticide spray (Max-Foliar) was applied to the leaves after plant germination. In Krayburg, the farmyard size is 24.711 Acres with 24.711 Acres of sunflower plantation landscape coverage. Maize was the only crop previously cultivated on the farmland. Soil amendments include the application of urea and organic manure.
Soil physicochemical analysis
The analyses of rhizosphere and bulk soil samples for physicochemical parameters were performed using standard procedures, and 30 g of pulverized and sieved soil was taken from each sample. The soil pH in distilled water was measured using a pH meter (ratio 1:2.5, soil to water), the organic matter (OM) present in the soils was determined using the Walkley–Black method (Walakley and Black 1934), while phosphorus (P) was extracted from the samples according to the method of (Bray and Kurtz 1945). Potassium (K) was evaluated using 1 M acetate at pH 7.0 (Gutierrez Boem et al. 2011). The soil’s total carbon (C) and total nitrogen (N) were determined using the dry combusting technique as described by (Craft et al. 1991). The nitrate (N–NO3) and ammonium-N (N–NH4) were determined using the KCl extraction method by (Nelson et al. 1996).
DNA extraction and 16S rRNA amplicon sequencing
The DNA was extracted from 5 g of each sieved rhizosphere and bulk soil samples using a Zymo DNA isolation kit (Zymo Research, Irvine, USA) following the manufacturer’s instructions. All the data are products of 16S amplicon sequencing at the Molecular Research Laboratory (MR DNA, Shallowater, TX. USA). The polymerase chain reactions (PCRs) were performed in a single-step PCR using the HotStarTaq Plus Master Mix Kit (Qiagen, USA) primer pairs 515F (5′- AATGATACGGCGACCACCACCGAGATCTAC AC TATGGTAATT GT GTGCCAGCMGCCGCG GTAA-3′) and 806R (5′-CAAGCAGAAGACGGCATAC GAGAT TCCCTTGTCTCCAGTCAGTCAG CC GGA CTACHVGGGTWTCTAAT-3′).
The PCR products from the DNA samples were quantified using PicoGreen dsDNA assay. The samples were pooled together in an equimolar concentration. Then, calibrated Ampure XP beads (Agencourt Bioscience Corporation, MA, USA) was used for purification. The Illumina DNA library was prepared from the pooled and purified PCR products. Sequencing was performed on an Illumina MiSeq 2000 using a paired-end approach to obtain 312 bp paired-ends reads.
The sequence read processing was performed using Quantitative Insights Into Microbial Ecology (QIIME 2) 16S pipeline (version 2020.11) (Caporaso et al. 2010) performed on Nephele microbial bioinformatics platform (version 1.8) (https://nephele.niaid.nih. gov/) (Weber et al. 2018). Preprocessing steps involve read pair joining using default parameters (a minimum overlap of 10, and percentage maximum difference of 25), an average Phred score of ≤ 20 was removed, while chimeras were removed using VSEARCH (Edgar et al. 2011), while clustering was done using Open Reference Method and SILVA 99 version 132 (Wang et al. 2007). SILVA version 132 was used to assign taxa, with a sequence similarity of 0.99, and then chimeric sequences, including mitochondria, singleton, and chloroplast reads were eliminated.
Statistical analysis
Microsoft excel sheet was used to derive the mean and standard errors of the soil physicochemical properties. Soil physicochemical data were transferred to the Statistical Package for the Social Sciences (SPSS), where one-way analysis of variance (ANOVA) and Duncan multiple tests were performed. The relative abundance graph of the bacterial community between the sunflower rhizosphere and bulk soil was plotted using the Shinyheatmap (version 0.12.2) online tool (www1.heatmapper.ca/expression/) (Khomtchouk et al. 2017). The alpha diversity (diversity within the samples) of the bacterial community structure across each sampling sites, diversity indices (Simpson, Evenness, and Shannon_H) and bacterial richness were assessed using a Kruskal–Wallis test in the paleontological statistics software package (PAST version 4.0) (Hammer et al. 2001). These indices were also compared the rhizosphere and bulk soils.
The beta diversity was determined using the principal coordinate analysis (PCoA) on a Bray–Curtis dissimilarity matrix and the one-way analysis of similarities (ANOSIM) was used to determine the variances in community structure and composition among the sites (Clarke and Green 1988). Principal component analysis (PCA) using the Euclidean matrix was employed to identify the distribution of bacteria across the sunflower sites. Also, PCA was used to evaluate the environmental variables that best described the composition of the obtained bacteria and we assessed the possible correlations between bacterial communities and the measured environmental variables.
We employed a forward selection of environmental variables to conduct a significance test. The PCoA and PCA plots were designed using CANOCO version 5 (Microcomputer Power, Ithaca, NY, USA) software. The predictive functional annotation of the bacterial categories in the sampling site was assessed on Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt); the predicted functional classifications at the different levels (i.e. first, second, and third) were obtained.
Results
Physical and chemical analysis of sunflower rhizosphere and bulk soils
Soil analysis showed that OM, N-NH4 and total N were higher in rhizosphere soils from Lichtenburg (LTR) than in rhizosphere soils from Krayburg (KRPR) as shown in Table 1. We observed that the pH values of the soil samples from the LT site had low pH values (acidic) compared to the pH values of the soils from the KRP site.
Sequence data and beta analyses of the rhizosphere and bulk soil samples
The taxonomic groups were assigned using the SILVA reference database. The total number of uploaded sequences varied between samples and across the sites. Sequence base pair count of 87,446 (LTR), 80,404 (KRPR), 100,988 (bulk soils from Lichtenburg- LTB) and 74,956 (bulk soils from Krayburg- KRPB) sequence reads for the soil samples. Consequently, quality control (QC) check revealed the sequence read count for LTR—47,471, LTB—9,628, KRPR- 16,621, and KRPB – 19,050 between the samples and across the sites. Sequences were clustered at 97% similarity according to their connection to one another by Operational Taxonomic Units (OTUs) and the different OTU abundances in all samples were obtained based on the similarity threshold.The PCoA graph of the bacterial diversity at the phyla level in the soil samples across the sites is presented in Fig. 1, which indicated that samples from LTB differ significantly from LTR, KRPR, and KRPB samples. The vector length of the PCA graph revealed the most dominant bacterial phyla in each soil niche. Specifically, this is the bacterial phyla having the longest vector length of PCA. The vector length was used as an indicator, notably, in LTR Acidobacteria, Planctomycetes, Chloroflexi, Armatimonadetes, Gemmatimonadetes and Cyanobacteria dominated, whereas Actinobacteria, Elusimicrobia and Nitrospirae were predominant in LTB, whereas Proteobacteria, Verrucomicrobia, Bacteroidetes and unclassified sequences were prevalent in KRP, and the main bacterial phyla in KRPB were Spirochaetes and unclassified bacteria (Fig. 2). The bacterial phyla selected for PCoA and PCA plots were established on the level of significance. Analysis of similarities (ANOSIM) revealed that the differences in the beta diversity of the bacterial communities across the sites differed significantly (P = 0.01 and R = 0.58).

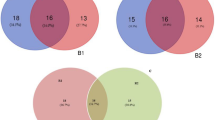
A Rarefaction curves used to determine the bacterial species richness sequences across the cropping sites. LTR, rhizosphere soil from Lichtenburg site; LTB, bulk soil from Lichtenburg site. KRPR, rhizosphere soil from Krayburg site; KRPB, bulk soil from Krayburg site. B Venn diagram of the distributed operation taxonomic units between the bacterial communities (at the phyla level) of the rhizosphere and bulk soils obtained from sunflower farms in Lichtenburg and Krayburg. LTR- Lichtenburg rhizosphere soil; LTB- Lichtenburg bulk soil; ` KRPR = Rhizosphere soils from Krayburg, KRPB = Bulk soils from Krayburg. C Principal coordinate analysis (PCoA) of shared OTUs between the rhizosphere and bulk soils from Lichtenburg and Krayburg at phylum level. (LTR = Rhizosphere soils from Lichtenburg, LTB = Bulk soils from Lichtenburg, KRPR = Rhizosphere soils from Krayburg, KRPB = Bulk soils from Krayburg)

Principal component analysis (PCA) of shared OTUs between the rhizosphere and bulk soils from Lichtenburg and Krayburg at phylum level. (LTR = Rhizosphere soils from Lichtenburg, LTB = Bulk soils from Lichtenburg, KRPR = Rhizosphere soils from Krayburg, KRPB = Bulk soils from Krayburg)
Structural composition of the bacterial community
At the phylum level, the dominant rhizospheric bacteria in LTR were Proteobacteria, Planctomycetes, Gemmatimonadetes, Acidobacteria, Armatimonadetes, and Cyanobacteria, while Actinobacteria, Nitrospirae and Elusimicrobia predominated LTB. Interestingly, unclassified bacteria dominated KRPR, while Firmicutes, Bateroidetes, Verrucomicrobia and unclassified sequences, and Spirochaetes were abundant in KRPB (Fig. 3).

A Taxonomic classification of the relative abundance of bacterial phylum from rhizosphere and bulk soils from Lichtenburg and Krayburg (LTR = Rhizosphere soils from Lichtenburg, LTB = Bulk soils from Lichtenburg, KRPR = Rhizosphere soils from Krayburg, KRPB = Bulk soils from Krayburg). The colour permeation gradient is designated as the scale bar based on the relative abundances; with a row z-score of the bacterial communities transformed relative abundance. B Taxonomic classification of the relative abundance of bacterial family from rhizosphere and bulk soils from Lichtenburg and Krayburg (LTR = Rhizosphere soils from Lichtenburg, LTB = Bulk soils from Lichtenburg, KRPR = Rhizosphere soils from Krayburg, KRPB = Bulk soils from Krayburg). The colour permeation gradient is designated as the scale bar based on the relative abundances; with a row z-score of the bacterial communities transformed relative abundance. C Taxonomic classification of the relative abundance of bacterial genus from rhizosphere and bulk soils from Lichtenburg and Krayburg (LTR = Rhizosphere soils from Lichtenburg, LTB = Bulk soils from Lichtenburg, KRPR = Rhizosphere soils from Krayburg, KRPB = Bulk soils from Krayburg). The colour permeation gradient is designated as the scale bar based on the relative abundances; with a row z-score of the bacterial communities transformed relative abundance
At the family level (Fig S1), Moraxellaceae, Caulobacteraceae, Geodermatophilaceae, Solirubrobacteraceae, Streptomycetaceae, Acetabacteraceae, Bradrhizobiaceae, Comamonadaceae, Micromonosporaceae, and Chitinophagaceae were predominant in LTR. Unknown bacteria, Micrococcaceae, Nocardioidaceae, Rhodospirillaceae, Pseudonocardiaceae, Sphingomonadaceae, Thermomonosporaceae, and Microbacteriaceae were dominant in LTB. Pseudomonadaceae, Streptomycetaceae, Paenibacillaceae and Planococcaceae influenced KRPR, while Baccilaceae, Rubrobacteraceae, Oxalobacteraceae, and Clostridiaceae dominated KRPB.
Influence of environmental factors on the bacterial community structure
The PCA (Fig. 4) was used to determine the correlation between the soil physical and chemical properties (Table 1) on the bacterial community distribution at the phylum level. The six best explained soil physical and chemical properties (Table 1) were considered for the PCA plot (Fig. 4). The PCA plot indicated that the bacterial community structure was influenced by the soil physicochemical properties. The total variation was 0.14385 and explanatory variable account for 100%. Using the vector length as an indicator, it is obvious that OM was at the mid-point. The vector lengths of total C, total N, and N-NH4 positively correlated with Planctomycetes, Armatimonadetes, Cyanobacteria, and Gemmatimonadetes from LTR. The vector length of N-NO3 and pH was positively correlated with Verrucomicrobia and unclassified sequences from KRPR.

Principal Component Analysis (PCA) plot of the bacterial phyla distribution and soil environmental variables of both rhizosphere and bulk soils from Lichtenburg and Krayburg. (OM = Organic matter, N-NH4 = Ammonium-N, N-NO3 = Nitrate, Total C = Total carbon, Total N = Total nitrogen)
Predictive functional information analysis associated with the bacterial community in the rhizosphere and bulk soils
The predictive functional categories of bacterial community composition with differences in their relative abundances across the sunflower farms at three different levels were analyzed employing PICRUSt. At level 1 functional classification, the bacterial predictive functions were categorized into 6 major predicted functions in both rhizosphere and bulk soils of the farms, including cellular processes, environmental information processing, genetic information processing, human diseases, metabolism, and organismal systems (Figs. 5a and 5b). Also, unclassified predicted functions were categorized (Fig. 5b).

a Major metabolisms of bacterial communities in the sunflower rhizosphere and bulk soils from Lichtenburg and Krayburg at level 1 and 2. (LTR = Rhizosphere soils from Lichtenburg, LTB = Bulk soils from Lichtenburg, KRPR = Rhizosphere soils from Krayburg, KRPB = Bulk soils from Krayburg). b Major metabolisms of bacterial communities in the sunflower rhizosphere and bulk soils from Lichtenburg and Krayburg at level 1 and 2. (LTR = Rhizosphere soils from Lichtenburg, LTB = Bulk soils from Lichtenburg, KRPR = Rhizosphere soils from Krayburg, KRPB = Bulk soils from Krayburg)
Furthermore, the predicted functions revealed at second-level classification (Figs. 5a and 5b), 16 predicted functions including cell communication, cell growth and death, replication and repair, immune system diseases, metabolic diseases, amino acid metabolism, biosynthesis of other secondary metabolites, carbohydrate metabolism, lipid metabolism, metabolism of cofactors and vitamins, metabolism of amino acids, xenobiotic biodegradation and metabolism, environmental adaptation and immune system were more predominant in LTR, whereas the predicted functions including cell motility, signal transduction, signal molecules and interaction, cancers, infectious diseases, neurodegenerative disease, N-Glycan biosynthesis and metabolism, circulatory system and nervous systems predominated the KRPR. The abundance of enzyme families in the soils across the sites were the same (1.86), except for LTB whose enzyme family’s relative abundance was 1.85. N-Glycan biosynthesis and metabolism relative abundance (1.47) were the same in LTR and LTB whereas KRPR and KRPB were 1.52 and 1.41 respectively.
The predicted functions revealed that at third-level selection (Fig. 6), the highest predicted functional profiling of bacteria was in KRPR. The abundance of bacterial motility proteins was predominant followed by ABC transporters (KRPR) whereas the least predicted function was the biosynthesis of steroid hormone (0.04) recorded in both the rhizosphere and bulk soils from Krayburg. Nitrogen (N) and sulfur (S) metabolism were higher in KRPR. Beta-Lactam resistance was the same (0.07) across all sites and samples.

Selected predictive metabolic pathways of bacterial communities in the rhizosphere and bulk soils of sunflower from Lichtenburg and Krayburg at level 3. (LTR = Rhizosphere soils from Lichtenburg, LTB = Bulk soils from Lichtenburg, KRPR = Rhizosphere soils from Krayburg, KRPB = Bulk soils from Krayburg)
We found that amino acids and derivatives pathways including alanine, aspartate and glutamate metabolism, phenylalanine metabolism, tryptophan, cyanoamino acid metabolism and taurine and hypotaurine metabolism, were more abundant in LTR than in other samples (Fig S2). Alternatively, the relative abundances of amino acid related enzymes, arginine and proline metabolism, cysteine and methionine metabolism, glycine, serine and threonine metabolism, histidine metabolism, lysine degradation, tyrosine metabolism, D-alanine metabolism, D-arginine and D-ornithine metabolism, D-glutamine and D-glutamate metabolism, glutathione metabolism, and phosphonate and phosphinate metabolism were more in KRPR than in other samples (Fig S2).
The predictive functions and bacterial community distribution in the rhizosphere and bulk samples
The PCA (Fig. 7) was used to illustrate the correlation between the predictive functional categories (Level 1) on the bacterial community distribution at the phylum level. The PCA plot indicated that axis 1 had 94.17% and axis 20.5%. The vector length of environmental information processing positively correlated with Spirochaetes, Verrucomicrobia, Firmucutes, unclassified sequences and unclassified bacterial community structure. The vector length of organismal systems positively correlated with Elusimicrobia, Nitrospirae, Acidobacteria and Actinobacteria.

Principal Component Analysis (PCA) of major predictive functional information (Level 1) of bacterial communities in the rhizosphere and bulk soils from Lichtenburg and Krayburg. The vector lengths depict the strength of the dominance of the bacterial metagenomes
The impact of soil physical and chemical properties on bacterial predictive functions
To determine the relationship between the predictive functional categories of bacterial communities in the samples from LT and KRP and soil physical and chemical properties, we used PCA (Fig S3). The forward selection result of environmental factors that best explain the variations in the bacterial structural composition and predictive functional categories revealed that only the p-value of N-NH4 at the structural categories was statistically significant (Table 2). The total variation was 0.00093 and the explanatory variable account for 100%. The results revealed that OM had the most explained variable and contribution of 93.4% at the structural classification, whereas pH had the most explained variable and contribution of 75.7% at the predictive functional categories, which is depicted by the length of the vector arrows, as shown in Fig S3 and Table 2.
Alpha diversity assessment of bacterial communities and predictive functions in the rhizosphere and bulk soil
The Simpson, Shannon_H and Evenness diversity index values within the samples were used to describe the alpha diversity of the bacterial communities at the taxonomic level presented in Table 3 across the sites. At the phylum and family levels, high Shannon_H diversity index values were obtained between the samples compared with other diversity indices measured across the farm sites (Table 3). These diversity indices at phylum and family levels demonstrated that there were no significant differences (p > 0.05) in the alpha diversity of the bacterial composition. Based on Shannon_H diversity indices, LTR had the highest alpha diversity index observed at the family level, and the least Shannon_H diversity index values were obtained LTB at the phylum level (Table 3).
Also, the result from the predictive functional categories analysis (Kruskal–Wallis, p-value = 0.51) (Table 3) showed that Shannon_H in LTR had a higher alpha diversity index compared to other samples. The alpha diversity showed that bacterial diversity and predictive functions were not significantly different (p-value > 0.05) between the LTR, LTB, KRPR and KRPB (Table 3).
Discussion
Sunflower is an important oil-seed crop, hence, increasing its production is a major step toward ensuring food availability and sustainable agriculture. Improving sunflower yield requires a better understanding of the structural, functional and metabolic potentials of the diverse bacterial communities abundant in their rhizosphere, especially those involved in biogeochemical cycles, plant-growth promotion, conservation of ecosystem function and sustainable agriculture (Li et al. 2022). In a bid to comprehend the activities in the plant rhizosphere, we employed a next-generation sequencing technique to evaluate the bacterial community structure in the rhizosphere and bulk soil of sunflower at the growing stage.
According to the information on the farm history, we postulated that soil physicochemical properties and agricultural practices, including the use of organic manure, chemical fertilizer, and cropping type (mono-cropping, and mixed cropping) may influence the bacterial diversity and their functions in the sunflower rhizosphere, which compelled the choice of the sampling sites. The application of organic manure and chemical fertilizer to enhance soil nutrients and plant growth, in reverse, may incite a shift in the bacterial community structure and soil properties (Li et al. 2017a). Our results demonstrated that agricultural practices altered both the structure and functional traits of the rhizosphere bacterial communities.
The use of next-generation sequencing technique (amplicon-based approach) has been employed in studies to evaluate the diversity of the bacterial communities in the rhizosphere soil of maize, soybean, as well as sunflower with success (Kielak et al. 2016; Naumoff and Dedysh 2012). In the present study, predominant bacterial phyla were identified in the rhizosphere and bulk soil of sunflower at the growing stage. The presence of these bacterial phyla might be due to their attraction to form a community within the rhizosphere. Most of the identified rhizosphere bacterial phyla have been previously reported in the rhizosphere of sunflower, soybean, wheat and maize (Alawiye and Babalola 2021; Igiehon et al. 2021; WEN et al. 2016). Firmicutes contributes a significant quantity of nitrogen to plant nutrition resulting to increase in agricultural crop yield (Ichihashi et al. 2020). Acidobacteria have previously reported to play a significant role in carbon cycling because of their potential to degrade complex plant tissues, as well as lignin and cellulose, though, their role in the rhizosphere is not well recorded (Ward et al. 2009), whereas Bacteroidetes contain some species that are involved in nitrogen cycling through denitrification (Chaparro et al. 2014).
The abundance of the identified bacterial phyla in sunflower soils from Lichtenburg has been reported to be important in improving soil health, plant growth and disease suppression (Kielak et al. 2016; Naumoff and Dedysh 2012; Li et al. 2017b). The phylum Armatimonadeteswas among the less abundant bacteria community identified in LT and it is relatively novel and was formerly recognized as a member phylum OP10. (Hu et al. 2014; Jiménez et al. 2020). There is limited information on its function in the rhizosphere or of the phylum (Jiménez et al. 2020). The unclassified bacterial phyla and identified unclassified sequences may create insights for further research in determining their novel distinctiveness.
The dominance the bacterial community in LTR compared to other samples may indicate the agricultural relevance of this bacterial family, whereas the bacterial community dominant in the KRPR site has been reported to be important plant growth-promoting bacteria. Similarly, the majority of these families have been shown to positively influence sunflower growth in the past (Tseng et al. 2021; WEN et al. 2016; Majeed et al. 2018).
Furthermore, differences in the relative abundance of the majority of bacterial community compositions in the rhizosphere soil of sorghum, maize, mustard, and cucumber plants have been shown to improve agrobiodiversity (Agomoh et al. 2020; Ali et al. 2019; Wang et al. 2012). Moreover, the effect of climatic conditions and soil management practices impact the distribution of bacterial communities in the rhizosphere (Igiehon and Babalola 2018). Interestingly, the variation in the bacterial community observed in the rhizosphere soil of Lichtenburg compared to the rhizosphere soil of Krayburg supports the study's hypothesis on the influence of mixed and crop rotational farming systems on the diversity of bacterial communities under diverse agricultural practices.
In the present study, the bacterial diversity indices at the phylum and family level showed a significant difference in the bacterial distribution across sites and this further explained how mixed cropping and crop rotational practice showed greater bacterial diversity in Lichtenburg than the mono-cropping system in Krayburg. The use of crop rotation systems in maintaining stable biodiversity and bacterial activities has been documented by (Gentsch et al. 2020). The series of mixed cropping system can increase nutrient acquisition and nutrient bioavailability, which directly increases rhizospheric bacteria and selectively attracts diverse plant growth-promoting bacteria into the region (Tyler 2021; Couëdel et al. 2018).
The bacterial phylum identified in this study, such as Elusimicrobia predominantly in soils from Lichtenburg, has not been reported in the soil of any oilseed crops, thus revealing its bioprospecting potential in agriculture. However, a study by (Gkarmiri et al. 2017) revealed an abundance of Verrucomicrobia, Gemmatimonadetes, Planctomycetes, Proteobacteria, Acidobacteria, Actinobacteria, and Chloroflexi in the rhizosphere soil of oilseed rape has been documented. The most active phylum Proteobacteria from the KRPR corroborates with that of (Saleem et al. 2016), who reported a similar bacterial phylum from the rhizosphere and roots of burley tobacco plants.
Intriguingly, the different families of rhizospheric bacteria in the LTR and KRPR can highlight their importance in agriculture in improving plant growth and health. Because of the large number of unclassified bacteria phyla found in sunflower using amplicon sequencing, the findings of this study can be used as a model in future studies of plant growth-promoting rhizospheric bacteria associated with oilseed crops, including sunflower. Continuous fertilization of farming soils may alter the bacterial diversity and nutritional profile (Zhang et al. 2020; Xiong et al. 2021).
Correspondingly, researchers have documented that OM is an important factor determining the diversity of bacterial in different soils, including sunflower and other agricultural soils (Cordero et al. 2020; WEN et al. 2016). The diversity, abundance, and richness of bacterial communities are largely dependent on the soil OM content. This study revealed that the soil OM influenced the relative abundances of the major phyla differently across the sites.
The vector lengths of the environmental variables in the PCA plot revealed that OM is not the only factor that influences the modelling of the bacterial communities and their functional diversity. The pH of the soil is a fundamental driver of the bacterial community structure (Qu et al. 2020). The pH values of sunflower rhizosphere and bulk soils ranged from 6.91 to 6.94 and this validates the findings of (Alawiye and Babalola 2021), who documented pH values, ranging from 5.8 to 6.6 on rhizosphere soils collected from four sunflower farms in South Africa. The effect of these factors on rhizospheric bacterial structure diversity and their functional potentials has been reported (Chen et al. 2021), though, may form the bacterial community structure and selection of soil for agricultural purposes.
In accordance to (Jacoby et al. 2017), phosphorus, sodium and potassium available in the rhizosphere also contribute to the soil microbial community structure and participate in the mineralization processes critical for plant nutrition in natural ecosystems. The secretion of root exudates released by plants is linked to the modulation of microbial communities and their functions in the rhizosphere (Bargaz et al. 2018). Also, root exudates initiate’s connections between the plant roots and soil microbes. The alpha diversity revealed no significant difference (p-value > 0.05) between bacterial diversity and predictive functions of the soils from LTR, LTB, KRPR and KRPB. In this study, the sunflower rhizosphere effect is the major driving force of alpha diversity.
Sunflower root exudates can influence bacterial diversity and functions in the rhizosphere and bulk soils after secreting different profiles of bioactive compounds and nutrients into the rhizosphere (Reavy et al. 2015; Wei et al. 2019). The alpha diversity indices (Shannon_H) also indicated that only the predictive functional diversity represented by the bacterial metagenomes of the rhizosphere and bulk soils passed its hypothetical limit of 2.81 (Dinsdale et al. 2008; Rygaard et al. 2017), suggesting that bacterial metagenomes were most characterized in both soils from LTR, LTB, KRPR, and KRPB. The Simpson and Evenness diversity indices for the metagenomes across all samples were < 1, indicating that there are a few predominant bacterial taxa, (e.g. Moraxellaceae, Solirubrobacteraceae, Chitinophagaceae, and Streptomycetaceae) and the predictive functional categories (At level 1, cell processing, environmental information processing, genetic information processing, organismal systems, metabolisms, and human diseases) in each soil samples.
The relative abundance of bacterial predictive functional categories at the second-level was used to distinguish the particular predictive functions that are of greater benefit to the bacteria present in a given habitat. Bacteria from soil use carbons as a source of energy for metabolism and growth, so they rely on diverse carbon sources such as maltose, inositol, glucose, and mannose for their growth and survival. Evidently, this is seen in the abundance of predictive functional categories involved in carbon dioxide, di- and oligosaccharides fixation and carbohydrate metabolism at level 2, as well as the presence of metabolic pathways related to sugar usage, galactose metabolism, fructose and mannose metabolism, starch and sucrose metabolism, pentose phosphate pathway and TCA cycle in our samples.
Also, bacteria use amino acids as an energy source for survival in environments with poor nutrient and in environments with little OM content (Gianoulis et al. 2009). This is in agreement with the results obtained for the soil physicochemical properties of our samples, where we found higher amounts of OM, N-NO3, P, and Na in the LTR samples than in the samples from the Krayburg site. The reason for the decline in soil nutrients in KRPR soil samples may be because of cropping system and land-use practices, this substantiate the report from previous studies that continuous cropping of a particular crop depletes soil nutrients (Kumar Behera et al. 2009; Dhaliwal et al. 2019; Foley et al. 2005; El-Fouly et al. 2015; Chen et al. 2018). Therefore, as a response mechanism for bacterial survival in nutrient-poor soils, the richness of genes linked with cell motility (bacterial mobility proteins and bacterial chemotaxis) is necessary.
Our findings demonstrated that the bacterial communities found in soil samples assist plants in acquiring carbon through various metabolic pathways (Wang et al. 2019; Xu et al. 2020; Prabha et al. 2019). In addition, sequences related to the metabolism of important mineral nutrients like S and N were discovered in soils. LTR and KRPR had larger relative abundances of these predictive functions than LTB and KRPB. Mineral nutrients are required for plant growth and health. As a result, our findings suggest that the bacterial communities found in our samples assist sunflower plants in obtaining essential nutrients for growth and development.
Similarly, at level 3, the presence of selected predicted metabolic functions such as ABC transporters, oxidative phosphorylation, glycerophospholipid, and nitrogen and sulfur metabolism in our samples indicate that bacterial communities inhabiting these soils play an important role in promoting nutrient cycling and plant growth (Tang et al. 2016; Reed et al. 2011; Lindsay et al. 2010). The metabolism of sulfur in level 3 revealed that the samples were also dominated by S. Studies have documented many critical roles played by bacteria found in various environments, including sunflower soil in the enzymatic sulfur metabolism (Wrighton et al. 2012; Yin et al. 2014; Zhang et al. 2016). Hence, the presence of genes involved in sulfur metabolism in our samples suggests that the bacteria present in our soil samples contribute to sustaining a balanced sulfur metabolism in their environments.
Moreover, we observed the presence of sequences involved in the biosynthesis of chelating and iron compounds such as siderophore in our results. Iron is a micronutrient that is important in the initiation of metabolic pathways and a constituent of the prosthetic group in living organisms (Dimkpa et al. 2009). Abiotic stresses in plants caused by iron can be alleviated by bacteria through high-affinity transport systems linking the biosynthesis of siderophores. The transport systems play critical roles in many soil environments, including aiding the competitive acquisition of iron for plant usage (Prabha et al. 2019; Mohapatra et al. 2021).
The sequences associated with metabolic activity, ABC transporters, were discovered at level 3. Most of these metabolic genes were found to be abundant in the Kraybrug samples. As a result, we predict that our samples will be dominated by bacteria that aid in the acquisition of minute iron, hence increasing iron bioavailability in our soil samples. At level 3, we observed the predicted metabolic processes, such as those involved in secondary metabolism, virulence, disease, and defense, stress response, and aromatic chemical metabolism. The abundance of sequences relevant to antibiotic and hazardous chemical resistance, siderophores, plant hormones, oxidative stress, and virulence regulation at level 3 support these findings.
The metabolic pathways involved in indole alkaloid biosynthesis, flavonoid biosynthesis, clavulanic acid biosynthesis, steroid hormone biosynthesis, inositol phosphate metabolism, linoleic acid metabolism, and N-Glycan biosynthesis were all shown to be abundant at level 3. Sequences associated to streptomycin biosynthesis and antibiosis resistance, particularly beta-lactam resistance, were discovered once more. Secondary metabolism, which includes the bacterial community's biosynthesis of several metabolites (low molecular-weight compounds) as a sign of metabolic complexity, is an important feature of bacteria from soil (Berdy 2005; Barka et al. 2016). Bacteria use this defense mechanism to defend themselves against pathogens. As a result, they are important in clinical practice, serving as antimicrobials and antibiotics (Newman and Cragg 2016).
Many of the bacterial phyla identified, such as Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes, produce a variety of bioactive compounds, such as siderophores, which act as antibacterial, antifungal, and biosurfactant (Hwang et al. 2014; O'Connor 2015; Ludwig-Müller 2015; Gómez Expósito et al. 2017). Because secondary metabolism is so important to plant growth, competent culturing methods must isolate and identify bacterial strains from the soil bacterial community that can perform secondary metabolic functions efficiently for increased crop yield. As a result, examining the soil bacterial community for bacteria that have these different genes can lead to the classification of novel secondary metabolic traits that can be used as biofertilizers in soils and plants to enhance resistance against pathogenic attacks. Our findings are in accordance with previous studies that show the diversity and abundance of genes linked to antibiotic resistance (Wang et al. 2013; Enagbonma and Babalola 2020).
The metabolic pathways of amino acids at level 3 revealed the samples were also dominated by amino acids and derivatives. Our results indicate that the bacterial communities inhabiting the fields can produce amino acids such as glutathione and Lysine involved in the protection against oxidative stress in the crops such as sunflower plants (Takagi and Ohtsu 2016). The high richness of the unclassified predicted functions and poorly characterized predicted functions at level 1 and level 2 respectively in our samples, show that there are many bacterial genes whose predicted functions in the soils are still uncharacterized. Though, the fact that they are present indicates that they contribute to significant functions in the soils that can be useful to the plants’ growth and health.
Another hypothesis of this study was that the physicochemical parameters would influence the predictive functional attributes of the bacterial communities in the samples. The aggregation of various bacterial communities occurs because of the pressure of selection of sunflower roots that constantly release exudates containing amino acids and carbohydrates into the rhizosphere. The effect of host plants on the bacterial diversity in the rhizosphere has been observed in various agricultural plants including, maize, wheat and peas (Mohammadi et al. 2011; Gentsch et al. 2020). (Hamel et al. 2006), documented that in a study of high-frequency pea production, there was a rise in the diversity of bacteria in pea rhizosphere soil with mineral nitrogen levels compared to the bulk soil.
Furthermore, metagenomic analysis of wheat rhizosphere and bulk soil found that the rhizosphere soil has higher bacterial diversity than the bulk soil (Priya et al. 2018; Velázquez-Sepúlveda et al. 2012). Moreover, several research focused mostly on selected rhizospheric isolates have found that the rhizosphere region of young plants is a more unpredictable environment than the rhizosphere region of mature plants during the developing phases of maize (Tiemann et al. 2015). This is consistent with the findings of (García‐Salamanca et al. 2013), who reported that the rhizosphere is a more nutrient-dense ecosystem than bulk soil, and that the activity levels of some enzymes from bacterial cells in the maize rhizosphere, such as dehydrogenase, β-glucosidase, and alkaline phosphatase, were higher than similar enzymatic activity tested in bulk soil.
We also discovered that physicochemical parameters influenced the predicted functionalities. The primary factors to the distinctiveness exhibited in soil bacterial structural diversity have been identified as soil physicochemical characteristics (Shi et al. 2011; Hanson et al. 2012). The functional diversity of bacterial communities is driven by soil characteristics, according to studies (Shi et al. 2011; Hanson et al. 2012). The present study's findings show that the physical and chemical properties of the soil impacted the relative abundance of bacterial predicted functions in the two study sites.
Conclusion
Understanding on the roles of various rhizospheric bacterial communities in the promotion of plant growth and health using 16S rRNA gene sequencing creates novel prospects for enhancing effective and eco-friendly methods for improving agricultural yield through the manipulation of microorganisms. Dissimilarities in predominant bacterial communities were documented between the rhizosphere and bulk soil across the sites. The dominance of unclassified bacteria and sequences in the samples proposes further studies in developing culturable approaches for their classification and discovery of new genes that can be harnessed as bioinoculants in developing environmentally friendly agriculture.
Across the sites, bacterial diversity was positively and negatively influenced by environmental variables. The predicted functional attributes of these bacteria propose their agricultural significance, which can be discovered in emerging biofertilizers as a substitute to chemical fertilizer. Because of the economic importance of sunflower, it is recommended to employ culture-dependent methods, invitro inoculation of seeds, and planting in the fields and greenhouse to further study the potential of rhizospheric bacteria on sunflower crops. Also, mining the metagenomes using more advanced techniques is important to identify novel genes that encode valuable metabolic pathways with numerous essential functions crucial for plant development and enhancement of sustainable agriculture [Klimek et al., 2016, Kumar and Dubey 2020].
Likewise, understanding plant-associated microorganisms under different cropping systems will help determine their functional roles in nutrient cycling, plant nutrition, development, and health. Interestingly, this study offers clear proof of the effect of agricultural practices, crop rotation and physicochemical properties on the bacterial diversity in sunflower soils from the two sites (Lichtenburg- LT and Krayburg- KRP). Conclusively, this study will enable the agricultural industry to enhance economic, agricultural and environmental sustainability by making critical soil management decisions.
Availability of data and materials
Sequence data obtained in this work have been deposited in the NCBI Sequence Read Archive under Accession Number PRJNA782103 and PRJNA672856.
References
Agomoh IV, Drury CF, Phillips LA, Reynolds WD, Yang X (2020) Increasing crop diversity in wheat rotations increases yields but decreases soil health. Soil Science Society of Amer J 84(1):170–181
Ai C, Liang G, Sun J, Wang X, Zhou W (2012) Responses of extracellular enzyme activities and microbial community in both the rhizosphere and bulk soil to long-term fertilization practices in a fluvo-aquic soil. Geoderma 173:330–338
Alawiye TT, Babalola OO (2021) Metagenomic insight into the community structure and functional genes in the sunflower rhizosphere microbiome. Agriculture 11(2):167
Ali A, Imran Ghani M, Li Y, Ding H, Meng H, Cheng Z (2019) Hiseq base molecular characterization of soil microbial community, diversity structure, and predictive functional profiling in continuous cucumber planted soil affected by diverse cropping systems in an intensive greenhouse region of northern China. Int J Mol Sci 20(11):2619
Bargaz A, Lyamlouli K, Chtouki M, Zeroual Y (2018) Dhiba D Soil microbial resources for improving fertilizers efficiency in an integrated plant nutrient management system. Front Microbiol 9:1606
Barka EA, Vatsa P, Sanchez L, Gaveau-Vaillant N, Jacquard C, Klenk H-P, Clément C, Ouhdouch Y, van Wezel GP (2016) Taxonomy, physiology, and natural products of Actinobacteria. Microbiol Mol Biol Rev 80(1):1–43
Berdy J (2005) Bioactive microbial metabolites. J Antibiot 58(1):1–26
Berlanas C, Berbegal M, Elena G, Laidani M, Cibriain JF, Sagües A, Gramaje D (2019) The fungal and bacterial rhizosphere microbiome associated with grapevine rootstock genotypes in mature and young vineyards. Fron Microbiol 10:1142
Bray RH, Kurtz LT (1945) Determination of total, organic, and available forms of phosphorus in soils. Soil Sci 59(1):39–46
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336
Chaparro JM, Badri DV, Vivanco JM (2014) Rhizosphere microbiome assemblage is affected by plant development. The ISME J 8(4):790–803
Chen S, Qi G, Luo T, Zhang H, Jiang Q, Wang R, Zhao X (2018) Continuous-cropping tobacco caused variance of chemical properties and structure of bacterial network in soils. Land Degrad Dev 29(11):4106–4120
Chen WC, Ko CH, Su YS, Lai WA, Shen FT (2021) Metabolic potential and community structure of bacteria in an organic tea plantation. Appl Soil Ecol 157:103762
Clarke K, Green R (1988) Statistical design and analysis for a’biological effects’ study. Mar Ecol Prog Ser. 46(1–3):213–226
Cordero J, de Freitas JR, Germida JJ (2020) Bacterial microbiome associated with the rhizosphere and root interior of crops in Saskatchewan. Canada Can J Microbiol 66(1):71–85
Couëdel A, Alletto L, Justes É (2018) Crucifer-legume cover crop mixtures provide effective sulphate catch crop and sulphur green manure services. Plant Soil 426(1):61–76
Craft C, Seneca E, Broome S (1991) Loss on ignition and Kjeldahl digestion for estimating organic carbon and total nitrogen in estuarine marsh soils: calibration with dry combustion. Estuaries 14(2):175–179
Dhaliwal S, Naresh R, Mandal A, Singh R, Dhaliwal M (2019) Dynamics and transformations of micronutrients in agricultural soils as influenced by organic matter build-up: A review. Environ Sustain Indicators 1:100007
Dimkpa C, Merten D, Svatoš A, Büchel G, Kothe E (2009) Siderophores mediate reduced and increased uptake of cadmium by Streptomyces tendae F4 and sunflower (Helianthus annuus), respectively. J Appl Microbiol 107(5):1687–1696
Dinsdale E, Edwards R, Hall D, Angly F, Breitbart M, Brulc J, Furlan M, Desnues C (2008) Functional metagenomic profiling of nine biomes. Nature 452:629–632
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27(16):2194–2200
El-Fouly MM, Fawzi A, Abou El-Nour E, Zeidan M, Firgany A (2015) Impact of long-term intensive cropping under continuous tillage and unbalanced use of fertilizers on soil nutrient contents in a small holding village. Afr J Agricult Res 10(53):4850–4857
Enagbonma BJ, Babalola OO (2020) Unveiling plant-beneficial function as seen in bacteria genes from termite mound soil. J Soil Sci Plant Nut 20(2):421–430
Foley JA, DeFries R, Asner GP, Barford C, Bonan G, Carpenter SR, Chapin FS, Coe MT, Daily GC, Gibbs HK (2005) Global consequences of land use. Science 309(5734):570–574
García-Salamanca A, Molina-Henares MA, van Dillewijn P, Solano J, Pizarro-Tobías P, Roca A, Duque E, Ramos JL (2013) Bacterial diversity in the rhizosphere of maize and the surrounding carbonate-rich bulk soil. Microb Biotechnol 6(1):36–44
Gentsch N, Boy J, Batalla JDK, Heuermann D, von Wirén N, Schweneker D, Feuerstein U, Groß J, Bauer B, Reinhold-Hurek B (2020) Catch crop diversity increases rhizosphere carbon input and soil microbial biomass. Biol Fert Soils 56(7):943–957
Gianoulis TA, Raes J, Patel PV, Bjornson R, Korbel JO, Letunic I, Yamada T, Paccanaro A, Jensen LJ, Snyder M (2009) Quantifying environmental adaptation of metabolic pathways in metagenomics. PNAS 106(5):1374–1379
Gkarmiri K, Mahmood S, Ekblad A, Alström S, Högberg N, Finlay R (2017) Identifying the active microbiome associated with roots and rhizosphere soil of oilseed rape. Appl Environ Microbiol 83(22):e01938-e1917
Gómez Expósito R, De Bruijn I, Postma J, Raaijmakers JM (2017) Current insights into the role of rhizosphere bacteria in disease suppressive soils. Front Microbiol 8:2529
Gutierrez Boem FH, Rubio G, Barbero D (2011) Soil phosphorus extracted by Bray 1 and Mehlich 3 soil tests as affected by the soil/solution ratio in Mollisols. Commun Soil Sci Plant Anal 42(2):220–230
Hamel C, Hanson K, Selles F, Cruz AF, Lemke R, McConkey B, Zentner R (2006) Seasonal and long-term resource-related variations in soil microbial communities in wheat-based rotations of the Canadian prairie. Soil Biol Biochem 38(8):2104–2116
Hammer Ø, Harper DA, Ryan PD (2001) PAST: Paleontological statistics software package for education and data analysis. Palaeontol Electron 4(1):9
Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JB (2012) Beyond biogeographic patterns: processes shaping the microbial landscape. Nature Rev Microbiol 10(7):497–506
Hu ZY, Wang YZ, Im WT, Wang SY, Zhao GP, Zheng HJ, Quan ZX (2014) The first complete genome sequence of the class Fimbriimonadia in the phylum Armatimonadetes. PLoS ONE 9(6):e100794
Hwang KS, Kim HU, Charusanti P, Palsson BØ, Lee SY (2014) Systems biology and biotechnology of Streptomyces species for the production of secondary metabolites. Biotechnol Adv 32(2):255–268
Ichihashi Y, Date Y, Shino A, Shimizu T, Shibata A, Kumaishi K, Funahashi F, Wakayama K, Yamazaki K, Umezawa A (2020) Multi-omics analysis on an agroecosystem reveals the significant role of organic nitrogen to increase agricultural crop yield. PNAS 117(25):14552–14560
Igiehon NO, Babalola OO (2018) Below-ground-above-ground plant-microbial interactions: focusing on soybean, rhizobacteria and mycorrhizal fungi. Open Microbiol J 12:261
Igiehon NO, Babalola OO, Aremu BR (2019) Genomic insights into plant growth promoting rhizobia capable of enhancing soybean germination under drought stress. BMC Microbiol 19(1):1–22
Igiehon NO, Babalola OO, Cheseto X, Torto B (2021) Effects of rhizobia and arbuscular mycorrhizal fungi on yield, size distribution and fatty acid of soybean seeds grown under drought stress. Microbiol Res 242:126640
Jacoby R, Peukert M, Succurro A, Koprivova A, Kopriva S (2017) The role of soil microorganisms in plant mineral nutrition—current knowledge and future directions. Front Plant Sci 8:1617
Jiménez JA, Novinscak A, Filion M (2020) Inoculation with the plant-growth-promoting rhizobacterium Pseudomonas fluorescens LBUM677 impacts the rhizosphere microbiome of three oilseed crops. Front Microbiol 11:2534
Khomtchouk BB, Hennessy JR, Wahlestedt C (2017) shinyheatmap: Ultra fast low memory heatmap web interface for big data genomics. PLoS ONE 12(5):e0176334
Kielak AM, Barreto CC, Kowalchuk GA, Van Veen JA, Kuramae EE (2016) The ecology of Acidobacteria: moving beyond genes and genomes. Front Microbiol 7:744
Klimek B, Chodak M, Jaźwa M, Niklińska M (2016) Functional diversity of soil microbial communities in boreal and temperate Scots pine forests. Euro J For Res. 135(4):731–742
Kumar A, Dubey A (2020) Rhizosphere microbiome: Engineering bacterial competitiveness for enhancing crop production. J Adv Res 24:337–352
Kumar Behera S, Singh D, Swaroop Dwivedi B (2009) Changes in fractions of iron, manganese, copper, and zinc in soil under continuous cropping for more than three decades. Commun Soil Sci Plant Anal 40(9–10):1380–1407
Li F, Chen L, Zhang J, Yin J, Huang S (2017a) Bacterial community structure after long-term organic and inorganic fertilization reveals important associations between soil nutrients and specific taxa involved in nutrient transformations. Front Microbiol 8:187
Li Y, Liu X, Hao T, Chen S (2017b) Colonization and maize growth promotion induced by phosphate solubilizing bacterial isolates. Int J Mol Sci 18(7):1253
Li Y, Wang C, Wang T, Liu Y, Jia S, Gao Y, Liu S (2020) Effects of different fertilizer treatments on rhizosphere soil microbiome composition and functions. Land 9(9):329
Li J, Dong L, Liu Y, Wu J, Wang J, Shangguan Z, Deng L (2022) Soil organic carbon variation determined by biogeographic patterns of microbial carbon and nutrient limitations across a 3, 000-km humidity gradient in China. CATENA 209:105849
Lindsay EA, Colloff MJ, Gibb NL, Wakelin SA (2010) The abundance of microbial functional genes in grassy woodlands is influenced more by soil nutrient enrichment than by recent weed invasion or livestock exclusion. Appl Environ Microbiol 76(16):5547–5555
Lu Y, Zhang E, Hong M, Yin X, Cai H, Yuan L, Yuan F, Li L, Zhao K, Lan X (2020) Analysis of endophytic and rhizosphere bacterial diversity and function in the endangered plant Paeonia ludlowii. Arch Microbiol 202(7):1717–1728
Ludwig-Müller J (2015) Plants and endophytes: equal partners in secondary metabolite production? Biotechnol Lett 37(7):1325–1334
Majeed A, Abbasi MK, Hameed S, Imran A, Naqqash T, Hanif MK (2018) Isolation and characterization of sunflower associated bacterial strain with broad spectrum plant growth promoting traits. Int J Biosci 13:110–123
Maquia IS, FareleiraVideira e CastroBrito PIDR, Soares R, Chaúque A, FerreiraPinto MM, Lumini A, Berruti E, Ribeiro NS (2020) Mining the microbiome of key species from African savanna woodlands: potential for soil health improvement and plant growth promotion. Microorganisms 8(9):1291
Meena VS, Maurya B, Verma JP (2014) Does a rhizospheric microorganism enhance K+ availability in agricultural soils? Microbiol Res 169(5–6):337–347
Mohammadi K, Heidari G, Khalesro S, Sohrabi Y (2011) Soil management, microorganisms and organic matter interactions: a review. Afr J Biotechnol 10(86):19840–19849
Mohapatra M, Yadav R, Rajput V, Dharne MS, Rastogi G (2021) Metagenomic analysis reveals genetic insights on biogeochemical cycling, xenobiotic degradation, and stress resistance in mudflat microbiome. J Environ Manag 292:112738
Naumoff DG, Dedysh SN (2012) Lateral gene transfer between the Bacteroidetes and Acidobacteria: the case of α-L-rhamnosidases. FEBS Lett 586(21):3843–3851
Nelson DW, Sommers LE (1996) Total carbon, organic carbon, and organic matter. In D. L. Sparks (ed) Methods of soil analysis: Part 3 Chemical methods. SSSA Washington DC 5: 961–1010
Newman DJ, Cragg GM (2016) Natural products as sources of new drugs from 1981 to 2014. J Nat Prod 79(3):629–661
Nwachukwu BC, Babalola OO (2021) Perspectives for sustainable agriculture from the microbiome in plant rhizosphere. Plant Biotechnol Rep. 15:1–20
Nwachukwu BC, Ayangbenro AS, Babalola OO (2021) Elucidating the rhizosphere associated bacteria for environmental sustainability. Agriculture 11(1):75
Oberholster T, Vikram S, Cowan D, Valverde A (2018) Key microbial taxa in the rhizosphere of sorghum and sunflower grown in crop rotation. Sci Tot Environ 624:530–539
O’Connor SE (2015) Engineering of secondary metabolism. Ann Rev Genet 49:71–94
Pandey R, Chavan P, Walokar N, Sharma N, Tripathi V, Khetmalas M (2013) Pseudomonas stutzeri RP1: a versatile plant growth promoting endorhizospheric bacteria inhabiting sunflower (Helianthus annus). J Biotechnol 8(7):48–55
Prabha R, Singh DP, Gupta S, Gupta VK, El-Enshasy HA, Verma MK (2019) Rhizosphere metagenomics of Paspalum scrobiculatum l.(kodo millet) reveals rhizobiome multifunctionalities. Microorganisms 7(12):608
Priya G, Lau N-S, Furusawa G, Dinesh B, Foong SY, Amirul AAA (2018) Metagenomic insights into the phylogenetic and functional profiles of soil microbiome from a managed mangrove in Malaysia. Agri Gene 9:5–15
Qu Z, Liu B, Ma Y, Sun H (2020) Differences in bacterial community structure and potential functions among Eucalyptus plantations with different ages and species of trees. Appl Soil Ecol 149:103515
Reavy B, Swanson MM, Cock PJ, Dawson L, Freitag TE, Singh BK, Torrance L, Mushegian AR, Taliansky M (2015) Distinct circular single-stranded DNA viruses exist in different soil types. Appl Environ Microbiol 81(12):3934–3945
Reed SC, Cleveland CC, Townsend AR (2011) Functional ecology of free-living nitrogen fixation: a contemporary perspective. Annu Rev Ecol Evol Syst 42:489–512
Rygaard AM, Thøgersen MS, Nielsen KF, Gram L, Bentzon-Tilia M (2017) Effects of gelling agent and extracellular signaling molecules on the culturability of marine bacteria. Appl Environ Microbiol 83(9):e00243-e217
Saleem M, Law AD, Moe LA (2016) Nicotiana roots recruit rare rhizosphere taxa as major root-inhabiting microbes. Microb Ecol 71(2):469–472
Shi JY, Yuan XF, Lin HR, Yang YQ, Li ZY (2011) Differences in soil properties and bacterial communities between the rhizosphere and bulk soil and among different production areas of the medicinal plant Fritillaria thunbergii. Inter J Mol Sci 12(6):3770–3785
Takagi H, Ohtsu I (2016) L-Cysteine metabolism and fermentation in microorganisms. In: Yokota A and Ikeda M (eds) Amino Acid Fermentation, Springer, Berlin 129–151
Tang Y, Zhang X, Li D, Wang H, Chen F, Fu X, Fang X, Sun X, Yu G (2016) Impacts of nitrogen and phosphorus additions on the abundance and community structure of ammonia oxidizers and denitrifying bacteria in Chinese fir plantations. Soil Biol Biochem 103:284–293
Tiemann L, Grandy A, Atkinson E, Marin-Spiotta E, McDaniel M (2015) Crop rotational diversity enhances belowground communities and functions in an agroecosystem. Ecol Lett 18(8):761–771
Tseng S-c, Liang C-m, Chia T, Ton S-s (2021) Changes in the composition of the soil bacterial community in heavy metal-contaminated farmland. Inter J Environ Res Pub Health 18(16):8661
Tyler HL (2021) Shifts in bacterial community in response to conservation management practices within a soybean production system. Biol Fert Soils 57(4):575–586
Velázquez-Sepúlveda I, Orozco-Mosqueda M, Prieto-Barajas C, Santoyo G (2012) Bacterial diversity associated with the rhizosphere of wheat plants (Triticum aestivum): Toward a metagenomic analysis. Phyton 81:81
Walakley A, Black C (1934) Estimation of organic carbon by chromic acid titration method. Soil Sci 37:29–38
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267
Wang D, Wang Y, Wu F (2012) Effect of different cultivation modes on cucumber growth and the numbers of culturable rhizosphere soil microorganisms. J Northeast Agric Univ 7:95–99
Wang Z, Zhang XX, Huang K, Miao Y, Shi P, Liu B, Long C, Li A (2013) Metagenomic profiling of antibiotic resistance genes and mobile genetic elements in a tannery wastewater treatment plant. PLoS ONE 8(10):e76079
Wang S, Li T, Zheng Z, Chen HY (2019) Soil aggregate-associated bacterial metabolic activity and community structure in different aged tea plantations. Sci Tot Environ 654:1023–1032
Ward NL, Challacombe JF, Janssen PH, Henrissat B, Coutinho PM, Wu M, Xie G, Haft DH, Sait M, Badger J (2009) Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl Environ Microbiol 75(7):2046–2056
Weber N, Liou D, Dommer J, MacMenamin P, Quiñones M, Misner I, Oler AJ, Wan J, Kim L, Coakley McCarthy M (2018) Nephele: a cloud platform for simplified, standardized and reproducible microbiome data analysis. Bioinformatics 34(8):1411–1413
Wei Y, Zhao X, Sun J, Liu H (2019) Fast repetition rate fluorometry (FRRF) derived phytoplankton primary productivity in the Bay of Bengal. Front Microbiol 10:1164
Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, Wilkins MJ, Hettich RL, Lipton MS, Williams KH (2012) Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337(6102):1661–1665
Xiong Q, Hu J, Wei H, Zhang H, Zhu J (2021) Relationship between plant roots, rhizosphere microorganisms, and nitrogen and its special focus on rice. Agriculture 11(3):234
Xu Y, Du A, Wang Z, Zhu W, Li C, Wu L (2020) Effects of different rotation periods of Eucalyptus plantations on soil physiochemical properties, enzyme activities, microbial biomass and microbial community structure and diversity. Forest Ecol Manag 456:117683
Xy WEN, Dubinsky E, Yao W, Rong Y, Fu C (2016) Wheat, maize and sunflower cropping systems selectively influence bacteria community structure and diversity in their and succeeding crop’s rhizosphere. J Integr Agric 15(8):1892–1902
Yadav AN, Verma P, Singh B, Chauhan VS, Suman A, Saxena AK (2017) Plant growth promoting bacteria: biodiversity and multifunctional attributes for sustainable agriculture. Adv Biotechnol Microbiol 5(5):1–16
Yin H, Zhang X, Li X, He Z, Liang Y, Guo X, Hu Q, Xiao Y, Cong J, Ma L (2014) Whole-genome sequencing reveals novel insights into sulfur oxidation in the extremophile Acidithiobacillus thiooxidans. BMC Microbiol 14(1):1–14
Zhang X, Niu J, Liang Y, Liu X, Yin H (2016) Metagenome-scale analysis yields insights into the structure and function of microbial communities in a copper bioleaching heap. BMC Genet 17(1):1–12
Zhang BH, Hong JP, Zhang Q, Jin DS, Gao CH (2020) Contrast in soil microbial metabolic functional diversity to fertilization and crop rotation under rhizosphere and non-rhizosphere in the coal gangue landfill reclamation area of Loess Hills. PLoS ONE 15(3):e0229341
Acknowledgements
BCN thanks the National Research Foundation (NRF), South Africa/The World Academy of Science African Renaissance Ph.D. scholarship (Ref: UID: 121772) for giving her a stipend. A. S. A. is grateful to the North-West University for postdoctoral bursary and research support. OOB. want to thank the National Research Foundation of South Africa for grants (Grant Refs: UID123634; UID 132595 granted to OOB) that have supported work in our laboratory.
Funding
Open access funding provided by North-West University. This work was supported by the National Research Foundation of South Africa [Grant Refs: UID123634; UID 132595 OOB].
Author information
Authors and Affiliations
Contributions
BCN handled the literature findings, carried out the laboratory work, performed all necessary analyses, interpreted the results, wrote the first draft and corrected the manuscript. ASA provided technical input and proofread the manuscript. OOB supervised all co-authors, provided academic and technical inputs, intensively critiqued the manuscript, and funded the research. All authors agreed that the manuscript is published.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Human and animal rights
No human subjects or livestock were included in this research.
Informed consent
The scientists certify that this study adhered to ethical and professional standards.
Competing of interests
No potential conflict of interest is declared by the author(s).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nwachukwu, B.C., Ayangbenro, A.S. & Babalola, O.O. Structural diversity of bacterial communities in two divergent sunflower rhizosphere soils. Ann Microbiol 73, 9 (2023). https://doi.org/10.1186/s13213-023-01713-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13213-023-01713-y