
Abstract
The aim of the study was to prepare elastic composites of epoxidized natural rubber with polylactide that was reinforced with lignocellulose. To improve the functional properties of an ENR/PLA blend, we used lignocellulose modified with silanes and silica. The addition of silanes, particularly U511-tetraethoxysilane, improved the strength characteristics of ENR/PLA composites, which were primarily shown as a considerable improvement in the elasticity of these materials. According to the thermogravimetric analysis and its derivative and differential scanning calorimetry curves for lignocellulose before and after modification with silane, changes were verified in its thermal profiles. Based on the tests of accelerated ageing, it is observed that all of the ENR/PLA composites with lignocellulose are characterized by a great degradability. The ageing coefficient (K) and parameters of oxidation induction time (OIT/DSC) show that the modification of lignocellulose with silanes increases the resistance of these materials to the action of climatic conditions, particularly UV radiation at 340 nm.
Similar content being viewed by others

Introduction
Conventional polymeric composites consist primarily of synthetic raw materials. Most of the polymers used do not exist in nature and do not undergo degradation. One can now observe an increase in the number of studies published on the technology of degradable polymeric materials.
Biodegradable materials have been produced and used in an increasingly wider range. The properties of all heterogeneous materials are determined by their structures, compositions and interfacial interactions. A direct influence on these components is exerted by synthesizing or modifying the materials used in the manufacturing processes of these materials or their types and origin. One can observe with increasing frequency the return to the use of raw materials from renewable sources [1–11].
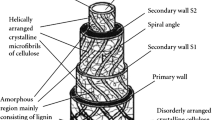
The desirable properties of lignocellulose materials encourage one to apply them to make polymeric composites that are designed for use in many industrial branches. One can observe a growing interest in renewable cellulose, hemicellulose or lignocellulose. Lignocellulose biomass is a fibrous portion of plant materials that is attractive because of its renewable origin and large resources. Lignocellulose fibres have some interesting mechanical and physical properties. The term lignocellulose is used to describe the three-dimensional polymeric composites that plants create as structural materials. It is composed of variable parts of cellulose, hemicellulose and lignin [12–24].
Renewable raw materials are also used to produce monomers and polymers. The basic biodegradable polymers include PHA, PLA, PHB, PHBV, PGA, PD, PHH, PBAT, PBS, PBSA, PHBH, PVOH, PCL, PES, PKA, BAT and TPS. Among the mentioned polymers, polylactide (PLA) plays a dominating role and constitutes approximately 40 % of all biodegradable polymers. This polymer is called doubly green because it is both biodegradable and is obtained from renewable sources [25–30].
Materials and methods
Reagents
The subject of the study, polylactide, was obtained from Nature Works™ (USA) (40phr). Epoxidized natural rubber (Epoxyprene 50; 50 mol% epoxidation) was obtained from Kumpulan Guthrie Berhad, Malaysia (60 phr). Dodecanoic acid (1 phr) 97 % obtained from Sigma-Aldrich was used as a cross-linking agent; 1,2-dimethylimidazole (0.5 phr) was used as a cross-linking catalyst (98 %). Lignocellulose (LC) (Lignocel C 120/HB 120) was characterized by length of fibre 70–150 μm, from Rettenmaier Poland Sp. z o.o.
Silanes obtained from Unisil Sp z.o.o. (Poland): U13 (3-aminopropyl)triethoxysilane, U15 [3-(2-aminoethylamino)propyl]trimethoxysilane, U511-tetraethoxysilane, U611-vinyltrimethoxysilane, were used as modificators (4 phr) of lignocellulose.
Symbols
Polylactide—PLA; epoxidized natural rubber—ENR; lignocellulose—LC, filler; silica Aerosil 380—A380, filler; polylactide/epoxidized natural rubber blend—ENR/PLA; oxygen induction time—OIT (min); a.u.—arbitrary unit; tensile strength—TS (MPa); elongation at the break—EB (%); energy–E (J g−1); ageing coefficient—K (a.u.).
Methods for modification of fibres and preparation of ENR/PLA blends with modified lignocellulose
Mechanochemical treatment of lignocellulose with silane coupling agents
Samples of lignocellulose fibres modified by using silane were obtained by intergrinding the specified mixtures in a laboratory planetary mill (Fritsch Co., Germany). Modification of the tested filler was performed in a ball mill at speeds of 500 rpm. The reaction time, i.e. the time to apply mechanochemical treatment of fibres, was 2 h. The dry mixture (lignocellulose with silane) in the amount of 10 g was ground in a steel beaker with steel balls (15 steel balls; individual diameter 10 mm and mass 4.08 g) in a Pulverisette 5 from Fritsch. The concentration of the silane coupling agent was 10 % by mass of lignocellulose.
The preparation of ENR/PLA biocomposites with biofillers
Homogenization of polymers (polylactide and epoxidized natural rubber) and cellulose was done in a Brabender Measuring Mixer N50 (Duisburg, Germany). The polymer blends were processed at 40 rpm rotors speed, and the initial temperature was set at 180 °C. After around 15 min of polymer mastication, filler (lignocellulose or silica Aerosil 380) was added and mixed for 15 min.
The vulcanization of rubber blends was carried out with the use of steel vulcanization moulds placed between the shelves of an electrically heated hydraulic press. Teflon films were used as spacers to prevent the adherence of the blends to the press plates. Samples were vulcanized at a temperature of 160 °C under a pressure of 15 MPa for 60 min.
Measurement methods
Mechanical properties of ENR/PLA composites
The tensile strength of vulcanizates was tested according to the standard PN-ISO 37:1998 by means of a ZWICK tester, model 1435, for dumbbell w-3.
Ageing characteristics were determined according to the standard PN-82/C-04216. Samples were exposed to air at elevated temperature (383 K) for 10 days in a dryer with thermocirculation. UV ageing was performed using a UV 2000 apparatus from Atlas. The measurement lasted 288 h and consisted of two alternately repeating segments with the following parameters: daily segment (radiation intensity 0.7 W m−2, temperature 60 °C, duration 8 h) and night segment (no UV radiation, temperature 50 °C, duration 4 h). Climatic ageing was carried out using a Weather Ometer (Atlas; Ci 4000). The test was based on two variable segments simulating day and night conditions, and the samples were subjected to two different cycles: daily cycle (radiation intensity 0.4 W m−2, temperature 60 °C, duration 240 min, humidity 80 %, rain water on) and night cycle (no radiation, temperature 50 °C, humidity 60 %, duration 120 min).
The ageing coefficient was calculated according to the relationship:
where TS indicates tensile strength, EB indicates elongation at break, and TS′ and EB′ are the corresponding values after ageing.
Scanning electron microscope (SEM) analysis
The dispersion degree of the fillers in the elastomeric matrix was assessed from images obtained using a LEO 1530 scanning electron microscope (SEM). The samples tested consisted of vulcanizate fractures in liquid nitrogen dusted with carbon.
Thermal analysis of fibre and composite
The oxygen induction time (OIT) test was performed on a Mettler Toledo DSC instrument. Samples with a mass of 4 mg were heated from room temperature to the test temperature, 220 °C, at a rate of 20 °C min−1 under a nitrogen atmosphere. After 5 min at 220 °C, the gas was switched from nitrogen to air at a flow rate of 60 mL min−1. When all antioxidants are consumed, the sample starts to oxidize, producing a deviation in the baseline. The OIT was measured as the time between the switch to air flow and the intersection with a tangent from the maximum derivative after oxidation has started. Two analyses of each sample were performed to ensure the accuracy of the results. The OIT was measured according to the national and international standards such as ASTM D 3895.
The crystallized specimens were characterized by using temperature-modulated DSC (TA 2920; TA Instruments) at the heating rate of 5 °C min−1 with a heating/cooling cycle of the modulation period of 60 s and an amplitude of ±0.769 °C, to determine the glass transition (T g) crystallization temperature (T c) and the melting temperature (T m). The DSC was calibrated with indium before use. For the measurement of T c, we used amorphous samples after quenching from malt state (∼200 °C).
Thermal decomposition (TG) was studied by using a Mettler Toledo Thermobalance. Samples of approximately 5 mg were placed in aluminium oxide crucible and heated from 20 to 700 °C under a dynamic flow of nitrogen (50 mL min−1). Five heating rates (5 °C min−1) were used.
Infrared spectra (FTIR) analysis
Infrared spectra (FTIR) were measured within the wavelength range of 3500–700 cm−1 using a FTIR Nicolet 6700 spectrophotometer and OMNIC 3.2 software (Thermo Scientific Products: Riviera Beach, FL, USA). ATR accessory equipped with a single reflection diamond ATR crystal on ZnSe plate was used for all of the analysis. Fourier transform infrared spectroscopy was performed with a DGTS/KBr detector, with the following measurement parameters: 128 scans, resolution 8 cm−1 and the speed of scanning equal to 0.6329 cm s−1.
Results and discussion
The first step of the present study was the thermal analysis of lignocellulose samples. Various thermal techniques (DSC, TG, OIT) were used to determine the effect of mechanochemical modifications on the thermal properties of natural fibres. The next step was the characteristics properties of the composites based on epoxidized natural rubber and polylactide with the addition of modified fillers. Another very helpful analysis to study the effectiveness of modification of natural fibres is undoubtedly FTIR spectroscopy. Modification with silane of lignocellulose fibre had helped achieve better interaction between the polymer matrix and filler. Better adhesion between polymer and filler results in better dispersion and what is associated with it is a better ability to transfer stress in composite.
Thermal analysis (differential scanning calorimetry and thermogravimetry)
The DSC curve of the lignocellulose shows an endothermic effect at 46–137 °C (E = 142 J g−1) and a similar effect at 42–144 °C (E = 144 J g−1) for the fibre that was modified with silane. These effects are connected to the dehydration and phase changes in the lignocellulose components: hemicellulose, lignin and cellulose. According to the literature data, the glass transition temperature of hemicellulose is approximately 400 °C and that of lignin is in the range of 50–100 °C. The mechanochemical modification with silane shifted the decomposition temperature of lignocellulose from 340–320 °C (Fig. 1).

DSC curves for lignocellulose and lignocellulose modified by silane obtained in the heating (from room temperature to 200 °C)
The TG curve illustrates the thermal stability of lignocellulose. The first mass change observed is connected to the thermal decomposition of hemicellulose and lignin. The second inflexion in the TG curve is observed at a temperature of approximately 340 °C and is connected to the cellulose degradation. The silane modification exerted no significant influence on the thermal stability of lignocellulose, and the differences observed in the thermogravimetric curves of the first transition connected to the material decomposition are within the limits of 2°–3° (Fig. 2).

TG curves of lignocellulose and lignocellulose modified by silane obtained during the temperature increase by 5 °C per minute in air static
The TG curve of ENR/PLA composites shows two peaks connected to the thermal degradation of the material. The decomposition of the first stage for ENR/PLA composites is observed at a temperature of 150 °C (46 %), whereas the second stage begins at 350 °C and has a mass loss of 47.16 %. The addition of lignocellulose decreases the mass loss in the first stage to 36.67 % at a temperature of 150 °C, whereas the second stage begins at a temperature of 320 °C and has a mass loss of 55.29 %. The first stage of thermal degradation is most likely connected to the decomposition of the epoxidized natural rubber, and the second stage results from the decomposition of polylactide with lignocellulose (hemicellulose, lignin and cellulose) (Fig. 3).

TG curves of ENR/PLA composites with lignocellulose and lignocellulose modified by silane obtained during the temperature increase by 5 °C per minute in air static
The increase in the ENR/PLA composites resistance is also confirmed by the oxidation induction times determined using the DSC method. The addition of lignocellulose itself prolonged the oxidation by 1.24 min (Figs. 4–7).

Oxidative stability of ENR/PLA composites containing mechanochemical modification lignocellulose. The oxidation stability of composites was determined via the oxidation induction time (OIT) by means of differential scanning calorimetry (DSC) in standardized procedures

Thermooxidation energy of ENR/PLA composites containing mechanochemical modification fillers

Values of tensile stress (TS/MPa) of ENR/PLA composites after thermooxidation, UV ageing and weathering. Shortcuts: (ENR) epoxidized natural rubber, (LC) lignocellulose, U13 (3-aminopropyl)triethoxysilane, U15 [3-(2-aminoethylamino)propyl]trimethoxysilane, U511-tetraethoxysilane, U611-vinyltrimethoxysilane, silica Aerosil 380 (A380)

Values of elongation at break (EB/%) of ENR/PLA composites after thermooxidation, UV ageing and weathering
The OIT value of the sample containing lignocellulose modified with 3-(2-aminoethylamino)propyl] trimethoxysilane amounted to 9.72 min.
Influence of modified lignocellulose on the mechanical and morphological properties of ENR/PLA composites
The composites of epoxidized natural rubber with polylactide are characterized by mechanical properties ranging from 9.30 to 16 MPa. The highest tensile strength is shown by the reference sample that contains unmodified lignocellulose. The modification of composites with the use of silanes, in most samples, decreases the tensile strength (TS), but there is a considerable increase in elongation at the break, which means that the elastic properties of the materials are improved. The highest elasticity of the material is observed in ENR/PLA samples that contain lignocellulose modified with U511-tetraethoxysilane, for which the value of elongation at the break amounts to 466 %, whereas that of the material modified with U611-vinyltrimethoxysilane is 382 %. Conversely, the reference sample of ENR/PLA with lignocellulose is characterized by a weak elongation at the break amounting to 210 %.
All of the ENR/PLA composites are degradable under both climatic conditions and under the influence of single factors such as UV or temperature. The addition of lignocellulose modified with silanes improves the resistance of the composites to ageing. The most susceptible to ageing are samples containing lignocellulose modified with (3-aminopropyl)triethoxysilane and 3-(2-aminoethylamino)propyl]trimethoxysilane, particularly when they are aged under the influence of synergic climatic conditions and concentrated radiation at 340 nm (Fig. 8).

Ageing coefficient (K) ENR/PLA composites containing mechanochemical modification fillers
The colour change of the composites under the influence of various degrading conditions was assessed based on the UV–Vis spectra. The highest values of parameter dE*ab over 40 a.u. are shown by composites after climatic ageing. UV irradiation resulted in thermooxidation, and it caused a similar and insignificant effect on the colour change of the composite samples (Fig. 9).

Colour difference (dE*ab) of ENR/PLA composites after thermooxidation, UV ageing and weathering was measured according to the Cie-Lab colour scale. Coordinate a* green/red colour component (a* > 0 red, a* < 0 green) and b* blue/yellow colour component (b* > 0 yellow, b* < 0 blue). (Color figure online)
It is observed that fibres are oriented and that their surface is not fuzzy or cracked. Thus, the fibres are very tough if they maintain their shape after modification. Because of the orientation and good quality of the fibres used, they can effectively stress in composites during the action of a greater force. On the fibre surface, one can observe a polymeric cover that is well adhered to lignocellulose because of an efficient transfer of stresses. However, photographs show a very pure fibre surface, which may indicate low adhesion on the fibre/matrix phase boundary. Therefore, the elongation at break is not great. The adhesion on the fibre/matrix phase boundary improved by modification allows us to obtain a positive composite reinforcement result.
Surface characterization by FTIR-ATR spectroscopy of lignocellulose treated with silane
On the FTIR spectrum visible are the most important bands of respondent fibres. The large band at 3300–3500 cm−1 is related to O–H groups or the C–H band at 2900 cm−1 (methyl and methylene groups). The band near 1240 cm−1, which appears in the untreated fibres of FTIR spectrum, corresponds to an axial asymmetric strain of =C–O–C. It is commonly observed when =C–O–, e.g. when ether, ester and phenol groups are present. The band near 1460 cm−1 appears to be from the –CH2 group of cellulose and lignin, but bands near 1370 and 1319 cm−1 are from the C–H group of cellulose (crystallized and amorphous) and hemicellulose.
The spectral band at 1635 cm−1 corresponds to water absorption because of the hydrophilic character of the lignocellulosic fibre. Bands near 1100 cm−1 are from Si–O–Si groups and absorption bands at 1059 cm−1 from the mSi–O–Si stretching vibrations, which can confirm the chemical modification of fibre.
The FTIR spectrum can have proposed reaction scheme for the interaction between silanized lignocellulose fibre and polymer. Epoxidized natural rubber can react with amine group from silane. Scheme 1 shows the possibilities of oxirane ring reaction with –NH2 group. The extent of silane reaction with ENR was estimated using semiquantitative FTIR approach. The spectrum in Fig. 10 exhibits the epoxide groups (C–O–C) of ENR-50 at 1248 and 880 cm−1. The moderate absorption peaks at 1060 and 1114 cm−1 are due to asymmetric and symmetric stretching of the C–O bond. The absorption band at 1627 cm−1 is a characteristic of –NH2 rocking vibration. Comparison of the FTIR spectra also indicates that there is a reduction in the epoxide band intensity after the reaction. Oxirane ring of ENR rubber can react with amine group of silane. The possible mechanism of this reaction is shown in Scheme 1 (Figs. 11, 12).

The probable mechanism of bonding between ENR rubber and silane

Scanning electron micrograph (SEM) of a lignocellulose; b lignocellulose treated by silane (U15); c lignocellulose treated by silica Aerosil 380; d ENR/PLA/LC/U611

Changes in the surface chemical structure of lignocellulose, before and after its modification with silane based on infrared spectra (FTIR)

FTIR of epoxidized natural rubber, silane and ENR/PLA composites modified by silane
Conclusions
Based on the studies performed, we can state that the addition of lignocellulose improves the strength characteristics of the composites of epoxidized natural rubber with polylactide. The mechanochemical modification of the filler with silanes has contributed to the improvement in ENR/PLA material’s elasticity according to the assumption concerning the design of elastic ENR/PLA composites. The material developed is characterized by high degradability under the influence of climatic conditions such as UV radiation, temperature or humidity. The modification of lignocellulose with silanes improves the adhesion at the fibre-matrix phase boundary by improving the dispersion of the ENR/PLA blend. At the same time, it also improves the stabilization of the ENR/PLA blend under the influence of UV radiation or thermooxidation.
References
Jiang L, Zhang J, Wolcott MP. Comparison of polylactide/nanosized calcium carbonate and polylactide/montmorillonite composites: reinforcing effects and toughening mechanisms. Polymer. 2007;48:7632–44.
Woodruff MA, Hutmacher W. The return of a forgotten polymer-polycaprolactone in the 21st century. Prog Polym Sci. 2010;35:1217–56.
Asokan P, Firdoous M, Sonal W. Properties and potential of bio fibres, bio binders, and bio composites. Rev Adv Mater Sci. 2012;30:254–61.
Wuzella G, Mahendran AR, Müller U, Kandelbauer A, Teischinger A. Photocrosslinking of an acrylated epoxidized linseed oil—kinetics and its application for optimized wood coatings. J Polym Environ. 2012;20:1063–74.
Zhang MQ, Rong MZ, Lu X. Fully biodegradable natural fibre composites from renewable resources: all-plant fibre composites. Comp Sci Technol. 2005;65:2514–25.
Khattak WA, Khan T, Ul-Islam M, Wahid F, Park JK. Production, characterization and physicomechanical properties of bacterial cellulose from industrial wastes. J Polym Environ. 2015;23:45–53.
Shirai MA, Müller CMO, Eiras Grossmann MV, Yamashita F. Adipate and citrate esters as plasticizers for poly(lactic acid)/thermoplastic starch sheets. J Polym Environ. 2015;23:54–61.
Khajeheian MB, Rosling A. Rheological and thermal properties of peroxide-modified poly(l-lactide)s for blending purposes. J Polym Environ. 2015;23:62–71.
Kalia S, Kaith BS, Kaur I. Cellulose fibres: bio- and nano-polymer composites. Berlin: Springer; 2011.
Faruk O, Bledzki AK, Fink HP, Sain M. Biocomposites reinforced with natural fibres: 2000–2010. Prog Polym Sci. 2012;11:1552–96.
Brebu M, Cornelia V. Thermal degradation of lignin. Cell Chem Technol. 2010;44:353–63.
Thakur VK, Thakur MK, Gupta RK. Graft copolymers from cellulose: synthesis, characterization and evaluation. Carbohydr Polym. 2013;97:18–25.
Solarski S, Ferreira M, Devaux E. Ageing of polylactide and polylactide nanocomposite filaments. Polym Degrad Stab. 2008;93:707–13.
Leszczynska A, Njuguna J, Pielichowski K, Banerjee JR. Polymer/montmorillonite nanocomposites with improved thermal properties Part I. Factors influencing thermal stability and mechanisms of thermal stability improvement. Thermochim Acta. 2007;453:75–96.
Tsuji H, Echizen Y, Nishimura Y. Photodegradation of biodegradable polyesters: a comprehensive study on poly(L-lactide) and poly(3-caprolactone). Polym Degrad Stab. 2006;91:1128–37.
Zou H, Yi Ch, Wang L, Liu H, Xu W. Thermal degradation of poly(lactic acid) measured by thermogravimetry coupled to Fourier transform infrared spectroscopy. J Therm Anal Calorim. 2009;97:929–35.
Masek A, Chrzescijanska E, Zaborski M, Piotrowska M. Dodecyl gallate as a pro-ecological antioxidant for food packing materials. CR Chim. 2014;17:1116–27.
Ahmed J, Zhang JX, Song Z, Varshney SK. Thermal properties of polylactides. Effect of molecular mass and nature of lactide isomer. J Therm Anal Calorim. 2009;95(3):957–64.
Garlotta D. A literature review of poly(lactic acid). J Polym Environ. 2001;9(2):63–84.
Manich AM, Carilla J, Miguel RAL, Lucas JM, Franco FGF, Montero LA, Cayuela D. Thermal transitions of polylactide false-twist textured multifilaments determined by DSC and TMA. J Therm Anal Calorim. 2010;99:723–31.
Masek A, Chrzescijanska E, Zaborski M, Piotrowska M. Controlled degradation of biocomposites ENR/PCL containing natural antioxidants. CR Chim. 2013;16:990–6.
Laskowska A, Zaborski M, Boiteux G, Gain O, Marzec A, Maniukiewicz W. Ionic elastomers based on carboxylated nitrile rubber (XNBR) and magnesium aluminum layered double hydroxide (hydrotalcite). Exp Polym Lett. 2014;8:374–86.
Mishra JK, Chang YW, Kim DK. Epoxidized natural rubber toughened polylactic acid/talc composites: mechanical, thermal, and morphological properties. Mater Lett. 2007;61:3551–4.
Laskowska A, Marzec A, Boiteux G, Zaborski M, Gain O, Serghei A. Effect of imidazolium ionic liquid type of nitrile rubber composites. Polym Int. 2013;62:1575–82.
Ljungberg N, Wesslen B. The effects of plasticizers on the dynamic mechanical and thermal properties of poly(lactic acid). J Appl Polym Sci. 2002;86:1227–34.
Yang HS, Gardnem DJ, Nader JW. Dispersion evaluation of microcrystalline cellulose/cellulose nanofibril polypropylene composites using thermogravimetric analysis. J Therm Anal Calorim. 2011;103:1007–15.
Marzec A, Laskowska A, Boiteux G, Zaborski M, Gain O, Serghei A. The impact of imidazolium ionic liquids on the properties of nitrile rubber composites. Eur Polym J. 2014;53:139–46.
Thakur VK, Thakur MK, Gupta RK. Development of functionalized cellulosic biopolymers by graft copolymerization. Int J Biol Macromol. 2013;62:44–51.
Agrawal M, Ray RB. Biodegradable polymeric scaffolds for musculoskeletal tissue engineering. J Biomed Mater Res. 2001;55:141–50.
Chrissafis K, Paraskevopoulos KM, Papageorgiou GZ, Bikiaris DN. Thermal and dynamic mechanical behavior of bionanocomposites: fumed silica nanoparticles dispersed in poly(vinylpyrrolidone), chitosan, and poly(vinyl alcohol). J Appl Polym Sci. 2008;110:1739–49.
Author contributions
Anna Masek conceived, designed and performed the experiments, analysed and interpreted the data and wrote the paper; Marian Zaborski and Karolina Diakowska performed the experiments and analysed and interpreted the data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Masek, A., Diakowska, K. & Zaborski, M. Physico-mechanical and thermal properties of epoxidized natural rubber/polylactide (ENR/PLA) composites reinforced with lignocellulose. J Therm Anal Calorim 125, 1467–1476 (2016). https://doi.org/10.1007/s10973-016-5682-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5682-5