
Abstract
We investigated Ocean sediments and seawater from inside the Fukushima exclusion zone and found radiocesium (134Cs and 137Cs) up to 800 Bq kg−1 as well as 90Sr up to 5.6 Bq kg−1. This is one of the first reports on radiostrontium in sea sediments from the Fukushima exclusion zone. Seawater exhibited contamination levels up to 5.3 Bq kg−1 radiocesium. Tap water from Tokyo from weeks after the accident exhibited detectable but harmless activities of radiocesium (well below the regulatory limit). Analysis of the Unit 5 reactor coolant (finding only 3H and even low 129I) leads to the conclusion that the purification techniques for reactor coolant employed at Fukushima Daiichi are very effective.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Following the earthquake on March 11, 2011, a gigantic tsunami destroyed the cooling systems of Fukushima Daiichi nuclear power plant (NPP)(Japan) and caused partial melt-down of 3 reactor cores. In the course of the nuclear accident a total of 520 PBq (excl. noble gases) were released to the atmosphere [1]. More than 99 % of the released substances were radionuclides of Kr, Te, I, Xe, and Cs. These nuclides have been monitored globally in air [2, 3]. Less volatile radionuclides such as radiostrontium [4, 5], plutonium [6–8] or radionuclides that are difficult to measure, such as 3H [9, 10], 135Cs [11] or 35S [12] have been monitored much less frequently [13]. In any case, at least 80 % of the airborne radionuclides were transported offshore by the wind [14, 15], and a yet unknown amount has leaked directly into the Pacific Ocean. Although several studies have addressed the impact of the accident on the Pacific Ocean and its organisms [16–18], our knowledge on the impact of the Fukushima nuclear accident on the marine environment is yet far from complete.
In the current study, we investigated several interesting and unique sample materials, many of which were taken directly after the accident and/or inside the “exclusion zone” around the crippled reactors. Target nuclides were radiocesium [134Cs (T 1/2 = 2.1 years) and 137Cs (T 1/2 = 30.2 years)] as well as the understudied nuclides 90Sr (T 1/2 = 28.6 years) and 3H (T 1/2 = 12.3 years). For two samples, also 129I analysis was performed. It is a long-lived (T 1/2 = 15.7 million years) fission product that can be used as an (environmental) tracer nuclide [19] that is also useful for retrospective dosimetry after the decay of short-lived 131I (T 1/2 = 8 days) [20] which accumulates in the thyroid [21] and is mostly responsible for thyroid cancer cases after Chernobyl.
Materials and methods
Environmental materials analyzed in this study included sediments and seawater from the Japanese Pacific Ocean coast inside the exclusion zone, tap water from Tokyo, seawater from Hawaii as well as a sample of the reactor coolant water of the undamaged Unit 5 of Fukushima Daiichi NPP. The coolant was investigated also for 129I to scrutinize the overall efficiency of the techniques currently employed for radionuclide removal from water at the Fukushima Daiichi NPP; most fission and activation products are cations that can be removed using natural or artificial cation exchanger. Iodine, however, is usually anionic (iodide or iodate) and thus more challenging to remove. A summary of all samples, the exact location and date of sampling, distance to the Fukushima Daiichi NPP as well as target nuclides is given in Table 1.
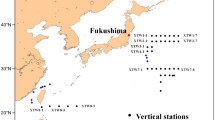
A map of the sample locations (excluding the seawater sample from Hawaii), is given in Fig. 1.

Geographical setting of the sampling sites in Japan
Gamma measurements for 134Cs and 137Cs were conducted with an ORTEC™ 364 cm3 HPGe detector with a 0.76 mm Be window (2.32 keV resolution at the 1332 keV 60Co peak; 87.4 % relative efficiency). The samples were weighed into 60 or 125 mL polypropylene containers (typically 100 mL for water samples; 70 g for sediment samples) that were placed on top of the detector. The samples were measured for at least 24 h. Detector efficiency calibration was done with an Eckert and Ziegler® multinuclide standard solution that was diluted in 2 M HCl (aqueous samples) or dispersed in quartz sand (sediment samples) (see [22]). Tritium (3H) and 90Sr was conducted with liquid scintillation counting (LSC). The LSC measurements were performed using a LabLogic 300SL Liquid Scintillation Counter with TDCR Technology (Hidex, Mustionkatu 2, FIN-20750 Turku, Finland). The user interface software is provided by MikroWin™ (Mikrotek Laborsysteme GmbH, Olper Strasse 35, D-51491 Overath, Germany). Instrument output was transferred to a vendor-provided PC-application spreadsheet with macros where the raw data could be analyzed and a graphic routine provided for convenient data visualization [23].
For radiocesium measurements, the sediment samples were dried at 100 °C, weighed and filled into calibrated geometries at the gamma detector station at Colorado State University. Water samples were weighed and measured for gamma emitters without further pre-treatment.
Pure beta emitters such as 90Sr or 3H require chemical separation and purification prior to beta counting in the LSC. Water samples were carefully distilled to dryness; the distillate was mixed with Ultima Gold™ low level tritium (LLT) scintillation cocktail for measurement. Ocean sediments were treated with nitric acid for Sr leaching and purified using the Eichrom™ SR resin (2 mL cartridges), as described elsewhere [4]. In brief, the separation process involved weighing of the dried sand into a flask, addition of 8 mL HNO3 (8 M), 2 mL H2O2 (30 %), 1 ml Sr carrier solution (c Sr = 1.2 mg mL−1), and 3 mL of concentrated HNO3 (70 %). This solution was boiled under reflux for 30 min and then filtered through a paper filter. The filtrate was loaded onto preconditioned (with 8 M HNO3) SR resin cartridges. The flask was rinsed with 3 × 2 mL HNO3 (8 M). The resin was then rinsed with 10 × 1 mL of a mixture of HNO3 (3 M) and oxalic acid (0.05 M). Then the Sr fraction was eluted with 0.01 M HNO3, which we found to be sufficiently acidic to not eluate any radiolead from the column. Water samples (50 mL) were either brought to 8 M HNO3 by addition of equal amounts of 16 m HNO3, or the solid residue after tritium distillation was taken up in 8 M HNO3. Since the natural Sr content in water is much lower than from leached sediments, the amount of Sr carrier was increased to 1.7 mg. The loading, rinsing and eluting processes were conducted as described above.
We found that the acidic eluate (0.01 M HNO3) does not mix well with the Ultima Gold™ LSC cocktail. In order to remove the acid, the eluate was evaporated to almost dryness and then taken up in 1 mL H2O again. This step was repeated 10 times. Then, the Sr fraction was transferred to LSC vials and the flask was rinsed with 4 × 0.5 mL H2O. Finally 18 mL of scintillation cocktail were added.
Addition of HNO3 to sediments should be performed carefully as the carbonate fraction of the sand will vigorously decompose under formation of CO2 gas which could cause the overflow of the flask with acidic foam. In our case, the sediments proved to have a very low carbonate content. Also organic substances may react violently upon exposure to the highly oxidizing HNO3/H2O2 mixture, especially when heated. If in doubt, the use of protective measures (such as gloves, face shields) is recommended [24–26].
For the accelerator mass spectrometry (AMS) measurements of 129I, iodine was separated from the matrix in two steps. First, Woodward iodine carrier was added and all the iodine was transferred into iodide. All iodine species were oxidized with Ca(OCl)2 to iodate and afterwards reduced with NH3OHCl and NaHSO3 to iodide. The second step consisted in an ion exchange separation using a DOWEX® 1 × 8 analytical grade ion exchange resin, which was preconditioned with KNO3. After rinsing the ion exchange columns with high purity water and a 0.5 mol L−1 KNO3 solution, the iodine was eluted with concentrated potassium nitrate solution (2.25 mol L−1). The iodine was precipitated as AgI, which was dried, mixed with silver powder (AgI:Ag 1:4 by weight), and pressed into titanium targets for AMS measurement. Measurements were performed with low-energy accelerator mass spectrometry (AMS) at the 0.5 MV TANDY facility at ETH Zurich, Switzerland.
For the ICP-MS measurement of 127I no iodine matrix separation was necessary. The samples were diluted and tetramethylammoniumhydroxide (TMAH) was added, which causes hydrolysis of any organic compounds and reduces the redox potential. The stability of I– increases in the sample relative to I2 and IO3 −. Moreover, iodide and iodate have not significantly different sensitivities in the ICP-MS measurements. A Thermo X7 (Thermo Fisher Scientific) was employed for the detection and quantification of stable 127I [27].
Results and discussion
Results of the radionuclide analysis are summarized in Table 2. Tritium, 90Sr, 134Cs, and 137Cs were obtained by radiometric means, 129I by AMS. Please note that the seawater sample from Hawaii was only analyzed for 129I, as radiometric methods were not deemed successful for the presumably very low contamination levels.
Ocean sediments
As expected, Pacific Ocean sediments from inside the exclusion zone exhibited the highest contamination levels. The correlation between radiostrontium and 134Cs as well as 137Cs is illustrated in Fig. 2.

Activity concentrations of 134Cs, 137Cs and 90Sr in Fukushima Daiichi exclusion zone Pacific Ocean sediments (dry mass)
Although a slight distance dependency can be observed (at least sample Sed 1 that was taken closest to the Fukushima Daiichi NPP exhibited the highest activity concentration), it was surprising to note that sample Sed 3 (taken 16.1 km north of the NPP) exhibited the lowest activities. It is also interesting that the radiocesium activity concentrations fluctuate by more than an order of magnitude within the samples, the 90Sr activity concentrations, however, only by a factor of 2. The 90Sr/137Cs activity ratios (where calculable) were 0.012 ± 0.002 (Sed 1), 0.036 ± 0.005 (Sed 2), 0.15 ± 0.03 (Sed 3), 0.012 ± 0.002 (Sed 4), and 0.024 ± 0.004 (Sed 5). It is unclear why Sed 3 has a ten times higher 90Sr/137Cs ratio than the other sediments (which fluctuate within a factor of 3). This odd ratio is not so much due to a high 90Sr concentration, which is in the range of the other samples, but rather due to an exceptionally low 137Cs concentration. The reason for this anomaly (which has been observed in similar manner in terrestrial environmental samples from north of the NPP [4] ) is yet unclear. Although, from an radioecological point of view, sediment is not comparable to food, the high 90Sr/137Cs ratio of 0.15 challenges the governmental assumption for the regulatory limits in food, namely a constant 90Sr/137Cs ratio in food of ≤0.1 [28]. Seafood organisms exposed to high-90Sr effluents may exhibit higher 90Sr concentrations than covered by this governmental assumption.
The 134Cs/137Cs activity ratios are reliable source identifiers [29]. The average 134Cs/137Cs ratio at the time of the accident (March 11, 2011) was found to be 0.98 [28]. Due to the shorter half-life of 134Cs, this average ratio has been calculated to have gone down to 0.77 by the time of sampling (Dec 20, 2011). The ratios found were 0.81 ± 0.05 (Sed 1), 0.91 ± 0.08 (Sed 2), 0.75 ± 0.25 (Sed 3), 0.77 ± 0.01 (Sed 4), and 0.76 ± 0.02 (Sed 5). Sed 2 seems to exhibit a somewhat higher 134Cs/137Cs ratio than the other sediments, however, with the given uncertainty margins, this outlier seems not significant. It would be interesting to study the 135Cs/137Cs ratio [11] to possibly identify the source of these contaminations.
Other studies [30] for radiocesium in sea sediments found up to 250 Bq kg−1-dry in Sendai (north of the Fukushima Daiichi NPP), which is naturally somewhat lower than the levels reported in this study, but in good agreement with our results. Our findings are also in good agreement with the activity concentrations reported by Yamamoto et al. [31]. A study on sediments from the Sea of Japan naturally found much lower radiocesium activity concentrations (0.25 Bq kg−1 137Cs and 0.15 Bq kg−1 134Cs in the Sado Basin) [32].
For comparison, it may be interesting to note that the activity levels found in these Ocean Sediments are roughly comparable to the radiocesium inventory in soil in Central Europe, e.g., in Austria [22], which is still significantly polluted from the Chernobyl accident and the fallout from 20th century’s atmospheric explosions.
Seawater
With only small volumes available for analysis, it was not expected to detect any radionuclides. However, low activities of radiocesium were found (with high uncertainties), as summarized in Table 2. Given the high uncertainties 134Cs/137Cs ratios are less meaningful; they are 0.89 ± 0.3 (OW 1), <1.08 (OW2), >4.5 (OW4), and 1.5 ± 0.7 (OW5). In any case, our findings for seawater (up to 2.8 Bq kg−1 137Cs and 2.5 Bq kg−1 134Cs) are in good agreement with previously reported data [30] for seawater.
No tritium was detected in any of the seawater samples. The 129I activity concentration in the seawater sample from Hawaii from 2012 (2.37 × 10−8 ± 1.9 × 10−9; 129I/127I ratio 1.10 × 10−10) are in the range of the pre-Fukushima background reported by Stan-Sion et al. [33].
Tap water
Tap water from Tokyo exhibited contamination levels that were of no health concern (up to 4.3 Bq kg−1 137Cs). The activity concentrations found in this study are in good agreement with previously reported tap water results [28, 29]. The radiocesium activity concentrations were clearly below the regulatory limit for radiocesium in tap water of 200 Bq kg−1. Previous studies [28] showed that 131I was probably also present in Tokyo’s tap water at the time of sampling; however, the measurements of this study were performed too late for the detection of this short-lived radionuclide. Strontium-90 and tritium could not be detected in tap water because of too low sample volumes and the limited detection limit of the LSC, respectively.
The 134Cs/137Cs activity ratios slightly deviated from the average ratio [28] of 0.98 at the time of the accident (0.97–0.96 at the time of sampling would have suggested): we found ratios of <0.2 (TW6), 0.90 ± 0.25 (TW7), 0.87 ± 0.09 (TW8), and 1.7 ± 0.6 (TW9). Like TW6, TW7, and TW8, also the ratios found in Tokyo tap water in a previous study [28] were consistently lower than the average; only TW9 exhibited a higher ratio, which raises the question of the source of these contaminations.
Reactor coolant
Without a doubt, the sample of reactor coolant was the most “unusual” sample of this study. The analysis of this sample allows the investigation of the performance of the water purification techniques currently employed at the Fukushima Daiichi NPP. Purification is performed in multiple steps, including the application of charcoal and zeolithe as adsorbent materials as well as reverse osmosis techniques. This purification method proves to be extremely efficient: No radiocesium and no radiostrontium could be detected. Also, the content of 129I is surprisingly low; it is just two orders of magnitude higher than 2012 Pacific Ocean water from Hawaii (see Table 2). By far the dominant activity in the reactor coolant is, as expected, tritium (HTO) that cannot be removed from water by chemical means.
Conclusions
The sea sediments investigated in this study were found to be contaminated with radiocesium (up to almost 800 Bq kg−1) and radiostrontium (up to 5.6 Bq kg−1). This is one of the first reports on radiostrontium in sea sediments from the Fukushima exclusion zone. Seawater exhibited radiocesium contamination levels up to 5.3 Bq kg−1, which is also in agreement with previous findings. Tap water from Tokyo weeks after the accident exhibited detectable but harmless activities of radiocesium (well below the regulatory limit). The investigation of the reactor coolant (finding only 3H and even low 129I) leads to the conclusion that the purification techniques for reactor coolant employed at Fukushima Daiichi are very effective.
References
Steinhauser G, Brandl A, Johnson TE (2014) Comparison of the Chernobyl and Fukushima nuclear accidents: a review of the environmental impacts. Sci Total Environ 470–471:800–817
Masson O, Baeza A, Bieringer J, Brudecki K, Bucci S, Cappai M, Carvalho FP, Connan O, Cosma C, Dalheimer A, Didier D, Depuydt G, De Geer LE, De Vismes A, Gini L, Groppi F, Gudnason K, Gurriaran R, Hainz D, Halldorsson O, Hammond D, Hanley O, Holey K, Homoki Z, Ioannidou A, Isajenko K, Jankovic M, Katzlberger C, Kettunen M, Kierepko R, Kontro R, Kwakman PJM, Lecomte M, Leon Vintro L, Leppanen AP, Lind B, Lujaniene G, McGinnity P, McMahon C, Mala H, Manenti S, Manolopoulou M, Mattila A, Mauring A, Mietelski JW, Moller B, Nielsen SP, Nikolic J, Overwater RMW, Palsson SE, Papastefanou C, Penev I, Pham MK, Povinec PP, Rameback H, Reis MC, Ringer W, Rodriguez A, Rulik P, Saey PRJ, Samsonov V, Schlosser C, Sgorbati G, Silobritiene BV, Soderstrom C, Sogni R, Solier L, Sonck M, Steinhauser G, Steinkopff T, Steinmann P, Stoulos S, Sykora I, Todorovic D, Tooloutalaie N, Tositti L, Tschiersch J, Ugron A, Vagena E, Vargas A, Wershofen H, Zhukova O (2011) Tracking of airborne radionuclides from the damaged Fukushima Dai-Ichi nuclear reactors by European networks. Environ Sci Technol 45:7670–7677
Thakur P, Ballard S, Nelson R (2013) An overview of Fukushima radionuclides measured in the northern hemisphere. Sci Total Environ 458–460:577–613
Steinhauser G, Schauer V, Shozugawa K (2013) Concentration of 90S at selected hot spots in Japan. PLoS ONE 8:e57760
Casacuberta N, Masque P, Garcia-Orellana J, Garcia-Tenorio R, Buesseler KO (2013) 90Sr and 89Sr in seawater off Japan as a consequence of the Fukushima Dai-ichi nuclear accident. Biogeosciences 10:3649–3659
Zheng J, Tagami K, Watanabe Y, Uchida S, Aono T, Ishii N, Yoshida S, Kubota Y, Fuma S, Ihara S (2012) Isotopic evidence of plutonium release into the environment from the Fukushima DNPP accident. Sci Rep 2:304
Zheng J, Tagami K, Uchida S (2013) Release of plutonium isotopes into the environment from the Fukushima Daiichi nuclear power plant accident: what is known and what needs to be known. Environ Sci Technol 47:9584–9595
Schneider S, Walther C, Bister S, Schauer V, Christl M, Synal H-A, Shozugawa K, Steinhauser G (2013) Plutonium release from Fukushima Daiichi fosters the need for more detailed investigations. Sci Rep 3:2988
Kakiuchi H, Akata N, Hasegawa H, Ueda S, Tokonami S, Yamada M, Hosoda M, Sorimachi A, Tazoe H, Noda K, Hisamatsu S (2012) Concentration of 3H in plants around Fukushima Daiichi nuclear power station. Sci Rep 2:947
Matsumoto T, Maruoka T, Shimoda G, Obata H, Kagi H, Suzuki K, Yamamoto K, Mitsuguchi T, Hagino K, Tomioka N, Sambandam C, Brummer D, Klaus PM, Aggarwal P (2013) Tritium in Japanese precipitation following the March 2011 Fukushima Daiichi nuclear plant accident. Sci Total Environ 445–446:365–370
Zheng J, Tagami K, Bu W, Uchida S, Watanabe Y, Kubota Y, Fuma S, Ihara S (2014) Isotopic ratio of 135Cs/137Cs as a new tracer of radiocesium released from the Fukushima nuclear accident. Environ Sci Technol 48:5433–5438
Priyadarshi A, Dominguez G, Thiemens MH (2011) Evidence of neutron leakage at the Fukushima nuclear plant from measurements of radioactive 35S in California. Proc Natl Acad Sci USA 108:14422–14425
Steinhauser G (2014) Fukushima’s forgotten radionuclides: a review of the understudied radioactive emissions. Environ Sci Technol 48:4649–4663
Morino Y, Ohara T, Nishisawa M (2011) Atmospheric behavior, deposition, and budget of radioactive materials from the Fukushima Daiichi nuclear power plant in March 2011. Geophys Res Lett 38:[L00G11/1-7]
Kawamura H, Kobayashi T, Furuno A, In T, Ishikawa Y, Nakayama T, Shima S, Awaji T (2011) Preliminary numerical experiments on oceanic dispersion of 131I and 137Cs discharged into the ocean because of the Fukushima Daiichi nuclear power plant disaster. J Nucl Sci Technol 48:1349–1356
Buesseler K, Aoyama M, Fukasawa M (2011) Impacts of the Fukushima nuclear power plants on marine radioactivity. Environ Sci Technol 45:9931–9935
Buesseler KO, Jayne SR, Fisher NS, Rypina II, Baumann H, Baumann Z, Breier CF, Douglass EM, George J, MacDonald AM, Miyamoto H, Nishikawa J, Pike SM, Yoshida S (2012) Fukushima-derived radionuclides in the ocean and biota off Japan. Proc Natl Acad Sci USA 109:5984–5988
Tumey SJ, Guilderson TP, Brown TA, Broek T, Buesseler KO (2013) Input of 129I into the western Pacific Ocean resulting from the Fukushima nuclear event. J Radioanal Nucl Chem 296:957–962
Povinec PP, Aoyama M, Biddulph D, Breier R, Buesseler K, Chang CC, Golser R, Hou XL, Ješkovský M, Jull AJT, Kaizer J, Nakano M, Nies H, Palcsu L, Papp L, Pham MK, Steier P, Zhang LY (2013) Cesium, iodine and tritium in NW Pacific waters-a comparison of the Fukushima impact with global fallout. Biogeosciences 10:5481–5496
Michel R, Handl J, Ernst T, Botsch W, Szidat S, Schmidt A, Jakob D, Beltz D, Romantschuk LD, Synal HA, Schnabel C, Lopez-Gutierrez JM (2005) 129I in soils from Northern Ukraine and the retrospective dosimetry of the 131I exposure after the Chernobyl accident. Sci Total Environ 340:35–55
Steinhauser G, Merz S, Kübber-Heiss A, Katzlberger C (2012) Using animal thyroids as ultra-sensitive biomonitors for environmental radioiodine. Environ Sci Technol 46:12890–12894
Steinhauser G, Merz S, Hainz D, Sterba JH (2013) Artificial radioactivity in environmental media (air, rainwater, soil, vegetation) in Austria after the Fukushima nuclear accident. Environ Sci Pollut Res 20:2527–2537
Atkinson R, Eddy T, Kuhne W, Jannik T, Brandl A (2014) Measurement of the tritium concentration in the fractionated distillate from environmental water samples. J Environ Radioact 135:113–119
Steinhauser G, Evers J, Jakob S, Klapötke TM, Oehlinger G (2008) A review on fulminating gold (Knallgold). Gold Bull 41:305–317
Klapötke TM, Krumm B, Steemann FX, Steinhauser G (2010) Hands on explosives: safety testing of protective measures. Saf Sci 48:28–34
Steinhauser G, Klapötke TM (2010) Using the chemistry of fireworks to engage students in learning basic chemical principles: a lesson in eco-friendly pyrotechnics. J Chem Educ 87:150–156
Michel R, Daraoui A, Gorny M, Jakob D, Sachse R, Tosch L, Nies H, Goroncy I, Herrmann J, Synal H-A, Stocker M, Alfimov V (2012) 129I and 127I in European seawaters and in precipitation from Northern Germany. Sci Total Environ 419:151–169
Merz S, Shozugawa K, Steinhauser G (2015) Analysis of Japanese radionuclide monitoring data of food before and after the Fukushima nuclear accident. Environ Sci Technol 49:2875–2885
Merz S, Steinhauser G, Hamada N (2013) Anthropogenic radionuclides in Japanese food: environmental and legal implications. Environ Sci Technol 47:1248–1256
Kofufi H (2015) In situ measurement of 134Cs and 137Cs in seabed using underwater gamma-spectrometry systems: application in surveys following the Fukushima Dai-ichi Nuclear Power Plant accident. J Radioanal Nucl Chem 303:1575–1579
Yamamoto K, Tagami K, Uchida S, Ishii N (2015) Model estimation of 137Cs concentration change with time in seawater and sediment around the Fukushima Daiichi Nuclear Power Plant site considering fast and slow reactions in the seawater-sediment systems. J Radioanal Nucl Chem 304:867–881
Inoue M, Yoneoka S, Ochiai S, Oikawa S, Fujimoto K, Yagi Y, Honda N, Nagao S, Yamamoto M, Hamajima Y, Murakami T, Kofuji H, Misonoo J (2015) Lateral and temporal variations in Fukushima Dai-ichi NPP-derived 134Cs and 137Cs in marine sediments in/around the Sado Basin, Sea of Japan. J Radioanal Nucl Chem 303:1313–1316
Stan-Sion C, Enachescu M, Petre AR (2015) AMS analyses of 129I from the Fukushima Daiichi nuclear accident in the pacific ocean water of the coast of La Jolla—San Diego, USA. Environ Sci Proc Impacts 17:932–938
Acknowledgments
This work was supported by JSPS KAKENHI (Grant Number 25870158), from CDC NIOSH Mountain and Plains Education and Research Center (Grant Number T42OH009229-07), and the US Nuclear Regulatory Commission (NRC) (Grant Number NRC-HQ-12-G-38-0044). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the CDC NIOSH and MAP ERC.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shozugawa, K., Riebe, B., Walther, C. et al. Fukushima-derived radionuclides in sediments of the Japanese Pacific Ocean coast and various Japanese water samples (seawater, tap water, and coolant water of Fukushima Daiichi reactor unit 5). J Radioanal Nucl Chem 307, 1787–1793 (2016). https://doi.org/10.1007/s10967-015-4386-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-015-4386-9