
Abstract
This paper addresses the preparation and characterization of protective anticorrosion silane- and polyrhodanine-based bilayer coatings on a tested electrode substrate. Polyrhodanine (pRh) coatings were electrodeposited using the cyclic voltammetry technique on glassy carbon and stainless steel substrates. Isobutyltriethoxysilane (IBTES) films were deposited by the simple dip-coating method on steel substrates and then were stored in air (“aged”). The formed novel bilayer pRh/IBTES coatings were applied on the electrode surface as moderately thin (ca. 6–8 nm thick) layers and exhibited very good adhesion to the steel substrate. The physicochemical properties of pRh, IBTES and bilayer pRh/IBTES coatings were determined with the use of electrochemical, X-ray photoelectron spectroscopy and scanning electron microscopy techniques. The bilayer coatings consisting of a conducting polymer—pRh and silane—IBTES most effectively hinder the access of pitting-causing aggressive anions (chlorides) to the surface of stainless steel and exhibit better barrier properties compared to single-component pRh and IBTES coatings.
Graphical Abstract

Similar content being viewed by others


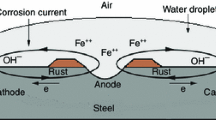
1 Introduction
The most widely used method for protecting metal surfaces against corrosion in different environments is to cover them with organic coatings (pre-treatment layers, paints, varnishes). Compatibility between the organic medium and the metal substrate is crucial for the stability and resistance of the organic coatings to atmospheric agents. For many decades, pre-treatments such as phosphate and chromate conversion coatings were usually employed before painting to improve the adhesion of organic coatings. Unfortunately, phosphate is not environmentally friendly and chromate (especially based on Cr(VI)) is highly toxic and carcinogenic [1–4]. For these reasons, in recent years many efforts have been made to develop alternative environmentally friendly treatments.
Among them, there are treatments based on organic–inorganic silicon chemicals (i.e. “trialkoxysilanes” or “silanes”) of the general formula R(CH2)nSi(OR)3, where –R is an organic functional group and –OR is an alkoxy group. These silanes were originally used as adhesion promoters between metal surfaces and organic resins [5–8]. Silane films are usually deposited by a simple conventional dip-coating method onto various metal substrates: aluminium and its alloys, iron and steels, galvanized steels, copper, zinc and magnesium alloys [9–15]. Silanol alkoxy groups are capable of hydrolysis in the presence of moisture and form silanols—Si(OH) [16], which may react with the metal hydroxide groups of the metallic substrate forming Si–O–Me—a covalent bonded layer on the metal surface [17]. The excess silanol groups (SiOH) also condense among themselves to form siloxane (SiOSi) chains. This results in the formation of a polymeric metallo-organic protective layer, resistant to the uptake of electrolytes and their chemical attack, thus constituting an effective barrier against corrosion of the underlying metal [18–22]. However, small defects such as micropores or cracks can be present in the silane film. These areas may be pathways for the diffusion of aggressive species to the metal substrate and consequently preferential sites for corrosion initiation [21, 23, 24].
In order to improve their protective properties, silane films have been modified by loading oxide nanoparticles (such as ceria, silica and zirconia) [13, 25, 26], doping with inorganic or organic corrosion inhibitors like rare earth metal ions (cerium, zirconium or lanthanum) [27–29], benzotriazole, tolyltriazole [29], or combining silane formulations with other corrosion protection systems such as conducting polymers [30, 31]. In particular, coatings made of conducting polymers are used to protect passivating metals such as Al [32], Fe [33, 34], Zn, Mg, Ti or stainless steel [35–38]. The polymer coating maintains the corrosion potential of the protected metal in the passive range, thus preventing its active dissolution. In many cases, however, conductive polymers deposited on the metal surface are characterized by high porosity and unsatisfactory adhesion to the substrate [39, 40]. One of the newest conducting polymers is polyrhodanine [41, 42]. Rhodanine and its derivatives have three kinds of heteroatoms as absorption centres (N, S, O) and can be used as anticorrosion compounds for protecting metals. It was found that rhodanine is an effective corrosion inhibitor for mild steel [43–45], copper [46] and stainless steel [47].
In view of the problems experienced individually by silane and conducting polymer coatings, the proposal of this study is to use both of them to adsorb a silane layer on a previously generated polyrhodanine coating. This paper shows the results of research on the protection of X20Cr13 stainless steel using a bilayer polyrhodanine/silane coating and compares the protective effects achieved by the individual layers.
2 Experimental
Analytically pure isobutyltriethoxysilane ≥95 % (IBTES) and rhodanine (Rh) were purchased from the Aldrich Chemistry company. Other reagents were also of an analytical reagent grade and were used in the as-received state. The solutions were prepared in doubly distilled water.
Samples of commercially available X20Cr13 stainless steel (C 0.17 %, Cr 12.6 %, Si 0.34 %, Ni 0.25 %, Mn 0.30 %, V 0.04 %, P 0.024 % and S < 0.005 %) were used in the form of discs (geometric area 0.2 cm2) embedded in epoxy resin as electrodes. Prior to each experiment, the electrode surface (stainless steel) was polished with waterproof emery paper (nos. 800, 1000 and 2000), rinsed with distilled water and degreased with ethyl alcohol. In order to characterize the basic electrochemical properties of the polyrhodanine (pRh) films, glassy carbon (Warsaw Mineral Expo, Poland) was used as an inert electrode substrate (geometric area, 0.13 cm2). The glassy carbon electrode was activated by polishing it with aqueous alumina slurries (grain size, 0.05 μm) on a polishing cloth and then the electrode was rinsed with distilled water.
Electrochemical experiments were carried out using the CH Instruments 660 electrochemical system (Austin, TX, USA) at a temperature of 20 ± 2 °C. The experiments were performed in the conventional three-electrode mode with glassy carbon or stainless steel working electrodes, a platinum wire as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. To characterize the corrosion behaviour of coated and uncoated stainless steel samples, potentiodynamic polarization curves were recorded at a 10 mVs−1 scan rate, by a potential move between −0.80 V and +1.5 V. (vs. SCE) in a chloride solution of 0.5 mol dm−3 NaCl.
Scanning electron microscopy (SEM) images were obtained using a JEOL model 6610LV microscope. Surface elemental analysis was performed with X-ray photoelectron spectroscopy (XPS). XPS measurements were carried out with Al Kα radiation at 10 kV and 10 mA. Depth profiles were obtained by Ar+ sputtering at 5 kV and an ion gun emission current in the range of 1–10 mA. The XPS spectra were analysed using the NIST XPS database [48, 49]. Adhesion of the coating to the substrate was checked by means of the adhesive tape test [50]. After depositing the coating, the surface was rinsed with distilled water and then dried, after which adhesive tape was applied to the whole specimen surface. Then, an attempt was made to tear off the tape together with the coating with a sharp movement. All of the coatings adhered well to the substrate; nevertheless, the best result of all the cases under discussion was obtained by the X20Cr13/pRh/IBTES coating. Only after the twentieth attempt, it was possible to remove the coating from the surface.
Modification of the tested electrode substrate (glassy carbon or stainless steel) was performed in the following ways:
-
1.
Preparation of silane film (X20Cr13/IBTES) The IBTES films on the steel surface were applied by dipping in a 2.0 mol dm−3 solution of IBTES in anhydrous ethyl alcohol for 20 min. After removing the samples from the silane solution, they were stored in air (“aged”) at ambient temperature (24 °C) for 72 h.
-
2.
Preparation of polyrhodanine coating (GCE/pRh or X20Cr13/pRh) The polyrhodanine coatings on glassy carbon and on stainless steel were electrodeposited from an anhydrous ethyl alcohol 0.025 mol dm−3 rhodanine monomer containing a 0.3 mol dm−3 ammonium oxalate aqueous solution, using the cyclic voltammetry (CV) technique with potential cycling between 0.00 and 1.40 V at a scan rate of 50 mVs−1. The colour of the electrode surface changed slightly from colourless to transparent yellow after 35 cycles. The polymerization mechanism of rhodanine on a Pt electrode and the structure of the polymer were proposed in [41].
-
3.
Preparation of bilayer coating (X20Cr13/pRh/IBTES) For the preparation of this bilayer coating, a pRh coating was deposited according to the method described in Sect. 2, and a silane film was adsorbed upon it using the same procedure (silane concentration, immersion time, aged time) specified in Sect. 1. The adsorption of silane upon the pRh coating is probably due to the interaction of the hydrolysed silane Si–OH group with the rhodanine S, O heteroatoms or N–H groups. Other authors [31, 51] proposed a similar model of interaction between organosilanes and polypyrrole or emeraldine.
The thickness of the bilayer coatings was determined using profilometry (Talysurf 50, Rank Taylor Hobson). The thickness of the pRh/IBTES coating on the stainless steel substrate was ca. 6–8 nm.
3 Results and discussion
3.1 Electrodeposition of polyrhodanine pRh films on glassy carbon and stainless steel substrate
This experiment was intended to determine the optimal potential at which rhodanine Rh polymerization would occur in a 0.025 mol dm−3 rhodanine monomer solution in anhydrous ethyl alcohol containing a 0.3 mol dm−3 ammonium oxalate aqueous solution. Figure 1A shows the chronoamperometric curves recorded during deposition of the pRh coating on the GCE glassy carbon surface in the potential range from 0.05 to 0.6 V. As can be seen, the shape of the chronoamperometric curves depends on the potential used for Rh polymerization. At low polymerization potentials (e.g. 0.050 V), the curves are flat and no polymerization process occurs on the electrode. The minimum potential needed for the Rh polymerization reaction is about 0.2 V.

A Chronoamperometric curves obtained during electrodeposition of pRh coating from ethyl alcohol 0.025 M rhodanine monomer containing 0.3 M ammonium oxalate aqueous solution on glassy carbon at several potentials in the range of 0.6–0.05 V. Inset B Chronoamperometric curve obtained for glassy carbon electrode during electrodeposition process of pRh coating at 0.3 V
After a few seconds at potentials from 0.2 to 0.3 V, the current density attains a minimal value and then starts to grow rapidly, which indicates the formation of a new phase and the formation of a coating on the substrate. The observed drop in current at the initial stage of the process might be caused by charging of the double layer and the redox processes of the adsorbed substance. The time from which the current density increase occurs is referred to as the nucleation duration (coating formation rate). The shorter the nucleation duration, the faster the coating forms; the nucleation duration, in turn, depends on the polymer deposition potential. [52].
Based on the chronoamperometric tests and microscopic observations, it can be stated that nucleation proceeds the fastest (with a nucleation duration of approx. 0.9 s) for the pRh coating (Fig. 1B) deposited on the GCE glassy carbon at the potential of 0.3 V. It was also observed that the coating obtained at this potential is regular and homogeneous. More positive potentials (0.4–0.6 V) do not favour the formation of a smooth, homogeneous film [53, 54]. At higher potentials, the GCE glassy carbon electrode becomes coated with an inhomogeneous thick deposit.
In the chronoamperometric curves shown in Fig. 1A, differences in nucleation duration and density of the current flowing in the initial coating formation period are observed. After the nucleation phase, a current density increase (current density maximum) is visible, which suggests the electrode being coated with the polymer (Fig. 1A—curves for 0.6–0.3 V). After the current density has reached a maximum value its drop follows, which might suggest a decrease in the polymerization rate (most probably caused by the diffusion of the monomer from the bulk of the solution). It can be expected that the pRh coating formation proceeds with instantaneous nucleation, followed by an increase limited by slow diffusion. Similar tests were carried out by Randramahazak et al. on platinum in a solution of acetonitrile and tetrabutylammonium phosphate with an EDOT monomer concentration of 0.1 mol dm−3 [55]. They found that nucleation occurred in an instantaneous manner. The processes of nucleation and growth of polymer coatings were investigated and confirmed by other authors [56].
Figure 2 shows the first cycle of pRh coating deposition on GCE. As can be seen, the increase in monomer oxidation occurs at the potential of 0.3 V. This is consistent with the course of the chronoamperometric curve at 0.3 V (Fig. 1A), for which instantaneous nucleation has been found to also occur at this potential.

Cyclic voltammograms illustrating the first cycle of electrodeposition of pRh film on GCE
Similar measurements were performed for the pRh coatings deposited on stainless steel X20Cr13. In this case, the determined nucleation potential was 0.75 V. This indicates that the nucleation potential value depends on the substrate of the electrode on which the polymer coating is being deposited.
The research presented in the previous section allowed us to ascertain the optimal conditions for the electrodeposition of pRh coatings on a stainless steel substrate. The electrodeposition was carried out in an ethyl alcohol–0.025 M rhodanine monomer solution containing a 0.3 M ammonium oxalate aqueous solution. The films were deposited from 0.0 to +1.4 V and at a potential sweep rate of 50 mVs−1. Figure 3 illustrates a comparison of the first cycles of the electrodeposition of pRh films on stainless steel (A) and glassy carbon (B) electrodes. An irreversible oxidation peak is observed at about 0.3 V for GCE and 0.75 V for stainless steel. These peaks are assigned to rhodanine monomer oxidation.

A Cyclic voltammograms illustrating the first cycle of electrodeposition of pRh film on stainless steel a in the potential range from −0.8 to 1.4 V, b in the potential range from 0.0 to 1.4 V. Inset B Cyclic voltammograms illustrating the first cycle of electrodeposition of pRh film on GCE c in the potential range from −0.8 to 1.4 V, d in the potential range from 0.0 to 1.4 V. Scan rate, 50 mVs−1
As can be seen in the first deposition cycle of pRh film on X20Cr13 steel in the potential range from −0.8 to 1.4 V (Fig. 3A-a), steel oxidation takes place at −0.5 V (reversible peak at −0.25 V). This peak corresponds to the active dissolution of steel followed by the formation of a passive layer (E > −0.37 V). At E > 0.75 V, the anodic current dramatically increases, which corresponds to the process of Rh monomer oxidation and the beginning of film deposition. A further increase in the anodic current (E > +1.1 V) is connected with the transpassivation of steel (oxidation of Cr(III) oxide to CrO4 2−) [57].
To avoid steel oxidation and ensure oxidation of the Rh monomer, the optimal potential range applied for pRh film deposition on stainless steel (Fig. 3A-b) and GCE (Fig. 3B-d) substrates has been narrowed from 0.0 to 1.4 V. The same range of potentials was applied to the deposition of pRh films on the surface of platinum [41].
3.2 Microscopic and XPS examination
Using SEM, we also examined and compared the morphologies of single-component pRh (Fig. 4A) and IBTES (Fig. 4B) coatings to the image of the bilayer pRh/IBTES coating (Fig. 4C) (all deposited on stainless steel). All the types of prepared coatings appeared to show uniform and compact morphologies; however, on the surface of the pRh coating electrodeposited on stainless steel by cyclic voltammetry (Fig. 4A), small defects such as microcracks and pits are visible. Considering the picture shown in Fig. 4B, different areas are observable. Probably, there are some accumulations of silane molecules in particular areas of the sample surface, despite the fact that the silane coating was applied over the whole surface (Fig. 4B). The bilayer pRh/IBTES coating demonstrates the most uniform morphology as well as good stability and compactness (Fig. 4C).

SEM images of (A) pRh, (B) IBTES and (C) pRh/IBTES coatings on stainless steel
The elemental composition of the coatings prepared on the stainless steel surface was investigated by XPS. Figure 5 shows the XPS spectra of the bare X20Cr13 stainless steel (A) and the steel surface modified with pRh (B), IBTES (C) and pRh/IBTES (D) coatings employing various sputter times. The sputter time of 0 mA × min denotes the spectra for the outer surface of the coating, whereas longer times correspond to the greater depths of the coating. The spectra obtained for the bare X20Cr13 steel surface (Fig. 5A) before sputtering revealed the presence of C, O, Si, Cr, N and Fe. After relatively mild sputtering (about 500 mA × min), the C and N peaks disappear, which suggests that these elements were surface contaminants. The still visible significant presence of oxygen can be attributed to Cr2O3 as a component of the steel surface oxide film. Figure 5B shows the spectra of the steel surface coated with polyrhodanine. Initially C, O, S and N signals are detected; however, there are no signals from Fe and Cr, indicating that the steel was entirely covered by the pRh coating. After relatively long sputtering (about 2400 mA × min), the analysis detected signals mainly from the steel (Fe, Cr). Nonetheless, the presence of oxygen is still considerable, due to its affinity for chromium.

XPS spectra for the uncoated X20Cr13 steel surface (A) and for the steel surface coated with pRh (B), IBTES (C), and pRh/IBTES (D) after various sputter times. Vertical bars indicate average electron binding energies (BE) for given species
The XPS spectra for the IBTES coating formed on steel surface samples after 72 h of ageing in air (Fig. 5C) indicate the presence of a relatively durable layer containing C, Si and O. Only after sputtering of about 200 mA × min, photoelectrons derived from Fe and Cr, i.e. from the substrate, begin to be visible. Nevertheless, the surface concentrations of these elements, even after such intense sputtering as 4800 mA × min, are still small. It means that under the applied Ar sputtering, complete removal of the IBTES coating is ineffective. The pRh/IBTES coating proved to be even more resistant to Ar ion bombardment (Fig. 5D). Undoubtedly, it was substantially thicker than the IBTES coating (Fig. 5C). The elemental composition of these two coatings is almost the same; however, characteristic peaks for S and N, which may be derived from polyrhodanine are not revealed. It means that the pRh/IBTES coating is dominated by silane.
3.3 Corrosion test
Measurements of the protective properties of the prepared coatings against local (pitting) corrosion were carried out in a chloride ion solution (0.5 mol dm−3 NaCl). The potentiodynamic polarization curves for the X20Cr13 steel uncovered and covered with the pRh, IBTES and pRh/IBTES coatings are shown in Fig. 6.

Potentiodynamic curve for bare stainless steel (a) and for stainless steel specimens coated with pRh (b), IBTES (c), pRh/IBTES (d)
In the case of bare steel, the presence of chloride ions in the solution caused a significant increase in the anodic current at E = −0.3 V (Fig. 6 curve a). Under such conditions, the bare steel was not passivated anymore. The Cl− anions adsorbed on the steel surface and formed soluble metal complexes causing destruction of the passive film and the formation of pits on the steel surface [58, 59]. The deposited pRh, IBTES and pRh/IBTES coatings on the steel protected the substrate against corrosion and resulted in shifting the corrosion potential towards a more positive value: by ca. 0.19 V for the pRh coating (Fig. 6 curve b), by ca. 0.32 V for the IBTES coating (Fig. 6 curve c) or by ca. 0.45 V for the pRh/IBTES bilayer coating (Fig. 6 curve d), relative to the value of the corrosion potential recorded for uncoated steel (−0.56 V) (Fig. 6 curve a). As is apparent from Fig. 6, the pRh and IBTES coatings inhibit anodic processes, whereas the pRh/IBTES bilayer coating inhibits both anodic and cathodic processes (the currents are two orders of magnitude lower compared to uncoated steel). The corrosion current densities are equal to 8 × 10−1 for the uncoated steel (a), as well as 8 × 10−2 (b), 6 × 10−2 (c) and 8 × 10−4 mA cm−2 (d) for the steel specimens covered with the pRh, IBTES and pRh/IBTES, respectively (Fig. 6). The pit nucleation potential (E pit ) equals ca. −0.3 V (Fig. 6 curve a) for bare steel, 0.06 V for steel covered with pRh (Fig. 6 curve b), 0.34 V for IBTES (Fig. 6 curve c) and 0.56 V for the steel specimen covered with pRh/IBTES (Fig. 6 curve d). The best pitting corrosion protection is demonstrated by the bilayer pRh/IBTES coating. The result is consistent with the ability of polyrhodanine coating, due to the presence of more than one active centre in the molecule, to block the transport of chloride anions to the steel substrate and to act as an additional chemical barrier to the corrosion process. These observations enable us to infer that the bilayer coating composed of the conducting polymer—pRh and silane—IBTES effectively hinders the access of aggressive anions to the substrate and exhibits better barrier properties compared to single-component pRh and IBTES coatings. An additional advantage of such a bilayer coating is its very good adhesion to the substrate.
The current–time amperometric approach was used to determine the resistance of bare and modified stainless steel samples against pitting corrosion in a 0.5 mol dm−3 NaCl solution. In each case, the time necessary to observe a rapid current increase upon application of the potential of −0.3 V was monitored (Fig. 7). The value of −0.3 V was selected on the basis of the potentiodynamic measurements in the 0.5 mol dm−3 NaCl solution. Furthermore, each observed current increase was attributed to the passive layer breakdown (initiation of pitting) under particular conditions, i.e. in the presence or absence of coatings. Figure 7 summarizes the amperometric responses recorded for (a) bare stainless steel and stainless steel covered with (b) pRh, (c) IBTES and (d) pRh/IBTES coatings. It can be observed from curve a, recorded for the bare stainless steel sample, that the current drastically increases even during a short time (100 s) of exposure to a chloride ion solution. This result is consistent with the initiation of localized corrosion leading to the growth of pits on the stainless steel surface. Curves b–d show the chronoamperometric characteristics of the X20Cr13 steel samples covered with pRh (curve b), IBTES (curve c) and pRh/IBTES (curve d) coatings, which were recorded in the same manner as before (curve a) for bare stainless steel. The current measured after 1 h for stainless steel modified with IBTES (curve b) and 25 h for steel coated with pRh (curve c) was lower than that for bare stainless steel (curve a). In other words, pRh and IBTES coatings show a capacity for inhibiting processes of local corrosion. Curve d shows the chronoamperometric characteristics of the stainless steel sample covered with the pRh/IBTES bilayer coating. In this case, the sample would have to be electrolysed for the longest time—470 h to reach the current value previously obtained after 100 s using bare stainless steel. The results indicate that the ability of the coatings to block the transport of chloride anions that are responsible for pitting corrosion of stainless steel is the best in the case of the pRh/IBTES bilayer coating.

Chronoamperometric curves recorded in chloride solution (0.5 M NaCl) at E = −0.3 V (a) for uncoated X20Cr13 stainless steel and X20Cr13 stainless steel covered with (b) pRh, (c) IBTES, (d) pRh/IBTES
To confirm the protective properties, measurements of the stationary potential of uncovered steel and steel covered with the pRh, IBTES and pRh/IBTES coatings were made as a function of the exposure time in the chloride ion solution (0.5 mol dm−3 NaCl). As can be seen from Fig. 8, the potential of uncoated steel drops down to −0.56 V after being immersed in the chloride solution. In identical conditions, steel covered with the pRh, IBTES or IBTES/Rh coating is characterized by a stationary potential within the passive range. After exposure in the solution for the samples coated with pRh (after achieving a potential ca. −0.18 V) or IBTES (after achieving potential ca. 0.15 V), a decrease in potential was observed, but not to the typical values for uncoated steel, whereas the pRh/IBTES bilayer coating is still stable under these conditions even after 150 h. There were no visible pits after removing the specimens covered with pRh, IBTES or pRh/IBTES/coatings on their surfaces from the chloride solution.

Open circuit potential vs. exposure times for bare stainless steel (a) and for stainless steel covered with pRh (b), IBTES (c), pRh/IBTES (d)
4 Summary
The formation of pRh, IBTES and pRh/IBTES coatings on carbon and stainless steel substrates was described. Polyrhodanine serves as a stable and conductive matrix with S and O heteroatoms or N–H groups providing adsorption of the hydrolysed silane Si–OH group. The pRh/IBTES coating was characterized by very good physicochemical stability and exhibited very good adhesion to the substrate that was confirmed in the tape test as well as SEM and XPS investigations.
The bilayer coating composed of the conducting polymer—pRh and silane—IBTES effectively hinders the access of aggressive anions to the substrate and exhibits better barrier properties compared to single-component pRh and IBTES coatings. The result is consistent with the ability of the polyrhodanine coating, due to the presence of more than one active centre in the molecule, to block the transport of chloride anions to the steel substrate and act as an additional chemical barrier to the corrosion process.
References
van Ooij WJ, Child TF (1998) Protecting metals with silanes coupling agents. Chem Tech 28:26–35
Child TF, van Ooij WJ (1999) Application of silane technology to prevent corrosion of metals and improve paint adhesion. Trans IMF 77:64–70
van Ooij WJ, Zhu D, Prasad G, Jayaseelan S, Fu Y, Teredesai N (2000) Silane based chromate replacements for corrosion control, paint adhesion, and rubber bonding. Surf Eng 16:386–396
van Ooij WJ, Song J, Subramanian V (1997) Silane-based pretreatments of aluminum and it alloys as chromate alternatives. ATB Metall 37:137–142
Mittal KL (2009) Silanes and other coupling agents. VSP, Leiden
Plueddemann EP (1991) Silane coupling agents, 2nd edn. Plenum Press, New York
Fink F (1991) Silicone surfactants. IV, Silicone surfactants as paint additives. Tensile Surface Deterg 28:306–312
Zhu D, van Ooij WJ (2002) Structural characterization of bis-[triethoxysilylpropyl]tetrasulfide and bis-[trimethoxysilylpropyl]amine silanes by Fourier-transform infrared spectroscopy and electrochemical impedance spectroscopy. J Adhes Sci Technol 16:1235–1260
van Schaftingen T, Le Pen C, Terryn H, Hörzenberger F (2004) Investigation of the barrier properties of silanes on cold rolled steel. Electrochim Acta 49:2997–3004
Subramanian V, van Ooij WJ (1998) Effect of the amine functional group on corrosion rate of iron coated with films of organofunctional silane. Corros 54:204–215
van Ooij WJ, Zhu D (2001) Electrochemical impedance spectroscopy of bis-[triethoxysilylpropyl]tetrasulfide on Al-2024-T3 substrates. Corros 157:413–427
Tremont R, De Jesus-Cardona H, Garcia-Orozco J, Castro RJ, Cabrera CR (2000) 3-mercaptopropyltrimethoxysilane as a Cu corrosion inhibitor in KCl solution. J Appl Electrochem 30:737–743
Montemor MF, Ferreira MGS (2007) Electrochemical study of modified bis-[triethoxysilylpropyl] tetrasulfide silane films applied on the AZ31 Mg alloy. Electrochim Acta 52:7486–7495
Suegama PH, de Melo HG, Benedetti AV, Aoki IV (2009) Influence of cerium (IV) ions on the mechanism of organosilane polymerization and on the improvement of its barrier properties. Electrochim Acta 54:2655–2662
Zucchi F, Frignani A, Grassi V, Balbo A, Trabanelli G (2008) Organo-silane coatings for AZ31 magnesium alloy corrosion protection. Mater Chem Phys 110:263–268
De Graeve I, Tourwe E, Biesemans M, Willem R, Terryn H (2008) Silane solution stability and film morphology of water-based bis-1,2-(triethoxysilyl)ethane for thin-film deposition on aluminium. Prog Org Coat 63:38–42
Franquet A, Le Pen C, Terryn H, Vereecken J (2003) Effect of bath concentration and curing time on the structure of non-functional thin organosilane layers on aluminium. Electrochim Acta 48:1245–1255
Franquet A, Terryn H, Vereecken J (2003) IRSE study on effect of thermal curing on the chemistry and thickness of organosilane films coated on aluminium. Appl Surf Sci 211:259–269
Schreiber F (2000) Structure and growth of self-assembling monolayers. Prog Surf Sci 65:151–257
Chico B, Galván JC, de la Fuente D, Morcillo M (2007) Electrochemical impedance spectroscopy study of the effect of curing time on the early barrier properties of silane systems applied on steel substrates. Prog Org Coat 60:45–53
van Ooij WJ, Zhu D, Stacy M, Seth A, Mugada T, Gandhi J, Puomi P (2005) Corrosion protection properties of organofunctional silanes—an overview. Tsinghua Sci Technol 10:639–664
Suegama PH, de Melo HG, Recco AAC, Tschiptschin AP, Aoki IV (2008) Corrosion behavior of carbon steel protected with single and bi-layer of silane films filled with silica nanoparticles. Surf Coat Technol 202:2850–2858
Quinet M, Neveu B, Moutarlier V, Audebert P, Ricq L (2007) Corrosion protection of sol–gel coatings doped with an organic corrosion inhibitor: chloranil. Prog Org Coat 58:46–53
Zhu D, van Ooij WJ (2004) Corrosion protection of metals by water-based silane mixtures of bis-[trimethoxysilylpropyl]amine and vinyltriacetoxysilane. Prog Org Coat 49:42–53
Montemor MF, Ferreira MGS (2008) Analytical characterization of silane films modified with cerium activated nanoparticles and its relation with the corrosion protection of galvanised steel substrates. Prog Org Coat 63:330–337
Montemor MF, Trabelsi W, Lamaka SV, Yasakau KA, Zheludkevich ML (2008) The synergistic combination of bis-silane and CeO2·ZrO2 nanoparticles on the electrochemical behaviour of galvanised steel in NaCl solutions. Electrochim Acta 53:5913–5922
Montemor MF, Trabelsi W, Zheludevich M, Ferreira MGS, Cecílio P (2006) Modification of bis-silane solutions with rare-earth cations for improved corrosion protection of galvanized steel substrates. Prog Org Coat 57:67–77
Trabelsi W, Cecilio P, Ferreira MGS, Montemor MF (2005) Electrochemical assessment of the self-healing properties of Ce-doped silane solutions for the pre-treatment of galvanised steel substrates. Prog Org Coat 54:276–284
Palanivel V, Huang Y, van Ooij WJ (2005) Effects of addition of corrosion inhibitors to silane films on the performance of AA2024-T3 in a 0.5 M NaCl solution. Prog Org Coat 53:153–168
Cecchetto L, Denoyelle A, Delabouglise D, Petit JP (2008) A silane pre- treatment for improving corrosion resistance performances of emeraldine base-coated aluminium samples in neutral environment. Appl Surf Sci 254:1736–1743
Correa-Borroel AL, Gutierrez S, Arce E, Cabrera-Sierra R, Herrasti P (2009) Organosilanes and polypyrrole as anticorrosive treatment of aluminium 2024. Appl Electrochem 39:2385–2395
Tallman DE, Spinks G, Dominis A, Wallace G (2002) Electroactive conducting polymers for corrosion control. J Solid State Electrochem 6:73–84
Le Nguyen Thi H, Garcia B, Deslouis C, Le Xuan Q (2001) Corrosion protection and conducting polymers: polypyrrole films on iron. Electrochim Acta 46:4259–4272
de Souza S, Pereira JE, da Silva SI, Cordoba de Torresi MLA, Temperini RM Torresi (2001) Polyaniline based acrylic blends for iron corrosion protection. Electrochem Solid-State Lett 4:B27–B30
Ren S, Barkley D (1992) Electrochemically prepared poly(3-methylthiophene) films for passivation of 430 stainless steel. J Electrochem Soc 139:1021–1026
Santos JR, Mattoso LHC, Motheo AJ (1998) Investigation of corrosion protection of steel by polyaniline films. Electrochim Acta 43:309–313
Tallman DE, Pae Y, Bierwagen GP (1999) Conducting polymers and corrosion: polyaniline on steel. Corros 55:779–786
Adamczyk L, Kulesza PJ (2011) Fabrication of composite coatings of 4-(pyrrole-1-yl) benzoate-modified poly-3,4-ethylenedioxythiophene with phosphomolybdate and their application in corrosion protection. Electrochim Acta 56:3649–3655
Posner R, Ozcan O, Grundmeier G (2013) Water and Ions at polymer/metal interfaces. Adv Struct Mater 25:21–52
Motheo AJ, Bisanha LD Aspects on fundaments and applications of conducting polymers, InTech Europe, www.intechopen.com
Kardas G, Solmaz R (2007) Synthesis and characterization of a new conducting polymer: polyrhodanine. Appl Surf Sci 253:3402–3407
Altunbas E, Solmaz R, Kardas G (2010) Corrosion behaviour of polyrhodanine coated copper electrode in 0.1 M H2SO4 solution. Mater Chem Phys 121:354–358
Solmaz R, Kardas G, Yazıcı B, Erbil M (2007) The rhodanine inhibition effect on the corrosion of a mild steel in acid along the exposure time. Prot Met 43:476–482
Solmaz R, Kardas G, Yazıcı B (2005) Inhibition effect of rhodanine for corrosion of mild steel in hydrochloric acid solution. Prot Met 41:581–585
Yüce AO, Solmaz R, Kardas G (2012) Investigation of inhibition effect of rhodanine-N-acetic acid on mild steel corrosion in HCl solution. Mater Chem Phys 131:615–620
Solmaz R, Altunbas E, Doner A, Kardas G (2011) The investigation of synergistic inhibition effect of rhodanine and iodide ion on the corrosion of copper in sulphuric acid solution. Corros Sci 53:3231–3240
Abdallah M (2002) Rhodanine azosulpha drugs as corrosion inhibitors for corrosion of 304 stainless steel in HCl solution. Corros Sci 44:717–728
Fitt: An XPS curve fitting. Fitt program with GTK library (ver. 1.2), introduced in 2000. Available from: http://www.gtk.org
Wagner CD, Naumkin AV, Kraut-Vass A, Allison JW, Powell CJ, Rumble Jr JR NIST X-Ray photoelectrob spectroskopy database. NIST Standard Reference Database 20, version 3.5. Available from: http://srdata.nist.gov/xps
Volinsky AA, Moody NR, Gerberich WW (2002) Interfacial toughness measurements for thin films on substrates. Acta Mater 50:441–466
Arenas MA, Gonzalez Bajos L, de Damborenea JJ, Ocon P (2008) Synthesis and electrochemical evaluation of polypyrrole coatings electrodeposited onto AA-2024 alloy. Prog Org Coat 62:79–86
Sonneveld PJ, Visscher W, Barendrecht E (1992) Nucleation and growth of zinc on a glassy carbon electrode from a zincate solution. Electrochim Acta 37:1199–1205
Arias C, Granstrom M, Petritsch K, Friend RH (1999) Organic photodiodes using polymeric anodes. Synth Met 102:953–954
Dietrich M, Heine J, Heywang G, Jonas F (1994) Electrochemical and spectroscopic characterisation of polyalkylenedioxythiophenes. J Electroanal Chem 369:87–92
Randriamahazaka H, Noel V, Chevrot C (1999) Nucleation and growth of poly(3,4-ethylenedioxythiophene) in acetonitrile on platinum under potentiostatic conditions. J Electroanal Chem 472:103–111
Romero M, del Valle MA, del Rio R, D´ıaz FR, Armijo F, Dalchiele EA (2013) Temperature effect on nucleation and growth mechanism of poly(o-anisidine) and poly(aniline) electro-synthesis. J Electrochem Soc 160(9):G125–G134
Jagielska-Wiaderek K, Bala H, Wieczorek P, Rudnicki J, Klimecka-Tatar D (2009) Depth characteristics of glow-discharge nitrided layer produced on AISI 4140 steel. Arch Metall Mater 54:115–120
Codaro EN, Nakazato RZ, Honwistiz AL, Ribero LMF, Ribero RB, Hein LRO (2002) An image processing method for morphology characterization and pitting corrosion evaluation. Mater Sci Eng A334:298–306
Williams DE, Zhu YY (2000) Explanation for initiation of pitting corrosion of stainless steel at sulfide inclusions. J Electrochem Soc 147:1763–1766
Acknowledgments
The research was supported by MNiSzW (Statutory Research BS/PB-207-301/2007) and the National Science Centre (Poland) under Project No. 2011/03/B/ST4/02413.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Owczarek, E., Adamczyk, L. Electrochemical and anticorrosion properties of bilayer polyrhodanine/isobutyltriethoxysilane coatings. J Appl Electrochem 46, 635–643 (2016). https://doi.org/10.1007/s10800-016-0946-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-016-0946-0