
Abstract
We describe a case of a boy with neurodevelopmental delay and a diffuse large B-cell lymphoma (DLBCL) in whom we discovered a germline de novo 2p16.3 deletion including MSH6 and part of the FBXO11 gene. A causative role for MSH6 in cancer development was excluded based on tumor characteristics. The constitutional FBXO11 deletion explains the neurodevelopmental delay in the patient. The FBXO11 protein is involved in BCL-6 ubiquitination and BCL-6 is required for the germinal center reaction resulting in B cell differentiation. Somatic loss of function alterations of FBXO11 result in BCL-6 overexpression which is a known driver in DLBCL. We therefore consider that a causative relationship between the germline FBXO11 deletion and the development of DLBCL in this boy is conceivable.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cancer in childhood is mostly sporadic, but in an estimated 6–10%, a genetic predisposition plays an important role [1,2,3]. Many of the children with underlying germline mutations have additional syndromic features, but the exact proportion of children with a cancer predisposition syndrome (CPS) is unknown. The identification of novel predisposing genes is an ongoing process [4, 5].
It is often challenging to discover or confirm the causal link between a (new) germline pathogenic variant in a gene and tumor development in an individual. Especially rare genetic syndromes with low penetrance for cancer are difficult to identify as a CPS. Examples are Weaver syndrome, caused by pathogenic variants in EZH2, and the Diets-Jongmans syndrome, caused by pathogenic variants in KDM3B [6,7,8,9]. Large case series are necessary to provide sufficient evidence that these patients indeed have an increased risk for tumor development. However, a first suspicion of a causative correlation often starts with cancer in a single case combined with knowledge of somatic cancer driving alterations in the gene of interest.
In this case report we describe a boy with developmental delay and a diffuse large B-cell lymphoma in whom we identified a de novo germline 2p16.3 deletion (134.1 kb), including MSH6 and part of FBXO11. The developmental delay can be explained by the FBXO11 deletion [10, 11]. We interrogate the role of this germline deletion in cancer development in this boy.
Case report
The patient is a 5-year-old boy who was born after an uncomplicated pregnancy and delivery. His parents are a healthy, non-consanguineous couple. The family history was unremarkable. The patient had a congenital tooth which was extracted at the age of 4 years. His cognitive and speech development are severely delayed for which he attends special education. The boy’s behavior is characterized by hyperactivity and a short attention span. His vision and hearing are normal. He had no history of frequent or severe infections. Physical examination showed normal height and head circumference for age. He has several facial dysmorphisms including a wide forehead, wide palpebral fissures, long eyelashes and a thin upper lip. He has no hyper- nor hypopigmentation of the skin and normal extremities.
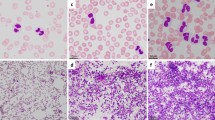
At the age of 5 years, the patient was referred to the hospital with pain in the lower right quadrant of the abdomen for 2.5 weeks that had worsened over time. Additional symptoms were frequent nosebleeds, easy bruising and weight loss. Physical examination revealed an enlarged spleen and liver. Laboratory tests showed hemoglobin 5.5 mmol/L, thrombocytes 36 × 109/L, leukocytes 67.8 × 109/L, ASAT 159 U/L, ALAT 20 U/L, LD 10,701 U/L, urine acid 0.9 mmol/L, CRP 68 mg/L. An ultrasound revealed hepatosplenomegaly and an ileocaecal intussusception. Conventional imaging showed no signs of a lymphoma. A hydrostatic reposition was performed but was insufficient. The next day, an ileal resection with primary anastomosis was performed. Evaluation of the resection material by the pathologist revealed a BCL-2 positive and BCL-6 negative high-grade B-cell lymphoma of the small intestine (Fig. 1). Based on morphology and BCL-6 negativity the preferential diagnosis was a DLBCL above a Burkitt lymphoma. Additional examination showed 72% blasts in the bone marrow and 3% blasts in the cerebrospinal fluid. Using FISH analysis, a complex t(8;14) IGH-MYC translocation was detected, but no translocations of the BCL2 and BCL6 genes were found. Immunohistochemistry for the mismatch repair proteins (MLH1, MSH2, MSH6, PMS2) was positive, and the tumor showed no microsatellite instability (BAT25, BAT26, BAT40, D17S250, D5S346, D2S123 markers).

Microscopic image of the DLBCL showing the tumor cells between the crypts in the lamina propria. a Hematoxylin and eosin (H&E) staining was used on the tissue. b The tumor cells were stained with BCL-6, showing loss of BCL-6 expression in the tumor of the patient
Considering the typical morphological and immunohistochemical aspect of the tumor, the diagnosis of a cMYC-IGH translocated diffuse large B-cell lymphoma (DLBCL) with bone marrow and central nervous system involvement was made. The patient was subsequently treated according to the EICNHL inter-B-NHL Ritux 2010 protocol (treatment arm C3). The treatment was complicated by multiple episodes of paralytic ileus and steroid induced diabetes mellitus. The patient finished his treatment according to the protocol and showed no sign of relapsed disease or long-term sequelae of treatment 1.5 years post treatment.
As part of the diagnostic process for DLBCL, a CytoSNP-850 K BeadChip SNP array (Illumina, San Diego, CA, USA) on lymphoma derived DNA was performed. This revealed a 134-kb deletion in 2p16.3 including the MSH6 gene and exons 2–23 of the FBXO11 gene (Fig. 2), and a copy-number-neutral loss-of-heterozygosity of part of the short arm of chromosome 17 (pter-p12) (not shown), suggesting a 17p13.1 TP53 mutation (see below). Because of the combination of developmental delay and DLBCL the patient was referred to a clinical geneticist for further evaluation. The 2p16.3 deletion was also detected in a remission sample from blood by SNP-array and therefore marked as a germline deletion. The deletion of MSH6 was confirmed by Multiplex Ligation-dependent Probe Amplification (MLPA). A second germline MSH6 variant was not detected using Sanger Sequencing, ruling out Constitutional Mismatch Repair Deficiency (CMMRD) underlying the DLBCL. Both parents tested negative for the deletion by SNP array analyses on peripheral blood DNA. Whole exome sequencing was performed on DNA from the tumor and a germline sample derived from blood. Analysis of somatic variants revealed a TP53 hotspot variant: c.742G > A (p.R248W) and several additional aberrations (Supplementary Table 1). No somatic alterations in FBXO11, MSH6 or BCL6 were present, nor did we find hypermutation in the tumor or a mutational signature related to MMR deficiency.

Illumina CytoSNP-850K SNP-array analyses revealed a de novo 134-kb 2p16.3 deletion including the MSH6 gene and the 3′ prime part of the FBXO11 gene in both DLBCL and germline DNA of the child. DLBCL DNA was derived from bone marrow at diagnosis. Germline DNA was derived from blood in remission
Discussion
In this case report we describe a child with developmental delay who developed a diffuse large B-cell lymphoma. Genetic testing revealed a de novo germline 2p16.3 deletion including the MSH6 gene and a large part of the FBXO11 gene.
MSH6 or mutS homolog 6 encodes for the DNA mismatch repair (MMR) protein MSH6, a member of the Mutator S family. Germline heterozygous loss-of-function variants in the MMR genes, MHL1, MSH2, MHS6, and PMS2 are associated with autosomal dominant Lynch syndrome (OMIM# 120435), which is associated with an increased risk of predominantly colorectal cancer and endometrial cancer in adults at a young age [12]. DLBCL is not a recurrent feature of Lynch syndrome, but DLBCL with microsatellite instability was reported in a patient with a germline MLH1 mutation [13]. Biallelic pathogenic germline variants in the MMR genes result in Constitutional Mismatch Repair Deficiency (CMMRD, OMIM# 276300) [14]. CMMRD presents with a much wider range of cancer types, particularly brain tumors, hematological malignancies and colorectal cancer, which mostly develop in childhood. Whereas lymphomas comprise ~ 15% of cancers in CMMRD, these are mainly T-cell lymphomas and reports of DLBCL are less frequent [15]. The patient we report here had no additional clinical features of CMMRD, nor did we find a second pathogenic germline or somatic variant in MSH6. The tumor did not show signs of hypermutation or a mismatch repair associated mutation spectrum. Furthermore, all MMR markers were expressed normally, and microsatellite instability was absent. Hence, we could not confirm a causal relation between loss of the MSH6 allele and development of the DLBCL. Because of the germline deletion of MSH6 the patient was diagnosed with Lynch syndrome, and colorectal cancer surveillance was recommended from the age of 25 years onwards.
Since the first description of a patient with a FBXO11 alteration and developmental delay in 2016, a total of 71 patients have been published [10, 11, 16,17,18,19,20,21,22,23,24,25]. Forty single nucleotide variants and indels were detected in 46 patients, including 18 missense, 5 nonsense, 10 frameshifts, 5 splice site variants, and 2 in-frame deletions [10, 11, 16,17,18,19]. All variants were de novo, except for one frameshift variant, which has recently been described in two sisters with developmental delay who inherited the variant from their mother [19]. In addition, 25 individuals have been described with a germline partial gene deletion of FBXO11 or a larger deletion including the FBXO11 gene [10, 11, 20,21,22,23,24,25,26]. Of these individuals, seven patients carry a deletion limited to the FBXO11 and MSH6 genes and four patients carry a partial deletion of FBXO11 only. Larger deletions often include the adjacent MSH6 and MSH2 genes. Common phenotypic presentations for all patients are developmental delay and behavior abnormality including autism, hyperactivity, and anxiety. Two of these patients, both affected by a large deletion in chromosome 2p16.3-p21 affecting multiple genes including FBXO11, MSH6 and MSH2, developed a malignancy. A male patient developed a prostate adenocarcinoma and a synchronous sigmoid adenocarcinoma at the age of 52. Both tumors showed loss of MSH2 and MSH6 expression [25]. A female patient developed a mucinous adenocarcinoma of the colon with loss of MSH2 [22]. The tumors in both patients were attributed to Lynch syndrome. No other malignancies have been reported in patients with a FBXO11 alteration.
So far, germline aberrations in FBXO11 have not been linked to cancer development. However, a relation between somatic FBXO11 alterations and cancer has been described. FBXO11 is part of the F-Box protein family, of which the members are the substrate recognition subunits of the SKPI-Cullin1-F-box (SCF) complex [27]. This complex is responsible for the catalyzation of ubiquitination and subsequent proteasomal degradation of substrates [28]. The SCF mediated process of protein degradation is important for genome stability and maintenance [29]. According to the cBioPortal and COSMIC databases, FBXO11 alterations have been detected in various adult neoplasms, including colon cancer, lung cancer, ovarian cancer, and head and neck cancer, as well as in diffuse large B-cell lymphoma (DLBCL) [30,31,32]. Recently, FBXO11 was also identified as a potential tumor suppressor in myelodysplastic syndrome and secondary acute myeloid leukemia [33, 34]. In approximately 4–8% of adult patients with DLBCL a somatic genetic alteration of the FBXO11 gene is present [35, 36]. According to the St. Jude Cloud PeCan database, somatic FBXO11 variants have been detected in eight pediatric oncology patients of whom seven were diagnosed with a Burkitt lymphoma (BL), but none with a DLBCL [37]. These variants include four missense variants, two frameshift variants located in the Beta Helix domain, and a stop-loss variant causing a 16-nucleotide extension of the open reading frame. Although DLBCL and BL are classified as two different types of lymphoma, similarities on both morphological and molecular level can make it difficult to distinguish them [38, 39].
Patients with DLBCL frequently carry somatic mutations in BCL6, a known proto-oncogene. BCL-6 is present in the germinal center and withholds premature B-cell activation and differentiation into mature plasma cells and memory B-cells [40]. BCL-6 expression is retained in most patients with DLBCL [41]. One of the mechanisms, resulting in retained BCL-6 expression is insufficient proteasomal degradation, which occurs as a result of diminished ubiquitination of BCL-6 by the mutated FBXO11 protein [35, 36]. Duan et al. found monoallelic deletions and mutations of FBXO11 in DLBCL, and suggested that FBXO11 is a haplo-insufficient tumor suppressor gene [35]. We hypothesize that the heterozygous germline FBXO11 deletion in the patient would result in retained BCL-6 expression in the tumor. However, immunohistochemistry of the tumor showed negative staining for BCL-6. No somatic aberration of BCL6 was discovered that could explain this phenomenon.
Based on the role of FBXO11 in DLBCL, we consider that the germline heterozygous FBXO11 deletion may have contributed to the DLBCL development in this child, although we cannot exclude that the development of DLBCL may be a coincidence. Thus far, no cancer attributed to the FBXO11 aberration has been reported in a total of 71 patients with germline FBXO11 alterations. This lack of cancer might be explained by reduced penetrance, as is the case for many other childhood cancer predisposing genes [42], and by the relatively low age of these patients at time of publication (median age of 9.25 years in a cohort of 61 individuals). Therefore, assessment of larger cohorts of individuals with constitutional FBXO11 aberrations is required to establish whether the incidence of lymphoma is indeed enriched in these individuals.
Conclusion
In conclusion we present a case of a boy with developmental delay resulting from a de novo germline 2p16.3 deletion including FBXO11 and MSH6, who developed a diffuse large B-cell lymphoma. We found no evidence of MSH6 inactivation nor MRR deficiency in the tumor, hence a causative role for the germline deletion of MSH6 in the development of the lymphoma was excluded. We consider a causative relationship with the germline deletion of FBXO11, a haplo-insufficient tumor suppressor in this cancer type, conceivable.
Data availability
Data is available upon request.
References
Zhang J, Walsh MF, Wu G et al (2015) Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 373(24):2336–2346. https://doi.org/10.1056/NEJMoa1508054
Parsons DW, Roy A, Yang Y et al (2016) Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol 2(5):616–624. https://doi.org/10.1001/jamaoncol.2015.5699
Grobner SN, Worst BC, Weischenfeldt J et al (2018) The landscape of genomic alterations across childhood cancers. Nature 555(7696):321–327. https://doi.org/10.1038/nature25480
Begemann M, Waszak SM, Robinson GW et al (2020) Germline GPR161 mutations predispose to pediatric medulloblastoma. J Clin Oncol 38(1):43–50. https://doi.org/10.1200/jco.19.00577
Waszak SM, Robinson GW, Gudenas BL et al (2020) Germline Elongator mutations in sonic hedgehog medulloblastoma. Nature 580(7803):396–401. https://doi.org/10.1038/s41586-020-2164-5
Tatton-Brown K, Hanks S, Ruark E et al (2011) Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget 2(12):1127–1133. https://doi.org/10.18632/oncotarget.385
Usemann J, Ernst T, Schafer V et al (2016) EZH2 mutation in an adolescent with Weaver syndrome developing acute myeloid leukemia and secondary hemophagocytic lymphohistiocytosis. Am J Med Genet A 170A(5):1274–1277. https://doi.org/10.1002/ajmg.a.37562
Diets IJ, van der Donk R, Baltrunaite K et al (2019) De novo and inherited pathogenic variants in KDM3B cause intellectual disability, short stature, and facial dysmorphism. Am J Hum Genet 104(4):758–766. https://doi.org/10.1016/j.ajhg.2019.02.023
Mahamdallie S, Yost S, Poyastro-Pearson E et al (2019) Identification of new Wilms tumour predisposition genes: an exome sequencing study. Lancet Child Adolesc Health 3(5):322–331. https://doi.org/10.1016/s2352-4642(19)30018-5
Gregor A, Sadleir LG, Asadollahi R et al (2018) De Novo Variants in the F-Box Protein FBXO11 in 20 Individuals with a Variable Neurodevelopmental Disorder. Am J Hum Genet 103(2):305–316. https://doi.org/10.1016/j.ajhg.2018.07.003
Jansen S, van der Werf IM, Innes AM et al (2019) De novo variants in FBXO11 cause a syndromic form of intellectual disability with behavioral problems and dysmorphisms. Eur J Hum Genet 27(5):738–746. https://doi.org/10.1038/s41431-018-0292-2
Kohlmann W, Gruber SB (1993) Lynch Syndrome. In: Adam MP, Ardinger HH, Pagon RA et al (eds) GeneReviews(®). University of Washington, Seattle. Copyright © 1993–2020, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved., Seattle (WA)
Cheah CY, Dsouza L, Taggart MW et al (2015) Diffuse large B-cell lymphoma with microsatellite instability developing in the setting of Muir-Torre variant hereditary non-polyposis colon cancer. J Clin Pathol 68(9):755–757. https://doi.org/10.1136/jclinpath-2015-203039
Wimmer K, Kratz CP, Vasen HF et al (2014) Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet 51(6):355–365. https://doi.org/10.1136/jmedgenet-2014-102284
Ripperger T, Schlegelberger B (2016) Acute lymphoblastic leukemia and lymphoma in the context of constitutional mismatch repair deficiency syndrome. Eur J Med Genet 59(3):133–142. https://doi.org/10.1016/j.ejmg.2015.12.014
Iossifov I, O’Roak BJ, Sanders SJ et al (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515(7526):216–221. https://doi.org/10.1038/nature13908
Martinez F, Caro-Llopis A, Rosello M et al (2017) High diagnostic yield of syndromic intellectual disability by targeted next-generation sequencing. J Med Genet 54(2):87–92. https://doi.org/10.1136/jmedgenet-2016-103964
Fritzen D, Kuechler A, Grimmel M et al (2018) De novo FBXO11 mutations are associated with intellectual disability and behavioural anomalies. Hum Genet 137(5):401–411. https://doi.org/10.1007/s00439-018-1892-1
Lee CG, Seol CA, Ki CS (2020) The first familial case of inherited intellectual developmental disorder with dysmorphic facies and behavioral abnormalities (IDDFBA) with a novel FBXO11 variant. Am J Med Genet A 182(11):2788–2792. https://doi.org/10.1002/ajmg.a.61828
Petit P, Fryns JP (1997) Interstitial deletion 2p accompanied by marker chromosome formation of the deleted segment resulting in a stable acentric marker chromosome. Genet Couns 8(4):341–343
Sanders SR, Dawson AJ, Vust A et al (2003) Interstitial deletion of chromosome 2p16.2p21. Clin Dysmorphol 12(3):183–185. https://doi.org/10.1097/01.mcd.0000065051.36236.e9
Lucci-Cordisco E, Zollino M, Baglioni S et al (2005) A novel microdeletion syndrome with loss of the MSH2 locus and hereditary non-polyposis colorectal cancer. Clin Genet 67(2):178–182. https://doi.org/10.1111/j.1399-0004.2004.00390.x
Firth HV, Richards SM, Bevan AP et al (2009) DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet 84(4):524–533. https://doi.org/10.1016/j.ajhg.2009.03.010
Collins DL, Schimke RN (2011) Regarding cancer predisposition detected by CHG arrays. Genet Med 13 (11):982; author reply 982–983. https://doi.org/10.1097/gim.0b013e31823552a8
Salo-Mullen EE, Lynn PB, Wang L et al (2018) Contiguous gene deletion of chromosome 2p16.3-p21 as a cause of Lynch syndrome. Fam Cancer 17(1):71–77. https://doi.org/10.1007/s10689-017-0006-x
Morton SA, Coppinger J, Ballif BC et al (2011) Response to the letter by Collins and Schimke. Genet Sci 13(11):982–983. https://doi.org/10.1097/GIM.0b013e31823552be
Yumimoto K, Yamauchi Y, Nakayama KI (2020) F-box proteins and cancer. Cancers (Basel) 12(5):1249. https://doi.org/10.3390/cancers12051249
Cardozo T, Pagano M (2004) The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol 5(9):739–751. https://doi.org/10.1038/nrm1471
Silverman JS, Skaar JR, Pagano M (2012) SCF ubiquitin ligases in the maintenance of genome stability. Trends Biochem Sci 37(2):66–73. https://doi.org/10.1016/j.tibs.2011.10.004
Tate JG, Bamford S, Jubb HC et al (2019) COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res 47(D1):D941–D947. https://doi.org/10.1093/nar/gky1015
Cerami E, Gao J, Dogrusoz U et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2(5):401–404. https://doi.org/10.1158/2159-8290.Cd-12-0095
Gao J, Aksoy BA, Dogrusoz U et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6(269):pl1. https://doi.org/10.1126/scisignal.2004088
Mo A, Chang L, Duns G et al (2019) Loss of FBXO11 functions drives acute myeloid leukemia. Exp Hematol 76:S78. https://doi.org/10.1016/j.exphem.2019.06.407
Schieber M, Marinaccio C, Bolanos LC et al (2020) FBXO11 is a candidate tumor suppressor in the leukemic transformation of myelodysplastic syndrome. Blood Cancer J 10(10):98. https://doi.org/10.1038/s41408-020-00362-7
Duan S, Cermak L, Pagan JK et al (2012) FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature 481(7379):90–93. https://doi.org/10.1038/nature10688
Schneider C, Kon N, Amadori L et al (2016) FBXO11 inactivation leads to abnormal germinal-center formation and lymphoproliferative disease. Blood 128(5):660–666. https://doi.org/10.1182/blood-2015-11-684357
Zhou X, Edmonson MN, Wilkinson MR et al (2016) Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet 48(1):4–6. https://doi.org/10.1038/ng.3466
Klapper W, Szczepanowski M, Burkhardt B et al (2008) Molecular profiling of pediatric mature B-cell lymphoma treated in population-based prospective clinical trials. Blood 112(4):1374–1381. https://doi.org/10.1182/blood-2008-01-136465
Miles RR, Shah RK, Frazer JK (2016) Molecular genetics of childhood, adolescent and young adult non-Hodgkin lymphoma. Br J Haematol 173(4):582–596. https://doi.org/10.1111/bjh.14011
Basso K, Dalla-Favera R (2012) Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev 247(1):172–183. https://doi.org/10.1111/j.1600-065X.2012.01112.x
Meriranta L, Pasanen A, Alkodsi A et al (2020) Molecular background delineates outcome of double protein expressor diffuse large B-cell lymphoma. Blood Adv 4(15):3742–3753. https://doi.org/10.1182/bloodadvances.2020001727
Taeubner J, Wieczorek D, Yasin L et al (2018) Penetrance and expressivity in inherited cancer predisposing syndromes. Trends Cancer 4(11):718–728. https://doi.org/10.1016/j.trecan.2018.09.002
den Dunnen JT, Dalgleish R, Maglott DR et al (2016) HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 37(6):564–569. https://doi.org/10.1002/humu.22981
Culp M, Johnson K, Michailidis G (2006) ada: An r package for stochastic boosting. J Stat Softw 17(2):9. https://doi.org/10.18637/jss.v017.i02
Lek M, Karczewski KJ, Minikel EV et al (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616):285–291. https://doi.org/10.1038/nature19057
Funding
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
The patient’s legal guardians gave consent for publication.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
van Engelen, N., van Dijk, F., Waanders, E. et al. Constitutional 2p16.3 deletion including MSH6 and FBXO11 in a boy with developmental delay and diffuse large B-cell lymphoma. Familial Cancer 20, 349–354 (2021). https://doi.org/10.1007/s10689-021-00244-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-021-00244-2