
Abstract
Knowledge of genetic diversity and population structuring represents a key component for the conservation of endangered species, especially where translocations and re-introduction operations are integral tools for population management. The blue duck (Hymenolaimus malacorhynchos) is a threatened riverine specialist that is endemic to New Zealand. Populations from the North and South Island form two distinct mitochondrial lineages, which currently necessitate separate conservation management. Here we examine the patterns of variability at 11 microsatellite loci and mitochondrial control region data to assess the range-wide genetic diversity and population structure of blue duck. Our data suggest that North and South Island blue duck populations likely diverged in the late Pleistocene with very limited gene flow, strongly reinforcing the current management strategy to avoid translocation between islands. Genetic diversity within both islands follows a pattern of isolation by distance with relatively high levels of gene flow among populations, likely driven by male–juvenile dispersal. The overall genetic diversity in blue duck is low and effective population size is small. These data will provide important information for conservation management of this species.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the general context of habitat loss and population fragmentation, understanding patterns of genetic diversity in declining species represents one of the key pieces of information needed for effective management programs (Avise 1989; Frankham et al. 2002). For instance, establishing the level of genetic diversity within endangered populations can give valuable clues as to the relative risks of inbreeding depression and loss of adaptive potential (Keller and Waller 2002). Establishing how genetic variation is geographically distributed further enables the identification of management units (populations with a low degree of connectivity that necessitate separate management) aiding decisions as to where management efforts might be best concentrated (Palsbøll et al. 2007). Such information is also critical when considering the benefits and risks of translocations or re-introduction operations. In particular, the prevalent principle in conservation translocations is the use of locally-adapted source populations and maintenance of existing population structure (Avise 1989). In contrast, mixing of individuals from different source populations can potentially enhance establishment and persistence of the translocated population (Sgrò et al. 2011), however, the potential risks of outbreeding depression (reduction in fitness due to interbreeding of distinct genetic lineages) have to be carefully weighed against the benefits of increased genetic diversity and adaptive potential (Weeks et al. 2011).
The New Zealand endemic blue duck or whio (Hymenolaimus malacorhynchos) is considered as ‘threatened’ under the New Zealand Threat Classification System (Miskelly et al. 2008) and is ranked as endangered in the IUCN red list, with its population trend decreasing (BirdLife International 2013). The blue duck remains one of the least well-known duck species (Williams 1991), with its phylogenetic affinities still debated (Robertson and Goldstien 2012). Most recent estimates indicate a total population size of 2500 to 3000 individuals (Glaser et al. 2010). Peculiar ecological traits make this species particularly vulnerable to some of the main drivers of species extinctions, such as habitat loss and introduced predators (Didham et al. 2007). Specifically, this species is one of only four specialized duck species worldwide that live permanently in rivers (Kear 2005). Blue ducks are long-lived, forming stable pairs that defend territories corresponding to river stretches of about 1.5 km, resulting in low-density populations (Williams 1991). Once widespread throughout most of the North and South Island of New Zealand, blue ducks are now limited to upstream parts of unmodified rivers, characterised by high water quality, high stability of the stream banks as well as abundant invertebrate prey (Collier et al. 1993). Stoats (Mustela erminea) have been identified as one of the main agents of decline at some sites (Adams et al. 1997; Whitehead et al. 2007), along with deforestation and the development of hydro dams for electricity generation (King et al. 2000; Williams 1991). Translocations, particularly the extraction of eggs from the wild and subsequent release of fully-fledged birds (Whio Operation Nest Egg) as well as the release of captive-bred birds, are being used as important management tools in this species (Glaser et al. 2010). Following a phylogeographic study of mitochondrial DNA (Robertson et al. 2007) that revealed a deep genetic split between North and South Island and significant genetic structuring within islands, blue duck translocations have been restricted to within island movements of birds. Currently information on range-wide nuclear genetic diversity, and thus male dispersal rates, is lacking for this species, but understanding fine scale population structuring, as well as natural dispersal, is crucial in maximising success of any future translocation actions.
Here we utilise mitochondrial and microsatellite marker data from extensive sampling of blue duck throughout the species’ current distribution to: (1) evaluate the level of blue duck genetic diversity and population structuring, and (2) infer population demographic history and current population size. These new data will have important implication for the conservation management, particularly translocation strategies, of the species.
Materials and methods
Sampling and DNA extraction
Seventy-five samples from six North Island (NI) and five South Island (SI) sites (Fig. 1) were obtained from an earlier study by Robertson et al. (2007) and prepared as described by the authors. Feathers were collected between 2002 and 2009 from an additional 194 birds and two NI and five SI sites (Fig. 1) and stored in paper envelopes or plastic bags with silica gel. Genomic DNA was extracted from feathers using a standard Chelex protocol (Walsh et al. 1991) followed by a LiCl and ethanol precipitation with the carrier linear polyacrylamide (for a detailed extraction protocol see Supplementary material 1). With the exception of a single sample obtained from the Waihi catchment (a potential vagrant animal), all individuals were sampled from known breeding population (Glaser et al. 2010).

Blue duck sampling locations (rivers). Numbers in parentheses represent numbers of samples analysed for mitochondrial DNA control region (listed first) and microsatellites (listed second). An asterisk marks samples obtained from an earlier study by Robertson et al. (2007)
Microsatellite genotyping
Of the 13 microsatellite markers previously developed for blue duck (Abdelkrim et al. 2009), Hmal12 was monomorphic in the present study and Hmal20 proved hard to score; both markers were subsequently removed from analyses. Touchdown PCR amplifications were performed independently for each marker. All reactions were performed in a final volume of 10 µL containing 0.1 µM of each reverse and fluorescent dye-labeled M13 (-21) primer (Table S1, Supplementary material 1), 0.05 µM of forward primer with the M13 (-21) universal sequence added (Schuelke 2000), 1× buffer, 1.5 mM of MgCl2, 0.2 mM of each dNTP, 4 mM of TMAC, 0.08 unit/µL of IMMOLASETM DNA polymerase (Bioline, London, UK), and 10 ng of DNA. Cycling conditions were as follows: 94 °C for 10 min; 38 cycles of 94 °C for 15 s, annealing temperature T (Table S1, Supplementary material 1) for 30 s (with T decreasing from 59 to 53 °C every two cycles then 51 °C for 4 cycles and 49 °C for 26 cycles) and 72 °C for 20 s; and a final extension of 72 °C for 20 min. PCR products were pooled into two sets and run separately (Table S1, Supplementary material 1) on an ABI 3730xl DNA Analyzer (Applied Biosystems). Genotypes were binned and scored manually using GeneMapper (Applied Biosystems).
Control region sequencing
Approximately 1000 bp of mitochondrial control region (mtDNA CR) were amplified in a 25 µL reaction volume containing 20 ng genomic DNA, 0.04 µM of each primer (BDCR1F, 5′-GGCTGGTGAATCGGGTAATA-3′; BDCR1R, 5′-CGGGGGTTATTTGGCTATGT-3′), 0.2 mM of each dNTP, 1.5 mM MgCl2, 1× buffer and 0.08 unit/µL of Taq polymerase (Bioline, London, UK). The thermal cycling profile was an initial 5 min at 94 °C, followed by 40 cycles at 94 °C for 15 sec, 45 °C for 25 sec and 72 °C for 60 sec. PCR reactions were purified with Whatman filter plates (GE Healthcare, UK) and sequenced using a Big Dye v. 3.1 sequencing kit (Applied Biosystems) as per manufacturer’s instructions with primer BDCR1R and internal primers, BDCR2F (5′-CATATACGCCAACCGTCTCA-3′) and BDCR2R (5′-CCTGCTACCTCACCAACCAT-3′), which yielded an 894 bp fragment. Sequencing products were purified using Sephadex-GS50 gel filtration (Amersham Bioscience, New Zealand) and run on an ABI 3730xl DNA Analyzer (Applied Biosystems).
Genetic variability and population differentiation
All sequences were imported into Sequencher 4.9 (Gene Codes Corp.) for alignment and manual editing. Each variable site was visually confirmed. Nucleotide diversity (π; average number of nucleotide differences per site in pairwise sequence comparisons), as well as haplotype diversity (h; probability that two randomly sampled alleles are different) were estimated using DNAsp 5.10.01 (Librado and Rozas 2009a, b), for all sampling locations combined, and for NI and SI separately. A median joining network was drawn to infer the evolutionary relationships between haplotypes using PopART (http://popart.otago.ac.nz).
Microsatellite loci were tested for linkage disequilibrium and deviation from Hardy-Weinberg expectations (HWE) in GenAlEx 6.4 (Peakall and Smouse 2006). P-values were adjusted for multiple comparisons using sequential Bonferroni corrections (Holm 1979). We used Micro-Checker 2.2.3 (van Oosterhout et al. 2004) to test loci for null alleles, large allele drop-out, and stutter-related miscalling in each population. Hmal10 and Hmal3 were removed from subsequent analyses (see Results) with nine loci remaining in the final dataset. The mean number of alleles per locus (A), the number of private alleles (Np), as well as observed (Ho) and expected heterozygosity (He) were calculated in GenAlEx 6.4 for different geographic levels, i.e. river catchment, region (geographic area encompassing several neighbouring rivers; see Fig. 1), and islands. The inbreeding coefficient FIS was calculated in Arlequin 3.5 (Excoffier and Lischer 2010) and significance tested with 1000 permutations.
As a first exploratory step in the genetic structure of blue duck populations, FST was estimated for microsatellite loci between each pair of sampling locations (catchments) using Arlequin. The significance of FST values was tested with 1000 permutations and p-values were adjusted for multiple comparisons using sequential Bonferroni corrections. A more recent alternative estimator of genetic differentiation, Jost’s Dest (Jost 2008), was also computed using SMOGD (Crawford 2010). For both estimators, populations with less than five individuals were excluded from the analysis.
In order to more precisely understand the geographic distribution of genetic variability in blue duck populations throughout their distribution range, we used a Bayesian clustering method to delimit genetic entities based on microsatellite allele frequencies as implemented in STRUCTURE 2.3.2 (Falush et al. 2003; Pritchard et al. 2000). The complete dataset was run for 500,000 chains following a 250,000 burn-in under the admixture and correlated allele frequency models for K = 1–10 and 10 iterations for each K. All other parameters were left at default settings. Change in alpha level was monitored to assess convergence of the runs. We then analysed the data for NI and SI separately using the same settings. Additionally, we analysed the NI and SI datasets including prior location information (the catchment each individual was sampled in) using LOCPRIOR (Hubisz et al. 2009), which aids the clustering process if the structure signal is weak. The best K value was evaluated by assessing the change in mean log likelihood of the data lnP(X|K) and Evanno’s ΔK method (Evanno et al. 2005) implemented in Structure Harvester 0.6.92 (Earl and vonHoldt 2012). Iterated runs were averaged with CLUMPP 1.1.2. (Jakobsson and Rosenberg 2007) and visualised with Distruct 1.1 (Rosenberg 2004).
Using microsatellite loci, we also performed a discriminant analysis of principal components (DAPC; Jombart et al. 2010; a clustering approach which, unlike STRUCTURE, does not rely on explicit model assumptions) with population prior information (catchment identity) using the “adegenet 1.3-1” package (Jombart 2008) in R (R Core Team 2015).
Analysis of molecular variance (AMOVA; Excoffier et al. 1992) was performed for microsatellite loci in Arlequin with 1000 permutations to test for hierarchical differentiation between blue duck regional groupings (NI vs. SI, within island groupings; see Table 3).
Finally, we tested for correlation between genetic (microsatellites) and geographic distances [isolation by distance (IBD)] using Mantel tests in Arlequin with 1000 randomisations to estimate the levels of significance. Calculations were based on linearised FST (FST/(1 − FST); Slatkin 1995) and log transformed shortest geographic distance between catchments. We restricted analyses to catchments within islands as NI and SI divergence is likely old and driven by mechanisms other than simple IBD.
Demographic history, population size and migration
We performed several analyses to investigate the demographic history of blue duck in the NI and SI. For mtDNA CR, we calculated Tajima’s D (Tajima 1989) and Fu’s F (Fu 1997); negative values of these statistics can indicate population expansion, whereas positive values are indicative of a reduction in population size. We further generated the mismatch distribution (distribution of the number of observed nucleotide differences between pairs of haplotypes). The shape of the distribution is expected to be unimodal in recently expanded populations, and ragged or multimodal when the population is stable or has experienced a recent bottleneck (Rogers and Harpending 1992; Slatkin and Hudson 1991). All analyses were performed in Arlequin with 1000 permutations to test for significance.
Microsatellite data were used to test for recent population bottlenecks based on the heterozygosity excess method implemented in Bottleneck 1.2.02 (Piry et al. 1999). Analyses were performed using the two-phase model (TPM; Di Rienzo et al. 1994) with 95% single-step mutations and a variance of 12 as recommended by Piry et al. (1999). TPM is thought to be most appropriate for microsatellite data. The Wilcoxon rank test, recommended for less than 20 loci (Piry et al. 1999), was used to assess statistical significance with 1000 replicates. We also performed a mode-shift test implemented in Bottleneck, which assesses the allele frequency distribution to identify signatures of recent bottlenecks (Luikart et al. 1998).
To estimate the effective number of breeders Nb for NI and SI populations from microsatellite data, we used the one-sample based linkage disequilibrium method (Hill 1981) implemented in NeEstimator 2.0 (Do et al. 2014). We specified mating system as monogamous (Triggs et al. 1992) and set the critical cut-off value for the frequency of rare alleles to 0.02. We also ran the programme without a minimum allele frequency cut-off. Confidence intervals for Nb were estimated using a jacknife method. Nb was also estimated for the SI genetic clusters (see STRUCTURE Results) Kahurangi, Central SI, and Fiordland separately, using the same settings.
We assessed the level of recent gene flow (within the last few generations) among islands, and between regions within islands using the Bayesian modelling approach implemented in BayesAss 3.0.4 (Wilson and Rannala 2003) using the microsatellite data. Within the NI, migration rates between Bay of Plenty and Central NI were calculated (there were too few samples for Ruahines). Within the SI, we calculated migration rates between Kahurangi, Central SI and Fiordland. Three replicate runs were performed for each analysis using different starting seeds. Each run consisted of 50 × 106 iterations with 10% burn-in, sampling every 5000 steps. Convergence was assessed using Tracer 1.6.0 (http://beast.bio.ed.ac.uk/Tracer) and by comparing results between replicate runs.
We also made use of a coalescent-based approach to assess migration as well as population divergence time and long-term population sizes using the isolation-with-migration model (Nielsen and Wakeley 2001) implemented in IMa2 (Hey and Nielsen 2004; Hey 2010). We used mtDNA CR sequences together with microsatellite genotypes to estimate six parameters in a full IM model (two contemporary and one ancestral population size (θ = 4Ne µ), divergence time, and migration rates between the two descendent populations). We used a random subset of 29 NI and 34 SI individuals for mtDNA CR and 72 NI and 90 SI individuals for nine microsatellite loci, which were analysed using the HKY and SMM mutation models, respectively. We ran separate analyses for two different mtDNA CR mutation rates (to account for uncertainty in the rate estimate for blue duck), with microsatellite rates scaled against these rates. First, we used the mean lineage specific substitution rate of 0.048 substitutions/site/million years (s/s/Myr) calibrated for several Anas species by Peters et al. (2005) (hereafter fast rate). Second, we used a lineage specific rate of 0.025 s/s/Myr, as a mutation rate of 5% per Myr has previously been suggested for avian mtDNA CR (Freeland and Boag 1999) (hereafter slow rate).
Upper limits for uniform priors were determined in several trial runs; divergence time—t5, θ—q15, and migration rate—m6. The generation time needs to be provided (in addition to mutation rates) to convert parameter estimates into demographic units. We calculated generation time G of blue duck using the equation G = α + [s/(1−s)] (Lande et al. 2003), where α is the age of first reproduction (2 years; Williams 1991) and s is the average adult survival (0.80 for females; Williams 1991).
For each mutation rate we started three independent runs with identical prior settings and different starting seeds. Eighty geometrically heated chains (–hfg–hn80–ha0.94–hb0.75) were run for ~600,000 steps before stationarity was reached (assessed based on proper heating of chains, high ESS values, lack of trends in trace, and similarity of the two parameter sets). Because parameter estimates were consistent between the replicated runs, we combined results to estimate joint posterior probabilities and perform nested model tests (L-mode). For fast and slow mutation rates, a total of 174,986 and 169,130 genealogies were recorded respectively, and 150,000 genealogies were randomly sampled from the three replicates for each L-mode run. We compared 24 nested models (provided in the IMa2 software package) to the full model using likelihood ratio tests. We calculated the Akaike information criterion and model support to assess best model fit (Burnham and Anderson 2002).
We also estimated timing of divergence between the NI and SI mitochondrial lineages using the coalescent-based approach implemented in BEAST 1.8.2 (Drummond et al. 2012) using a strict molecular clock with rate prior. Mallard duck (GenBank accession: AY112938) was used as an outgroup. The HKY+I model of sequence evolution was chosen, as determined by jModeltest 2.1.8 (Posada 2008). We used a coalescent tree prior due to the intraspecific nature of the analysis. We ran analyses for a slow (0.025 s/s/Myr, standard deviation (SD) 0.009) and a fast (0.048 s/s/Myr, SD 0.015) substitution rate prior (see IMa2 analysis) with normal distribution. The MCMC chains were run for 100 × 106 generations sampling every 10,000 generations with 10% discarded as burn-in. Convergence was confirmed by assessing trace plots and ESS in Tracer.
Results
There was no evidence for linkage disequilibrium among the 11 tested microsatellite markers. Significant departure from HWE after sequential Bonferroni corrections was observed for Hmal15 in Waimana River, Hmal3 and Hmal10 in Oparara River, and Hmal3 and Hmal14 in Clinton River. Micro-Checker suggested possible null alleles for Hmal3 and Hmal10 in five of the 17 populations, possible null alleles for Hmal23 in Styx River, and stutter related miscalling or null alleles for Hmal2 in Arthur River. Because null alleles and deviations from HWE were suggested for Hmal3 and Hmal10 in several populations, we removed these markers from subsequent analyses but retained all other markers.
Genetic diversity
For mitochondrial control region (mtDNA CR), we found 12 haplotypes, defined by 29 variable sites (Table S2, Supplementary material 1). Despite extra sampling at seven new sites (Fig. 1), as well as more extensive sampling in previously sampled areas, we only found the haplotypes previously identified by Robertson et al. (2007). MtDNA diversity in the NI (h = 0.576 and π = 0.0020) was lower than in the SI (h = 0.763 and π = 0.0024). The same trend was observed for microsatellite data (Table 1). Despite differences in sample sizes, the overall genetic diversity within regions was generally low and similar across the species’ range (Table 1).
Population structure
The median joining network for mtDNA CR shows that NI and SI haplotypes form two distinct groups, separated by a deep split of 17 evolutionary steps (Fig. 2). Within the NI and northern SI, with two exceptions (Whanganui, Wangapeka), only one haplotype was found within each catchment, whereas in the central and southern SI two or more haplotypes were detected in each catchment.

Distribution and median joining network of blue duck mitochondrial control region haplotypes. Colours represent haplotype identity. Circle size is proportional to haplotype frequency as indicated by the legend. Edges connecting haplotypes represent single mutational steps and missing haplotypes are indicated by small black circles. (Color figure online)
Genetic differentiation between pairs of populations, as measured by FST and Jost’s Dest for 9 microsatellite markers, is presented in Table 2. Both estimators show that genetic differentiation within regions (e.g., Bay of Plenty) was low, with pairwise comparisons of FST generally being not significantly different from zero. Among regions within islands, differentiation was higher than within regions and several comparisons yielded significant pairwise FST values. Intra-island comparisons resulted in high and mostly significant pairwise differentiation. This gradual increase in genetic differentiation across increasingly larger geographical areas observed within islands is suggestive of isolation by distance. Mantel tests indeed revealed significant correlation between genetic (FST/(1−FST)) and log transformed geographic distances (Fig. S1) within the NI (r = 0.67, p = 0.006) and SI (r = 0.41, p = 0.035).
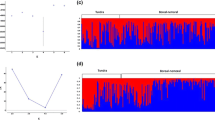
STRUCTURE analysis found most support for two distinct clusters, within the complete dataset, corresponding to NI and SI populations (Fig. 3; Fig. S2A, Supplementary material 1), with some individuals from Bay of Plenty and southern SI showing high assignment probability to the other cluster. Within the NI, no further subdivision was supported (Fig. S2B, Supplementary material 1; analysis with LOCPRIOR did not converge). Within the SI a northern SI and southern SI cluster could be distinguished with some suggestion of a clinal change in allele frequencies from North to South (Fig. S2B, Supplementary material 1). Using a LOCPRIOR, the northern SI and southern SI clusters could be more clearly distinguished, with Central SI forming a mixed assemblage (for K = 2; Fig. 3; Fig. S2C, Supplementary material 1). For K = 3, Central SI formed a separate cluster (Fig. S2C, Supplementary material 1). Change in mean log likelihood of the data lnP(X|K) and the Evanno’s ΔK method supported K = 2 as the most likely number of SI clusters. AMOVA analysis based on microsatellites also revealed significant hierarchical population structure corresponding to the same clusters identified with STRUCTURE for the complete dataset and SI (Table 3). Within the NI, AMOVA of microsatellites suggested significant structuring in concordance with the distribution of the mtDNA haplotypes (Table 3).

Bayesian clustering (STRUCTURE) of blue duck samples based on nine microsatellite loci for K = 2. The inner layer represents the analysis of the complete dataset (without LOCPRIOR) and the outer layer shows the results for the separate South Island analysis (with LOCPRIOR). Horizontal lines represent individuals and their proportional membership to each cluster (indicated by colours). Black lines separate individuals of different sampling location. (Color figure online)
Clustering using discriminant analysis of principal components also supported the separation of NI and SI populations into two distinct units, with some individuals from Bay of Plenty and southern SI showing affinities for the other cluster (Fig. S3, Supplementary material 1). The SI cluster was subdivided into two clusters corresponding to Kahurangi and Fiordland, with Central SI populations connecting the two clusters. Overlap was observed between the SI clusters. Within the NI cluster, populations were largely overlapping, but with the majority of Manganui o te Ao individuals forming a separate group.
Demographic analyses
Reliable inferences about expansions or contractions of populations were not possible because of non-significant values for Tajima’s D and Fu’s F statistics across all tested regions (Table 4). Similarly, ragged mismatch distributions for NI, as well as Central SI and Fiordland, suggested stable populations (Fig. 4), but the low diversity in mtDNA CR sequences made results difficult to interpret. The overall SI mismatch distribution showed a reasonable fit to the expected distribution under a rapid expansion model. The analysis did not converge for Central NI and Kahurangi. Bottleneck analysis based on microsatellites suggested a recent population reduction in Kahurangi (Wilcoxon test p value = 0.03, and shifted mode of allele frequencies) and a shifted mode was also observed for Central NI.

Mismatch distribution of mitochondrial control region haplotypes for North Island and South Island blue duck lineages as well as regional groupings within lineages. Bars represent observed values and the dashed line indicates simulated values under a model of sudden population expansion
Estimates for the effective number of breeders, Nb, are shown in Table 5. The summed estimate over the three SI regions was slightly larger than the single SI estimate, but the Central SI confidence intervals included infinity, suggesting this estimate is unreliable due to sampling limitations.
Gene flow between and within islands
The three replicate BayesAss runs were highly consistent. The analysis suggests little gene flow between NI and SI with a migrant fraction of 0.6% (0.5% SD) and 0.7% (0.6% SD), respectively (Table S3, Supplementary material 1). Two individuals from Styx and Arthur River were identified as possible first-generation migrants from the North Island with a probability of 20 and 16%, respectively. High STRUCTURE q-values (>0.96) similarly assign these individuals to the North Island cluster. One individual from Whirinaki River had a 10% probability of being a second-generation migrant from the South Island, also in agreement with STRUCTURE results. Thirteen other SI and NI blue duck showed low probabilities (3–5%) of being either first- or second-generation migrants (concurring with STRUCTURE; Supplementary material 2). Substantial gene flow was detected between regions within islands. Approximately 20% (9% SD) of Bay of Plenty and Central NI individuals were identified as migrants. However, because of difference in sample sizes, migration is potentially higher towards Central NI.
Within the SI, the fraction of migrants received by Kahurangi from Central SI and Fiordland was 12% (8% SD) each. Central SI received 13% (8% SD) from Kahurangi and 12% (8% SD) from Fiordland, and 14% (8% SD) and 12% (7% SD) of the Fiordland populations were migrants from Kahurangi and Central SI, respectively. Again, Central SI sample size was about half that of the other two regions, which suggests a higher proportion of migrants overall.
Coalescent analysis
Bayesian coalescent analysis of mtDNA haplotypes and microsatellites in IMa2 suggests that these lineages likely diverged in the late Pleistocene. Depending on whether a fast or a slow mutation rate was used in the analysis, the divergence time was estimated as 26,500 or 50,800 ya, respectively, but 95% highest posterior density (HPD) intervals are very large (~9000–2,000,000 and 10,000–4,000,000 ya, respectively) reflecting the high uncertainty in this estimate. All parameter estimates for slow and fast mutation rates converted to demographic units are given in Table 6. Unconverted estimates and diagrams of posterior distribution are shown in Table S4 and Fig. S4 (Supplementary material 1). Posterior distributions of divergence time, ancestral population size, and both population migration rates did not reach zero, hence 95% HPD intervals should be interpreted with caution. The model most supported in the nested model analysis (Table S5, Supplementary material 1) was a model with zero migration that allows population sizes to vary. The next five best models (including the full model) all included migration.
Coalescent analysis in BEAST suggests that the NI and SI mitochondrial lineages diverged approximately 0.28 million year ago (Mya; 0.13–0.62 95% HPD), and 0.54 Mya (0.20–1.38 95% HPD) for a substitution rate of 0.048 and 0.025 s/s/Myr, respectively.
Discussion
Our work represents the most extensive genetic study of the threatened blue duck undertaken to date. Analyses based on polymorphic microsatellite loci and mitochondrial control region revealed two highly distinct genetic units, which likely diverged in the late Pleistocene and correspond to New Zealand’s North and South Island. Overall genetic diversity was low across the blue duck range and comparable to several other endangered avian species endemic to New Zealand (see below). We detected a high level of gene flow among river catchments within islands, resulting in only weak or moderate population structuring in the NI and SI, respectively, and genetic similarity declined with increasing geographic distance (isolation by distance).
NI and SI blue duck represent two genetically distinct lineages
Our analysis of nine microsatellite markers provides genome-wide support for distinct NI and SI blue duck lineages that were previously detected based on mtDNA control region variation (Robertson et al. 2007). Our coalescent analyses estimated the divergence time of these lineages to be in the mid to late Pleistocene, however, the BEAST estimate was tenfold higher than the estimate obtained by IMa2. This discrepancy between estimates might be due to differences in the underlying models used by each programme or the additional markers (microsatellites) employed in the IMa2 analysis.
It is unclear whether apparent admixture of NI and SI lineages, as suggested by STRUCTURE analysis (Fig. 3), reflects gene flow between the islands or might be explained by other mechanisms, such as shared ancestral polymorphism or size homoplasy (Estoup et al. 2002). BayesAss identified three individuals as potential migrants between islands, but a coalescent model with zero migration received most support in IMa2 nested model analysis. However, posterior distributions of migration parameters showed clear peaks at non-zero values, hence migration between both island lineages cannot be easily discounted. To our knowledge there are no records of Cook Strait crossings by blue duck, but Buller (1888) reported sightings of blue duck in coastal waters when populations were substantially larger than today. Sampling of populations in the immediate vicinity to Cook Strait (historically populations are known from the Rimutaka ranges, NI, and Pelorus River, SI) might help to determine if blue duck occasionally cross this large body of water.
Gene flow within islands is potentially driven by male-biased dispersal
Within the NI, we observed a non-random distribution of mtDNA haplotypes concordant with the main geographic regions; this finding was also supported by microsatellite based AMOVA. However, STRUCTURE analysis did not detect any population structuring, and analysis of migration rates between the Bay of Plenty and Central NI suggested a substantial amount of gene flow between these regions, which is not reflected in the distribution of mtDNA haplotypes. Such discordance between patterns of mitochondrial and nuclear genetic structure can be indicative of male biased dispersal (Prugnolle and de Meeus 2002), which is common among waterfowl species (Peters et al. 2012), and congruent with field observations indicating that male juveniles are the main dispersers in blue duck (Whitehead et al. 2007).
Within the SI, analyses based on microsatellite loci (AMOVA, STRUCTURE) support the existence of two distinct genetic clusters—Kahurangi and southern SI (including Central SI and Fiordland). However, Central SI populations appear to be an admixed pool of northern and southern populations and hence distinct from Fiordland populations. This is also supported by the presence of shared, as well as unique mitochondrial haplotypes in this region. Because of sampling gaps between regions that correspond to genetic clusters, and the longitudinal distribution of these clusters within the SI, we cannot exclude the possibility of clinal genetic variation rather than discrete genetic units (STRUCTURE analysis has known limitations in the presence of allelic clines; Pritchard et al. 2000). Indeed, clinal genetic variation is supported by a significant result from our test for isolation by distance. Analyses of migration rates further indicate high connectivity between SI regions. Similar patterns of population structure have recently been discovered in New Zealand’s alpine parrot, the kea (Nestor notabilis; Dussex et al. 2014), which likely represent a history of postglacial expansion out of a single refugium followed by extensive gene flow between populations in this highly dispersive taxon.
Overall, our analyses suggest that blue duck dispersal between catchments, and on wider geographic scales, is greater than what had been assumed for many years. Previous field observations (Williams 1991) and genetic analyses based on DNA fingerprints and small geographical scales (Triggs et al. 1991, 1992) suggested that blue duck dispersal was limited and restricted to their natal ranges. For instance, Williams (1991) reported only one inter-catchment dispersal event in a 10-year period for the Manganui o te Ao River. Interestingly, several recent studies, involving increased field observation efforts, have detected blue duck dispersal events on larger scales. Specifically, Whitehead et al. (2007) reported juvenile males moving up to 24 km away from their natal area in Fiordland. Similarly, Bristol et al. (2008) reported a juvenile banded in the Whanganui catchment was observed 30 km away in the Tongariro River. Indeed, the Tongariro blue duck population has also previously been shown to be a sink population receiving a large number of migrants from surrounding catchments (King et al. 2000). Blue duck dispersal has also been observed between catchments on either side of several mountain passes in Central SI (Harding 1994). Observations of long distance juvenile dispersal have further been reported for the Bay of Plenty region (Glaser 2003).
Blue duck have low genetic diversity and small effective population size
Despite including new locations and increasing the sample size of previously studied populations, we identified no further mitochondrial haplotypes than Robertson et al. (2007). While mtDNA diversity was low, it was similar to levels of diversity observed in other duck species (e.g., Kulikova et al. 2005), many of which are not in decline. Nuclear genetic diversity in blue duck was very low and comparable, albeit on the lower end, to several other threatened avian species in New Zealand that have persisted on the mainland in small fragmented populations, e.g., Mohua (Mohoua ochrocephala), South Island robin (Pachyornis australis), and Kokako (C. wilsoni), New Zealand rock wren (Xenicus gilviventris) (Grueber et al. 2015; Weston and Robertson 2015).
Estimates of blue duck contemporary effective population size revealed that a substantially smaller number of breeders genetically contribute to NI and SI populations (~100 individuals each) than previous breeding pair counts imply (1200 pairs; Glaser et al. 2010). Given this small effective population size, blue duck are potentially at risk of further loss of genetic diversity due to genetic drift as well as inbreeding.
Blue duck demographic history
Our classical demographic analyses did not allow us to draw reliable conclusions about blue duck population history. The heterozygosity excess test suggested a recent population reduction in northern SI, potentially corresponding to anthropogenic impacts. Indeed the blue duck population has declined more than 60% within the last century, mostly due to introduced predators and habitat degradation (Glaser et al. 2010). However, this trend likely applies to the entire species’ range rather than to localised bottleneck events. Comparison of long-term effective population sizes (IMa2 estimates: NI ~7000 and SI ~9000) with contemporary estimates (see above) also suggests a strong overall decline in recent centuries.
In the SI, the unimodal mismatch distribution might be indicative of past population growth consistent with range expansion out of glacial refugia after the last glacial maximum (14,000 years ago; McGlone 1985). Post glacial range expansion was recently inferred for kea (N. notabilis; Dussex et al. 2014) and alpine New Zealand rock wren (X. gilviventris; Weston and Robertson 2015), which both show genetic patterns of population structure similar to those observed in blue duck. In contrast, the phylogeographic distribution of many NI species is thought to have been shaped by ongoing volcanism in the Taupo Volcanic Zone (McDowall 1996; Worthy and Holdaway 2002) with genetic breaks across the Central Plateau region reported in New Zealand short-tailed bats (Mystacina tuberculata; Lloyd 2003), ornate skink (Cyclodina ornata; Chapple et al. 2008) and NI brown kiwi (Apteryx mantelli; Shepherd 2006). Such a phylogeographic pattern was not observed in NI blue duck and the impact of large volcanic events like the last eruption of the Taupo Volcanic Zone approximately 2000 years ago remains unknown.
Conservation implications
The latest of the Department of Conservation’s 10-year recovery plans for blue duck considers two priorities (Glaser et al. 2010). First, a 5-year security phase concentrates on protecting blue duck at eight secure sites, each of which must provide protection for at least 50 pairs in one or several neighbouring rivers. Four in the NI (two in Bay of Plenty, two in Central NI) and four in the SI (two in Kahurangi, one in Central SI and one in Fiordland). In this context, it is important to understand how the predefined security sites might correspond to the genetic boundaries and diversity identified between populations in this study.
Overall, we found strong concordance between these predefined sites and results reported here. However, the detected higher genetic diversity and identification of at least two genetic clusters in the SI warrants the establishment of additional security sites, particularly in Central SI and Fiordland. Our results also suggest that more than 50 breeding pairs should be protected within a security site, as a much smaller number of individuals is likely to genetically contribute to the population than are protected within a site.
Our results, based on both nuclear and mitochondrial markers, strongly reinforce the recommendation to avoid translocations between NI and SI populations (Robertson et al. 2007). Indeed, it is hard to anticipate the balance between potential positive effects by introgression of a highly distant genetic pool (i.e. increase of genetic diversity, reduction of inbreeding depression) and the negative effects linked to the perturbation of local adaptation and co-adapted genes (see Weeks et al. 2011 for a recent review). Studies related to adaptation and inbreeding depression need to be conducted in order to have a better understanding of the outcome of such management strategies, and as long as NI and SI translocations are not a necessity to prevent extinction of one or both lineages, the precautionary principle should be applied. Similarly, within the SI we suggest limiting translocations to individuals sourced locally in the first instance. This strategy should also apply to Whio Nest Egg (WHIONE)—where wild eggs are harvested, chicks raised in captivity and subsequently released into managed sites.
Given the overall low genetic diversity and small effective population size of blue duck, continuing conservation management will be of utmost importance to ensure the survival of this species. Future studies would benefit from more fine scale geographic sampling and genomic approaches (e.g. double digest RADseq; Peterson et al. 2012) to assess the magnitude and effects of inbreeding and determine inter-island population boundaries. Analysis of historic population structure and genetic diversity using ancient DNA approaches may also provide better insights that would assist the management of the species going forward (Leonard 2008; Shepherd and Lambert 2008; Waters and Grosser 2016).
References
Abdelkrim J, Robertson BC, Stanton JAL, Gemmell NJ (2009) Fast, cost-effective development of species-specific microsatellite markers by genomic sequencing. Biotechniques 46:185–192
Adams J, Cunningham D, Molloy J, Phillipson S (1997) Blue duck (Whio), Hymenolaimus malacorhynchos recovery plan 1997–2007, vol 22. Department of Conservation, Wellington
Avise JC (1989) A role for molecular genetics in the recognition and conservation of endangered species. Trends Ecol Evol 4:279–281
BirdLife International (2013) Hymenolaimus malacorhynchos
Bristol R, Campbell J, Beath A (2008) Securing blue duck in the Central North Island, vol 4. Department of Conservation, Wanganui Conservancy, Wanganui
Buller WL (1888) A history of the birds of New Zealand. Buller, London
Burnham KP, Anderson DR (2002) Model selection and inference: a practical information-theoretic approach, 2nd edn. Springer-Verlag, New York, p 353
Chapple DG, Daugherty CH, Ritchie PA (2008) Comparative phylogeography reveals pre-decline population structure of New Zealand Cyclodina (Reptilia: Scincidae) species. Biol J Linn Soc 95:388–408
Collier KJ, Moralee SJ, Wakelin MD (1993) Factors affecting the distribution of blue duck Hymenolaimus malacorhynchos on New Zealand Rivers. Biol Conserv 63:119–126
Crawford NG (2010) Software for the measurement of genetic diversity. Mol Ecol Resour 10:556–557
Di Rienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci 91:3166–3170
Didham RK, Tylianakis JM, Gemmell NJ, Rand TA, Ewers RM (2007) Interactive effects of habitat modification and species invasion on native species decline. Trends Ecol Evol 22:489–496
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29(8):1969–1973
Dussex N, Wegmann D, Robertson BC (2014) Postglacial expansion and not human influence best explains the population structure in the endangered kea (Nestor notabilis). Mol Ecol 23:2193–2209
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Estoup A, Jarne P, Cornuet J-M (2002) Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol Ecol 11:1591–1604
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Freeland JR, Boag PT (1999) Phylogenetics of Darwin’s finches: paraphyly in the tree-finches, and two divergent lineages in the Warbler Finch. Auk 116:577–588
Fu Y-X (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Glaser A (2003) Northern Te Urewera ecosystem restoration project. Department of Conservation, East Coast Hawke’s Bay Conservancy, Gisborne
Glaser A, Van Klink P, Elliott G, Edge K-A (2010) Whio/blue duck (Hymenolaimus malacorhynchos) recovery plan: 2009–2019, vol 62. Department of Conservation, Wellington
Grueber CE et al (2015) Toll-like receptor diversity in 10 threatened bird species: relationship with microsatellite heterozygosity. Conserv Genet 16:595–611
Harding MA (1994) Blue duck dispersal at Arthur’s Pass. Notornis 41:293–295
Hey J (2010) Isolation with migration models for more than two populations. Mol Biol Evol 27:905–920
Hey J, Nielsen R (2004) Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167:747–760
Hill WG (1981) Estimation of effective population-size from data on linkage disequilibrium. Genet Res 38:209–216
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Stat 6:65–70
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:1–15
Jost LOU (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Kear J (2005) Ducks, gees and swans. Oxford University, Oxford
Keller LF, Waller DM (2002) Inbreeding effects in wild populations. Trends Ecol Evol 17:230–241
King TM, Williams M, Lambert DM (2000) Dams, ducks and DNA: identifying the effects of a hydro-electric scheme on New Zealand’s endangered blue duck. Conserv Genet 1:103–113
Kulikova IV et al (2005) Phylogeography of the mallard (Anas platyrhynchos): hybridization, dispersal, and lineage sorting contribute to complex geographic structure. Auk 122:949–965
Lande R, Engen S, Seather B-E (2003) Stochastic population dynamics in ecology and conservation. Oxford University, Oxford
Leonard JA (2008) Ancient DNA applications for wildlife conservation. Mol Ecol 17:4186–4196
Librado P, Rozas J (2009a) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Librado P, Rozas J (2009b) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lloyd BD (2003) The demographic history of the New Zealand short-tailed bat Mystacina tuberculata inferred from modified control region sequences. Mol Ecol 12:1895–1911
Luikart G, Allendorf F, Cornuet J-M, Sherwin W (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Heredity 89:238–247
McDowall RM (1996) Volcanism and freshwater fish biogeography in the northeastern North Island of New Zealand. J Biogeogr 23:139–148
McGlone MS (1985) Plant biogeography and the late Cenozoic history of New Zealand. New Zeal J Bot 23:723–749
Miskelly CM et al (2008) Conservation status of New Zealand birds. Notornis 55:117–135
Nielsen R, Wakeley J (2001) Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics 158:885–896
Palsbøll PJ, Bérubé M, Allendorf FW (2007) Identification of management units using population genetic data. Trends Ecol Evol 22:11–16
Peakall ROD, Smouse PE (2006) GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peters JL, Gretes W, Omland KE (2005) Late Pleistocene divergence between eastern and western populations of wood ducks (Aix sponsa) inferred by the ‘isolation with migration’ coalescent method. Mol Ecol 14:3407–3418
Peters JL, Bolender KA, Pearce JM (2012) Behavioural vs. molecular sources of conflict between nuclear and mitochondrial DNA: the role of male-biased dispersal in a Holarctic sea duck. Mol Ecol 21:3562–3575
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double Digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-Model species. PLoS ONE 7:e37135
Piry S, Luikart G, Cornuet J-M (1999) Computer note. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Heredity 90:502–503
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Prugnolle F, de Meeus T (2002) Inferring sex-biased dispersal from population genetic tools: a review. Heredity 88:161–165
R Core Team (2015) A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Robertson BC, Goldstien SJ (2012) Phylogenetic affinities of the New Zealand blue duck (Hymenolaimus malacorhynchos). Notornis 59:49–59
Robertson BC, Steeves TE, McBride KP, Goldstien SJ, Williams M, Gemmell NJ (2007) Phylogeography of the New Zealand blue duck (Hymenolaimus malacorhynchos): implications for translocation and species recovery. Conserv Genet 8:1431–1440
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Resour 4:137–138
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 1:233–234
Sgro` CM, Lowe AJ, Hoffmann AA (2011) Building evolutionary resilience for conserving biodiversity under climate change. Evol Appl 4:326–337
Shepherd LD (2006) Ancient DNA studies of the New Zealand kiwi and wattlebirds: evolution, conservation and culture. Massey University, Albany
Shepherd LD, Lambert DM (2008) Ancient DNA and conservation: lessons from the endangered kiwi of New Zealand. Mol Ecol 17:2174–2184
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129:555–562
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Triggs SJ, Williams MJ, Marshall SJ, Chambers GK (1991) Genetic relationships within a population of Blue Duck Hymenolaimus malacorhynchos. Wildfowl 42:87–93
Triggs SJ, Williams MJ, Marshall SJ, Chamber GK (1992) Genetic structure of blue duck (Hymenolaimus malachorhynchos) populations revealed by DNA fingerprinting. Auk 109:80–89
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Walsh PS, Metzger DA, Higuchi R (1991) Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513
Waters JM, Grosser S (2016) Managing shifting species: ancient DNA reveals conservation conundrums in a dynamic world. BioEssays. doi:10.1002/bies.201600044
Weeks AR et al (2011) Assessing the benefits and risks of translocations in changing environments: a genetic perspective. Evol Appl 4:709–725
Weston KA, Robertson BC (2015) Population structure within an alpine archipelago: strong signature of past climate change in the New Zealand rock wren (Xenicus gilviventris). Mol Ecol 24:4778–4794
Whitehead A, Smart A, Edge K-A, Williams MJ, Hill G (2007) Status of blue duck (whio) populations in Fiordland, New Zealand. Response to stoat control: a review of productivity, survival, and juvenile dispersal. Department of Conservation, Southland Conservancy, Invercargill, pp 2000–2006
Williams MJ (1991) Social and demographic characteristics of blue duck Hymenolaimus malacorhynchos. Wildfowl 42:65–86
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Worthy TH, Holdaway RN (2002) The lost world of the moa: prehistoric life of New Zealand. Indiana University Press, Bloomington
Acknowledgements
We thank New Zealand Department of Conservation staff and the blue duck recovery group for collecting the blood and feather samples used in this analysis, particularly Andrew Glaser and Dr Murray Williams. We also thank anonymous reviewers for helpful comments on an earlier version of the manuscript. Samples were collected and held under permits ECHB-23214-RES and TT-23326-FAU. This work was funded by a subcontract to NJG through the Sustaining and Restoring Biodiversity OBI FRST contract C09X0503 to Landcare Research together with funding from the University of Otago.
Author information
Authors and Affiliations
Corresponding author
Additional information
Stefanie Grosser and Jawad Abdelkrim are joint first auhtors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Grosser, S., Abdelkrim, J., Wing, J. et al. Strong isolation by distance argues for separate population management of endangered blue duck (Hymenolaimus malacorhynchos). Conserv Genet 18, 327–341 (2017). https://doi.org/10.1007/s10592-016-0908-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-016-0908-4