
Abstract
Disorders of sex development (DSD) are congenital conditions in which the development of chromosomal, gonadal, or anatomical sex is atypical. Many of the genes required for gonad development have been identified by analysis of DSD patients. However, the use of knockout and transgenic mouse strains have contributed enormously to the study of gonad gene function and interactions within the development network. Although the genetic basis of mammalian sex determination and differentiation has advanced considerably in recent years, a majority of 46,XY gonadal dysgenesis patients still cannot be provided with an accurate diagnosis. Some of these unexplained DSD cases may be due to mutations in novel DSD genes or genomic rearrangements affecting regulatory regions that lead to atypical gene expression. Here, we review our current knowledge of mammalian sex determination drawing on insights from human DSD patients and mouse models.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In mammals, biological differences between males and females are determined genetically during embryonic development. These differences have a significant impact on the physical, reproductive, psychological, and social life of an individual. Sex development can be divided into two processes, “sex determination”, which is the developmental decision that directs the undifferentiated gonad to develop as a testis or ovary and “sex differentiation”, which occurs once the gonad has developed and is induced by the products of the gonad to establish the phenotypic sex. In mammals, sex determination equates to gonad development. Factors influencing sex determination tend to be transcriptional regulators, whereas factors influencing sex differentiation are most often secreted hormones and their receptors.
Most of our current knowledge on genes and their proteins involved in sex development comes from mutation studies in human patients with disorders of sex development (DSD) and mouse models. Here, we will discuss the current knowledge of gonad development and how changes to this complex developmental network result in cases of DSD. We will highlight genes that have been implicated in embryonic mouse gonad development and finally, discuss how whole genome sequencing approaches are going to improve our current understanding of the etiology of DSD.
Disorders of sex development
DSD are congenital conditions in which development of chromosomal, gonadal, or anatomical sex is atypical (Hughes et al. 2006). DSD covers a wide spectrum of different phenotypes with hypospadias being the most common defect with an average of 1 in 250–350 male births. In addition, 1 in 4,500 babies worldwide is born with significant ambiguous genitalia (Hughes et al. 2006) and significantly, DSDs account for 7.5% of all birth defects. Furthermore, DSD phenotypes are often associated with other syndromes, such as Mayer–Rokitansky–Kuster–Hauser syndrome, Smith–Lemli–Opitz syndrome or genito-palato-cardiac syndrome (Porter 2008; Sultan et al. 2009). Since the discovery of the sex-determining region Y (SRY) in 1990 (Sinclair et al. 1990), there have been considerable advances in understanding the genetic factors involved in gonad differentiation. Nevertheless, it has been estimated that a molecular diagnosis is made in only approximately 20% of DSD cases (Hughes et al. 2006), and that up to 50% of 46,XY DSD patients cannot be provided with an accurate diagnosis. Furthermore, for approximately 80% of 46,XY DSD complete gonadal dysgenesis patients and about 20% of 46,XX testicular DSD patients, the causative mutation remains unknown (Hughes et al. 2006). DSD represents a major pediatric concern, due to the difficulty of clinical management of these complex conditions and their common sequelae of gonad cancer and infertility. The cause of these DSD conditions is most often the breakdown of the complex network of gene regulation and gene expression, essential for proper development of testes or ovaries in the embryo.
Gonad development in humans and mice and the molecular basis of DSD
In humans, both males and females have 22 pairs of autosomal chromosomes (mice have 19 pairs of autosomal chromosomes) but differ in their sex chromosome complement. Females in both humans and mice have two X chromosomes (46,XX in humans, 40,XX in mice), whereas males have one X and one Y chromosome (46,XY in humans and 40,XY in mice).
At the beginning of gestation (first and second week in humans), embryos of the two sexes differ only by their sex chromosomes. The first visible sign of sexual dimorphism in mammalian embryos is when the bi-potential gonad starts to develop into either a testis or an ovary in XY and XX individuals, respectively. This decision is made around embryonic day 10.5 (E10.5) in mice and 6 weeks of development in humans. Differentiation of the gonads leads to testicular and ovarian hormone production and subsequent induction of anatomical and psychological differences.
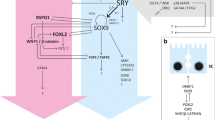
Both testis and ovarian development involve sex-specific pathways that appear to act antagonistically to one another (see Fig. 1). The normal role of SRY in XY gonads is to tip the balance in favor of the testis-specific pathway (Sinclair et al. 1990; Lovell-Badge 1992). In mouse, Sry is transiently expressed in the bi-potential XY gonad (Koopman et al. 1990) and in both species, SRY expression initiates an up-regulation of SOX9 expression. In mice, SOX9 has been shown to stimulate Fgf9 expression and subsequently, both FGF9 and SOX9 act together in a positive feedback loop, which are thought to suppresses Wnt4 (by unknown mechanisms) and leads to the establishment of the testis-specific pathway.

Overview of the key genes and regulatory networks leading from the bi-potential gonad to either testis or ovary development in mouse. a Genes that are essential for the development of the bi-potential gonad have been identified due to the total lack of gonads in the corresponding knockout mouse strain and they have been shown to be expressed in the bi-potential gonad. Functional studies revealed that specifically the WT1 −KTS isoform binds to and activates the Nr5a1 promoter in conjunction with LHX9. In the spleen and adrenal gland, CBX2 has been shown to regulate Nr5a1, leading to the hypothesis of a similar function in the bi-potential gonad. b Various genes have been implicated in the pathways leading to testis development in mouse. In XY mouse embryos, Sry is transiently expressed in the bi-potential gonad, whereas in human embryos, SRY expression is periodic. However, in both species, Sry expression initiates an increase of Sox9 expression, which then stimulates Fgf9 expression. Both FGF9 and SOX9 act in a positive feedback loop, which act to suppress the female specific genes, especially Wnt4 and subsequently lead to the manifestation of the testis-specific program. Numerous other genes and their gene products, such as Gata4, Fog2, Wt1 (+KTS isoform), Nr5a1, Pgsd, Fgfr1, Cbx2, Sox8, Amh, Dax1, and Dhh are necessary for the regulation (positive as well as negative) and maintenance of this crucial male determining pathway. Dmrt1 has recently been shown to be required for the maintenance of gonadal sex, especially to prevent female reprogramming in postnatal mouse testis. c In XX individuals, Sry is absent and ovary-specific genes, such as Rspo1, Wnt4, and Foxl2 are expressed. Rspo1 −/− ovaries show reduced levels of WNT4, suggesting that Rspo1 acts upstream of Wnt4. However, a synergistic action of WNT4 and RSPO1 to activate β-catenin has also been suggested. WNT4/β-catenin have been proposed to suppress the SOX9/FGF9 positive feedback loop, allowing the ovarian-specific pathway to progress. WNT4 and FOXL2 are also involved in the positive regulation of Bmp2. Together, FOXL2, RSPO1, and WNT4 activate Fst expression. Genes of the female pathway that have been shown or suggested to interact with the male pathway are shown in red, genes of the male pathway interacting with the female pathway are highlighted in blue. In this figure, solid lines do not necessarily imply direct interactions. Question marks indicate that the position of that gene and the interaction with other genes has been proposed. Many pathways shown in this figure are similar or even identical between mouse and human; however, for some of them, there might be differences between the two species
In the absence of SRY in XX individuals, RSPO1 and WNT4 are expressed at high levels and stabilize cytoplasmic β-catenin, which is then translocated into the nucleus, where it binds to the TCF/LEF (transcription factor/lymphoid enhancer-binding factor) and activates the transcription of target genes. Both WNT4 and β-catenin suppress (by unknown mechanisms) the SOX9/FGF9 positive feedback loop, allowing the ovarian-specific pathway to progress.
Genes required for the development of the bi-potential gonad
Mammalian gonads arise in both sexes from a bilateral, bi-potential gonad (also called genital ridges), an organ that has the potential to develop either as an ovary or as a testis, depending on differentially expressed genes (reviewed by (Capel 1998; Swain and Lovell-Badge 1999; Capel 2000)). In mouse, the bi-potential gonads are first visible at E9.5, 1 day before the onset of Sry expression, which is critical for initiating testis development in XY individuals. A number of genes have been shown to be required for the development of the bi-potential gonad (Fig. 1a; see also Table 1 for a summary of mouse and human phenotypes of genes described in this section).
Empty spiracles homeobox 2
Empty spiracles homeobox 2 (Emx2) is a homolog of the Drosophila head gap gene empty spiracles (ems) and is essential for the development of dorsal telencephalon in mice (Yoshida et al. 1997). In addition, Emx2 is expressed in the epithelial components of the developing urogenital system. The kidneys, ureters, gonads, and genital tracts are completely absent in Emx2 −/− mice, while the adrenal glands and bladder develop normally. However, a study by Taylor et al. (1999) failed to identify mutations within EMX2 in 120 patients with either Kallmann syndrome or idiopathic hypogonadotrophic hypogonadism (IHH).
LIM homeobox 9
The LIM homeobox 9 (Lhx9) is a member of the LIM homeobox gene family. In Lhx −/− mice, germ cells migrate normally, but somatic cells of the genital ridge fail to proliferate and a discrete gonad fails to form. As a result of the absence of testosterone and anti-Müllerian hormone, genetically male mice develop as phenotypic females (Luo et al. 1994; Birk et al. 2000). The expression of steroidogenic factor 1 (Sf1 also known as nuclear receptor subfamily 5 group A member 1 (Nr5a1)), a nuclear receptor essential for gonadogenesis, is reduced to minimal levels in the Lhx9-deficient genital ridge, indicating that Lhx9 may lie upstream of Nr5a1 in a developmental cascade in mouse (Birk et al. 2000). Furthermore, an in vitro biochemical analysis showed that LHX9 has the potential to bind and trans-activate the Nr5a1 promoter in conjunction with WT1 (Wilhelm and Englert 2002). However, mutation analysis of LHX9 in a range of human DSD patients (including bilateral gonadal agenesis) did not reveal any mutations (Ottolenghi et al. 2001).
Mouse polycomb group member M33/chromobox homolog 2
Mouse polycomb group member M33 (M33) or chromobox homolog 2 (Cbx2) is the mouse ortholog of the Drosophila Polycomb gene. Cbx2 −/− mice show male-to-female sex reversal in most XY animals and XX animals have either smaller or absent ovaries (Katoh-Fukui et al. 1998). Katoh-Fukui et al. (1998) suggested a role for Cbx2 early during gonad development, prior to the critical time of sex determination and before expression of Sry. As CBX2 has been implicated in the regulation of Nr5a1 expression in the adrenal gland (Katoh-Fukui et al. 2005), it has been speculated that CBX2 may play a similar role in gonad development. A recent study by Biason-Lauber et al. (2009) identified a compound heterozygous loss-of-function mutation in the coding sequence of CBX2 in a 46,XY girl with female external and internal genitalia (including ovaries). This study also supports a role for CBX2 in the trans-activation of NR5A1, and thus a role early during gonad development.
Nuclear receptor subfamily 5 group A member 1
NR5A1 encodes the nuclear receptor family member nuclear receptor subfamily 5 group A member 1, also known as steroidogenic factor-1 (SF1), which plays a role in gonadal and adrenal development (Val and Swain 2010). NR5A1 is expressed in the urogenital ridge, the developing hypothalamus, and the anterior pituitary gland. Expression in the bi-potential gonad can be detected prior to SRY onset and functional reporter studies suggest that NR5A1 acts to mediate the up-regulation of SRY (de Santa Barbara et al. 2001). Like the Lhx9 −/− mice, Nr5a1 −/− mice lack adrenal and gonadal development. Gonad development does not proceed beyond the early indifferent stage, which results in complete male-to-female sex reversal of XY animals, with the Müllerian ducts differentiating into uteri, oviducts, and upper vagina (Luo et al. 1994; Shinoda et al. 1995; Sadovsky and Dorn 2000). NR5A1 also has additional roles in the gonad post-differentiation (see Fig. 2). NR5A1 is expressed in Sertoli, and Leydig cells of the developing testis. Together with SOX9, NR5A1 regulates the levels of AMH (Arango et al. 1999) and other genes associated with gonad development (Burris et al. 1995). Furthermore, Sekido and Lovell-Badge (2008) have shown that NR5A1 binds synergistically with SRY to a testis specific enhancer region (TESCO) to up-regulate SOX9 expression. SOX9 then displaces SRY and, together with NR5A1 causes further up-regulation of itself. Mutations in and around NR5A1 cause 46,XY DSD gonadal dysgenesis (Achermann et al. 1999), whereas mutations in NR5A1 in 46,XX individuals are associated with primary ovarian insufficiency (Lourenco et al. 2009).

Known genes and pathways of the different cell types of the developing testis in mouse (modified from Wainwright and Wilhelm 2010). a Postulated molecular pathways underlying Sertoli cell specification, including the regulation of Sry, induction of Sox9 expression, and the maintenance of Sox9 expression. b Differentiation of peritubular myoid (PM) cells is regulated by signaling from Sertoli cells via desert hedgehog (DHH) and its receptor patched 1 (PTCH1). In addition, Dax1 expression in Sertoli cells is required for PM cell differentiation, but the molecular mechanism still remains to be elucidated. The interaction between Sertoli cells and PM cells results in the secretion of extracellular matrix (ECM) molecules by both cell types, which finally leads to the formation of basement membrane (BM). Both cell types contribute distinct components to the ECM, with PM cells secreting fibronectin, collagens, and proteoglycans. c–f The regulation of Leydig cell development via Sertoli cell and Leydig cell interactions. The morphogen DHH is secreted by Sertoli cells and induces Leydig cell specification through its receptor PTCH1. Signaling via the receptor NOTCH3 and its effector HES1 is crucial for the maintenance of the progenitor population and the restriction of their differentiation to fetal Leydig cells. Platelet-derived growth factor A (PDGFA) signaling via the receptor PDGFRα plays a role in Leydig cell differentiation. In addition, Sertoli cell expressed DAX1/NR0B1 has been implicated in Leydig cell survival. As Leydig cells start to differentiate, they start expressing genes required for steroid synthesis, such as side chain cleavage (Scc). Differentiated Leydig cells synthesize testosterone using the four enzymes P450 side chain cleavage (SCC), 3-β-hydroxysteroid dehydrogenase/Delta-5-4 isomerase type 2 (HSD3B2), cytochrome P450 17-hydroxylase (CYP17), and 17β-hydroxysteroid dehydrogenase 3 (17βHSDIII). The biosynthesis of testosterone leads to the masculinization of the developing embryo. Furthermore, fetal Leydig cells express insulin-like factor 3 (INSL3), which regulates testis decent. Both INSL3 and SCC are regulated by SF1/NR5A1 at the transcriptional level. Gene products from the female pathways known to interact with the male-specific pathway are shown in red. Solid lines in this figure do not indicate if the interactions occur in a direct or indirect manner. Genes/gene products shown in gray are not mentioned in the text, but have been included to provide a more accurate summary of or current knowledge
A large deletion (3 Mb in size) of chromosome 9q34 that includes NR5A1 and LMX1B (LIM homeobox transcription factor 1-beta) was identified in a 46,XY girl with clinical features of genitopatellar syndrome and ovotesticular DSD (Schlaubitz et al. 2007). van Silfhout et al. (2009) found a smaller, 970 kb deletion which includes NR5A1 in a 46,XY DSD female without skeletal features.
Wilms' tumor 1
Wilms' tumor 1 (WT1) encodes a zinc finger transcription factor, which is primarily expressed in embryonic mesodermal tissues such as the urogenital ridge, gonads, and mesonephros (Armstrong et al. 1993). WT1 mutations in humans were first identified in patients with Wilms' tumor, a form of kidney cancer occurring primarily in children (Haber et al. 1990). This tumor is also seen as part of WAGR syndrome (Wilms' tumor, Aniridia, genitourinary anomalies, and mental retardation), a more complex syndrome including other clinical features such as aniridia, genitourinary anomalies, and mental retardation. The gonadal phenotype is recapitulated in other disorders, where mutations in WT1 have been described such as Denys-Drash syndrome (including gonadal abnormalities and renal failure) (Pelletier et al. 1991; Lee et al. 2011) and Frasier syndrome (46,XY gonadal dysgenesis together with glomerolupathy) (Barbaux et al. 1997). The Frasier patients have been found to carry mutations that results in the loss of the WT1 + KTS isoform. This isoform has an additional three amino acids (K—lysine; T—threonine, and S—serine), which are located between the third and fourth zinc finger of WT1. The two different isoforms −KTS and +KTS have been shown to play distinct roles during embryogenesis. The −KTS isoform has been shown to bind to the SRY promoter region, leading to the transactivation of SRY (Hossain and Saunders 2001). Furthermore, the −KTS isoform of WT1 was shown to bind sequences within the Nr5a1 promoter (Wilhelm and Englert 2002). Conversely, the +KTS isoform seems to play a role in the regulation of the SRY transcript. Knocking out this +KTS isoform of WT1 specifically, results in reduced Sry levels (Hammes et al. 2001) presumably mediated by its RNA binding affinity. There is also evidence that this isoform may function synergistically with NR5A1 to increase Amh expression (Arango et al. 1999), which is essential for inhibiting the development of the female Mullerian structures. Mice carrying a mutated Wt1 gene fail to develop kidneys and gonads and often die at embryonic stages due to heart defects (Kreidberg et al. 1993).
Genes required for testis development
The following section will summarize key genes involved in testis development and their corresponding DSD phenotypes (see Table 1 for a summary of the human and mouse phenotypes of the genes described below). Figure 1b illustrates known molecular pathways (from mouse studies) that are important for testis development and putative inhibitory interactions between testis and ovary pathways. Figure 2 shows genes involved in testis determination and maintenance of different testis cell populations.
GATA binding protein 4 and zinc finger protein multitype 2
The evolutionary conserved GATA family of tissue- and organ-specific vertebrate transcription factors (GATA-binding proteins 1 to 6; GATA1 to GATA6) consists of two zinc finger domains. The two zinc fingers have been shown to be necessary for DNA recognition and binding (C-terminal zinc finger), the stabilization of the protein–DNA interaction (N-terminal zinc finger) and the protein–protein interactions of the GATA family members with other transcription co-factors (Evans and Felsenfeld 1989; Molkentin 2000). All family members recognize the consensus target sequence (T/A)GATA(A/G). Of these six family members, two genes, Gata2 and Gata4, are known to be expressed in the fetal mouse gonads. Gata2 expression is detected between E10.5–15.5 in XX gonads and the mesonephros of XX and XY gonads, but its expression is missing in XX gonads lacking germ cells. At E13.5 the expression is restricted to germ cells of XX gonads, suggesting a role for GATA2 in ovarian germ cell development (Siggers et al. 2002). Gata4 is the only GATA family member that is expressed in somatic cells (and not germ cells) in the bi-potential gonad (Heikinheimo et al. 1997; Viger et al. 1998). At E11.5, Gata4 is expressed in the somatic cells of the bi-potential gonad of both sexes but becomes sexually dimorphic at E13.5, with its expression being up-regulated in XY Sertoli cells and down-regulated in interstitial cells. By contrast, Gata4 expression is down-regulated in all cell types in XX gonads. Gata4 expression is maintained in Sertoli cells postnatally and regained in adult ovaries (predominantly in granulosa cells) (Heikinheimo et al. 1997; Viger et al. 1998). GATA4 cooperatively interacts with several proteins, including NR5A1 and zinc finger protein multitype 2 (FOG2) (friend of GATA protein 2 or zinc finger protein, multitype 2; encoded by the Zfpm2 gene) to regulate the expression of several sex-determining genes. Amongst those are SRY, SOX9, and AMH. Those complexes regulate key steroidogenic genes, such as STAR (encoding steroidogenic acute regulatory protein), CYP19A1 (encoding aromatase), INHA (encoding inhibin α-subunit), and HSD3B2 (encoding 3 beta-hydroxysteroid dehydrogenase/delta 5-4-isomerase type 2) (Viger et al. 2008; Nishida et al. 2008; Miyamoto et al. 2008).
Gata4 −/− mice die at around E7.0–E9.5 due to abnormalities in ventral morphogenesis and heart tube formation (Kuo et al. 1997; Molkentin et al. 1997) so analysis of gonadal differentiation is not possible. The role of GATA4 in gonad development has been highlighted by the use of Gata4ki mice that have a p.V217G mutation in the N-terminal zinc finger domain, which disrupts the protein–protein interaction of GATA4 with its cofactor FOG2 (Bouma et al. 2007). These mice have severe anomalies of testis development (Crispino et al. 2001; Tevosian et al. 2002).
In humans, mutations in GATA4 have been associated with congenital heart defects (CHD), whereas other organs were described as being normal in all cases (Garg et al. 2003; Hirayama-Yamada et al. 2005; Nemer et al. 2006; Tomita-Mitchell et al. 2007). A recent study describes a heterozygous loss-of-function mutation in GATA4 (p.G221R) in a family with three 46,XY DSD cases (with either ambiguous genitalia or reduced phallus length) and two 46,XX females with CHD. Functional studies showed failure of the mutated protein to bind and activate the AHM promoter and to bind to FOG2 (Lourenco et al. 2011). A 35-kb deletion downstream of GATA4 was identified by White et al. (2011) in a patient with 46,XY complete gonadal dysgenesis and adrenal hypoplasia congenita. The authors argue that the deletion might affect a regulatory region of GATA4, which could in turn explain the lack of cardiac malformations in this patient.
In vitro, FOG2 represses GATA4-dependent trans-activation of AMH in primary Sertoli cell cultures (Tremblay et al. 2001). Whether FOG2 acts as a transcriptional repressor or activator in the context of gonad development in vivo, in mouse as well as in human, still has to be elucidated, but a role in early gonadogenesis in mouse has been determined using Fog2 null mice. These mice die mid gestation (around E14.5) from a cardiac defect and exhibit a block of gonad development (Crispino et al. 2001; Tevosian et al. 2002). Finelli et al. (2007) identified a translocation including FOG2 in a male patient with hypergonadotropic hypogonadism, supporting a role for FOG2 in mammalian sex determination.
Sex-determining region Y
Testis development is characterized by the expression of SRY, the testis-determining gene on the Y chromosome, in the supporting cell lineage of the genital ridge (Bullejos and Koopman 2001; Wilhelm et al. 2005). SRY was identified using 46,XX testicular DSD male patients, who each had small translocations of material from the Y to the X chromosome (Sinclair et al. 1990). Further studies with 46,XY DSD gonadal dysgenesis female patients revealed numerous point mutations in the SRY gene, demonstrating it was required for testis development. Finally, studies of transgenic mice, showed Sry was sufficient, on its own, to cause XX sex reversal (Koopman et al. 1991). Since then, numerous studies have shown that translocations of the SRY gene account for approximately 90% of 46,XX testicular DSD, whereas point mutations in SRY account for approximately 15% of 46,XY DSD gonadal dysgenesis cases (Cameron and Sinclair 1997). 46,XX testicular DSD is a condition in which genotypically female individuals develop as males. That is the gonads develop varying degrees of testicular tissue and may produce testosterone, leading to virilization of external genitalia. In 46,XY DSD gonadal dysgenesis patients, a dysgenic streak gonad develops instead of a testis.
The expression pattern of SRY and its resulting protein differs between the two species. In mice, Sry expression is limited to a transient period in the genital ridge of male embryos from E10.5 to E12.5, whereas SRY expression in human embryos starts at E41, peaks shortly after, and then remains present at low levels beyond embryogenesis (Hanley et al. 2000). In all mammals studied, SRY is a single-exon gene, but the protein can vary in size. SRY is part of the SOX gene family (SRY-related HMG box), a family of 20 genes in human and mice (Bowles et al. 2000; Schepers et al. 2002), which contain a highly conserved high mobility group (HMG) DNA-binding domain. Outside the HMG domain, SRY shows limited sequence conservation between species, suggesting that this DNA-binding domain is of critical importance. In DSD patients, the vast majority of SRY mutations are located within the HMG domain (Cameron and Sinclair 1997), suggesting that this is the most important domain. The SRY HMG domain contains signal sequences that can direct SRY to the nucleus, where it is assumed to play a role in transcriptional regulation (Poulat et al., 1995). This domain also mediates the sequence-specific DNA-binding ability of SRY, which results in the alteration of chromatin conformation (Giese et al. 1992). However, an additional C-terminal CAG repeat domain, which is not present in the human homolog, has been shown to be essential for male sex determination in the mouse (Bowles et al. 1999).
SRY-related HMG-box genes 9, 8, and 10—the SoxE genes
The SOX gene family consists of 20 members, which all encode for transcription factors. These genes have been divided into 10 subgroups, A to J, according to their sequence homologies (Bowles et al. 2000). Besides SRY, three other members of this gene family are expressed in the XY gonad (Cory et al. 2007; Polanco et al. 2010; Schepers et al. 2003). These members are SOX8, SOX9, and SOX10, which together form the subgroup E within this gene family.
SOX9 was first shown to be associated with testis development in 1994, when mutations in and around SOX9 were identified in human DSD patients (Foster et al. 1994; Wagner et al. 1994).
In mice, Sox9 is expressed in the genital ridge at low levels at E10.5 (Morais da Silva et al. 1996) shortly after the expression of Sry in XY embryos and then shows a dramatic increase in expression levels. This temporal and spatial expression profile in mouse suggested that Sox9 is either a direct or indirect downstream target of SRY. This is supported by the findings that only Sry expressing precursor cells of the bi-potential gonad show an increase in Sox9 expression and subsequently develop into Sertoli cells (Sekido et al. 2004). Odsex (Ods) mice carry a 150-kb deletion, 1.3 Mb upstream of Sox9 (Bishop et al. 2000). These mice show XX female-to-male sex reversal (dependent on the genetic background of mice) (Poirier et al. 2004) mediated by the up-regulation of Sox9 and failed down-regulation in XX gonads, suggesting the presence of a distal repressor element that is deleted in Odsex mice. The existence of distal, long-range regulatory elements of Sox9/SOX9 was confirmed by Sekido and Lovell-Badge 2008, who identified a testis-specific regulatory element in mouse. Subsequent findings in human DSD patients with genomic re-arrangements upstream of SOX9 also point to novel gonad-specific SOX9 regulatory elements (see below).
Downstream targets of SOX9 have been identified, including the genes encoding anti-Müllerian hormone (Amh; (Arango et al. 1999)), Vanin-1 (Vnn; (Wilson et al. 2005)), Pgds (Wilhelm et al. 2007) and cerebellin 4 precursor (Cbln4; (Bradford et al. 2009)) (see Fig. 2).
Due to its role in chondrocyte differentiation, SOX9 loss of function mutations lead to a combined phenotype of 46,XY DSD gonadal dysgenesis and campomelic dysplasia, a syndrome characterized by various severe skeletal malformations (Foster et al. 1994; Wagner et al. 1994; Hanley et al. 2000). Duplications including SOX9 have also been described in a 46,XX testicular DSD patient with complete sex reversal (Huang et al. 1999). 46,XY ovotesticular DSD associated with a 12;17 translocation upstream of SOX9 has also been identified by Refai et al. (2010). Recent genome-wide studies, using a CGH array approach, have identified further genomic rearrangements upstream of the SOX9 gene in patients with varying severity of both 46,XX and 46,XY DSD. In 46,XY DSD patients, a number of deletions upstream of SOX9 have been identified. Pop et al. (2004) identified a 1.5-Mb de novo deletion in a patient affected by the acampomelic form of campomelic dysplasia (ACD) and 46,XY DSD (complete female phenotype) and Lecointre et al. (2009) identified an inherited 960 kb deletion 517 kb to 1.477 Mb upstream of SOX9 in a family affected by ACD combined with 46, XY DSD gonadal dysgenesis in a 46,XY DSD patient and the ACD-affected mother. White et al. (2011) identified a 1.2-Mb deletion in a 46,XY DSD patient with gonadal dysgenesis but without skeletal malformations. Benko et al. (2011) identified a 240-kb deletion upstream of SOX9 in two cousins affected by isolated 46,XY DSD (one with a complete female phenotype, the other one with ambiguous genitalia). In this familial case, the deletion was inherited by the mothers (who were sisters and both normal females).
In 46,XX DSD cases, a gain in copy number (duplications, as well as a triplication) upstream of SOX9 has been described in a number of patients. Cox et al. (2011) presented an extremely rare familial case of 46,XX complete sex reversal, where two brothers and their parental uncle were diagnosed with 46,XX testicular DSD (SRY negative and without any other developmental or skeletal phenotypes). Genetic analysis revealed a 178-kb duplication 600 kb upstream of SOX9. Vetro and colleagues (2011) identified a 96-kb triplication approximately 500 kb upstream of SOX9 in two 46,XX azoospermic brothers (both SRY negative) and Benko et al. (2011) identified three familial cases of isolated 46,XX DSD with duplications upstream of SOX9 of varying size. It should be noted that XX humans and mice show the opposite phenotypes where there is a loss of copy number upstream of SOX9/Sox9. In humans, a gain of copy number results in female-to-male sex reversal, whereas the Odsex mouse model shows female-to-male sex reversal when they lose one copy of the Sox9 upstream region (see above). The 1.9-Mb upstream region of SOX9 is a gene desert, which is extremely conserved in mammals. Sekido and Lovell-Badge defined a 3.2-kb testis-specific enhancer of Sox9 (TES) in mouse (Sekido and Lovell-Badge 2008), which is located in this gene desert, but not affected by the above mentioned CNVs in DSD patients. Further studies in mouse helped to refine this testis-specific region further to 1.4 kb, which is referred to as Testis Enhancer Sequence core element (TESCO). Functional analysis showed that SRY and NR5A1 were required to bind to this enhancer region in order to initiate Sox9 up-regulation. TESCO seems to play an important role in the testis-specific expression of Sox9 in mice. However, in human DSD patients, the range of deletions, duplications, and translocations described above (see references above and review by (Gordon et al. 2009)), suggests the involvement of multiple testis-specific regulatory elements that are not functionally conserved between human and mouse. Aligning all the CNVs that have currently been identified in DSD patients, results in a 78-kb minimal overlap region, which is located 517–595 kb upstream of the SOX9 promoter (Benko et al. 2011). This region is a highly likely to contain one or several gonad specific regulatory elements, which, when affected by CNVs, lead to DSD of variable severity, depending on the number and nature of the regulatory elements being affected. However, another possible scenario may be that the changes, especially when occurring, e. g., in tandem, influence the genomic architecture needed to activate SOX9 in the proper spacio-temporal manner essential for gonad development. A recent study screened 66 patients with 46,XY DSD gonadal dysgenesis for mutations in the human, TESCO-corresponding region, but failed to identify point mutations in this regulatory region (Georg et al. 2010).
Conditional knockout of Sox9 leads to ovary development in XY mouse embryos (Chaboissier et al. 2004; Barrionuevo et al. 2006) and over-expression of Sox9 in XX embryos results in the development of testis (Bishop et al. 2000; Vidal et al. 2001). The importance of Sox9 is highlighted in that all reported cases of XY sex reversal show a disturbance in the expression level or function of SOX9. Therefore, it is surprising that Sox9 expression does not seem important for the maintenance of Sertoli cell fate and testis cord integrity. XY embryos with null mutations of Sox9 show normal embryonic testis development after E13.5 and are initially fertile, but become infertile at the age of about 5 months (Barrionuevo et al. 2009). This demonstrates that SOX9 is dispensable at later stages of testis development, which may be due to redundancy of the related factors SOX8 and SOX10. The importance of Sox9 in testis differentiation, along with the close evolutionary relationship and the expression patterns it shares with the two other SoxE family members, raises questions about the functional role of Sox8 and Sox10 during gonadogenesis.
Initially, Sox8 was shown to be specifically expressed in the testis cords at E13.5. This and its ability to induce Amh (anti-Muellarian hormone) expression in vitro, suggested a role of this member of the SOX gene family member in male sex determination, testicular differentiation, or germ cell development (Schepers et al. 2002, 2003). However, the first studies of Sox8 ablated mice failed to result in abnormal sexual development and resulted only in idiopathic weight loss and reduced bone density phenotypes (Sock et al. 2001). Closer analysis of Sox8 −/− mice by O'Bryan et al. (2008) showed that Sox8 −/− males rarely produced litters, while Sox8 +/− males and Sox8 −/− females appeared reproductively normal. This study shows an essential role for Sox8 in the maintenance of male fertility beyond the first wave of spermatogenesis. Loss of Sox8 resulted in progressive degeneration of the seminiferous epithelium through perturbed physical interactions between Sertoli cells and the developing germ cells. Double knockout studies of Sox8 and Sox9 showed that SOX8 reinforces SOX9s role in testis differentiation (Chaboissier et al. 2004; Barrionuevo et al. 2006). Double knockout testis showed mainly complete absence of testis cord formation (tissue-specific Sox9 flox/flox testis showed a variable degree testis cord formation and abnormal coelomic vessel formation) (Chaboissier et al. 2004). These results, together with the above mentioned in vitro studies regarding SOX8s ability to bind and synergistically activate Amh expression alongside NR5A1, support the idea of redundancy for SOX8 and SOX9 in testis differentiation. It has been postulated that the interchangeable roles of SOX8 and SOX9 are due to their shared ancestry and high sequence, as well as structural homology (Koopman 2005).
In Sox10-null mice, no testis phenotype has been described. Sox10, over-expression studies in XX gonads showed that SOX10 is sufficient to induce testis differentiation (Polanco et al. 2010), suggesting that, although it might not be necessary for testis formation, Sox10 still can function as a testis-determining gene. Human 46,XX testicular DSD patients who are masculinized or incompletely feminized and who have a duplication of the region encompassing SOX10, amongst a number of other genes, have been described (Aleck et al. 1999; Cantu et al. 1981; Nicholl et al. 1994; Seeherunvong et al. 2004). The possibility that other genes within this duplicated region are responsible for this phenotype, either solely or in combination with SOX10, cannot be excluded. However, SOX10 is a strong candidate for the causative gene within this region. It is interesting to note that Sox9 is upregulated in Sox10-transgenic XX gonads. It would be worthwhile investigating whether overexpression of Sox8 and or Sox10 would be sufficient to rescue the Sox9 knockout phenotype.
Desert hedgehog
Desert hedgehog (DHH) is a member of the hedgehog family of signaling molecules, which also includes sonic hedgehog (SHH) and indian hedgehog (IHH) (Ingham 1998). Of the three family members, Dhh is the only one that is expressed in somatic cell population of the developing XY mouse gonad from E11.5 and continues later in Sertoli cells. No expression can be detected in XX ovaries at any stage (Bitgood et al. 1996; Yao and Capel 2002; Beverdam and Koopman 2006). DHH binds to its receptor Patched 1 (PTCH1), which is expressed shortly after DHH. PTCH1 is bound to the membrane of Leydig and peritubular myoid cells, and its expression is up-regulated via DHH (Clark et al. 2000; Yao and Capel 2002). Dhh null mice show disrupted testis cord formation due to abnormal peritubular tissue (Clark et al. 2000; Pierucci-Alves et al. 2001). DHH seems to be necessary for the up-regulation of Sf1 in Leydig cells (Yao et al. 2002). Several mutations of DHH have been described in patients affected by 46,XY partial or complete gonadal dysgenesis. The first case was described by Umehara et al. (2000). The patient was affected by 46,XY partial gonadal dysgenesis and minifascicular neuropathy. A homozygous missense mutation at the initiation codon in exon 1 (c.T2C; p.M1T) was identified in the patient. The father carried the same mutation in a heterozygous state, showing that the phenotype displays a recessive mode of inheritance. Further studies identified a homozygous substitution in exon 2 (c.T485C; p.L162P) in one patient with 46,XY complete gonadal dysgenesis (CGD) and a homozygous frame-shift deletion in exon 3 (c.1086delG), which results in a premature stop codon four codons after the deletion in two patients with 46,XY CGD (Canto et al. 2004). The later mutation has also been identified in two patients with 46,XY partial gonadal dysgenesis (PGD), but in a heterozygous state (Canto et al. 2005). Recently, Das et al. (2011) described two novel heterozygous mutations (c.271_273delGAG; p.D60del and c.57_60dupAGCC resulting in premature translational termination) in DHH in two patients affected by 46,XY CGD.
Nuclear receptor subfamily 0 group B, member 1
The dosage sensitive gene nuclear receptor subfamily 0 group B, member 1 (NR0B1) (dosage sensitive sex reversal, adrenal hypoplasia critical region, X chromosome gene 1), encodes DAX1 on chromosome Xp21.2. Mutations and deletions of NR0B1 result in congenital adrenal hypoplasia (CAH), whereas duplications of NR0B1 in 46,XY individuals lead to gonadal dysgenesis and a female phenotype (Bardoni et al. 1994). CAH patients develop testis, but the testis cords appear to be disorganized and hypogonadotropic hypogonadism occurs in these patients. Duplications of NR0B1 have been described in a small number of other 46,XY gonadal dysgenesis cases. Two studies identified duplications of NR0B1 in patients with isolated 46,XY DSD complete gonadal dysgenesis (Barbaro et al. 2007; White et al. 2011) and a unique 257 kb deletion located 250 kb upstream of NR0B1 was reported in a 46,XY female with complete gonadal dysgenesis (Smyk et al. 2007). This deletion presumably affects regulatory elements of NR0B1 and thus results in dysregulation of its expression. As duplications of NR0B1 resulted in 46,XY male-to-female sex reversal, it led to the concept of NR0B1 as an “anti-testis” gene. However, XY Nr0b1 null mice also show sex reversal, which suggest a potential “pro-testis” role of Nr0b1. Ludbrook and Harley (2004) propose a dosage-based mechanism that allows both a pro- and anti-testis function in an attempt to resolve these apparently contradictory roles of NR0B1(Ludbrook and Harley 2004).
Doublesex and mab-3 related, transcription factor 1
Doublesex and mab-3 related, transcription factor 1 (DMRT1) is located at the tip of chromosome 9p in human and encodes the transcription factor DMRT1 (doublesex and mab-3 related transcription factor 1). In some vertebrates, DMRT1 is up-regulated in the developing genital ridge of XY embryos (Moniot et al. 2000; Smith et al. 1999) with continued expression confined to Sertoli cells. Interestingly, DMRT1 represents one of the few sex determining genes that is shared between different species and phyla. Orthologous genes are found in a medaka species (Dmy) on the Y chromosome (Lutfalla et al. 2003) and in chicken (Dmrt1) on the Z sex chromosome (Nanda et al. 1999). Both, Dmy in medaka and Dmrt1 in chicken have been shown to be testis-determining genes (Matsuda et al. 2002; Smith et al. 2009). Interestingly, in platypus, a single copy of Dmrt1 is located on chromosome X5 (Grutzner et al. 2004) although its function, if any, in sex determination is unknown. All these genes encode a protein containing a DM domain as a DNA-binding motif (Raymond et al. 1998). In chicken and medaka, these genes are believed to initiate the molecular network leading to testis development (Smith et al. 2009; Matsuda et al. 2002; Nanda et al. 2002). In humans and mice, DMRT1 is required for the postnatal maintenance of Sertoli and germ cells. Matson et al. (2011) have recently shown in mouse that Dmrt1 and Foxl2 form another regulatory network necessary for maintenance of the testis during adulthood. Loss of Dmrt1 in mouse Sertoli cells leads to the upregulation of Foxl2, amongst other genes. This induces the reprogramming of Sertoli cells into granulosa cells. Subsequently theca cells form, oestrogen is produced and germ cells appear feminized.
Dmrt1-null mice develop normal gonads but show severely impaired testis development from postnatal day 2 resulting in dysgenic testes (Kim et al. 2007a; Raymond et al. 2000) and feminized germ cells (Matson et al. 2011). Similarly, human patients with DMRT1 hemizygosity are associated with dysgenic testis and 46,XY DSD gonadal dysgenesis (Crocker et al. 1988; Raymond et al. 1998; Veitia et al. 1998). In humans, heterozygosity of DMRT1 is associated with DSD. Deletions of chromosome 9p24 are associated with varying degrees of 46,XY DSD gonadal dysgenesis in about 70% of cases with this chromosomal rearrangement (Barbaro et al. 2009; Raymond et al. 1999; Calvari et al. 2000). This region contains: DMRT1, DMRT2, and DMRT3 and other genes, such as FOXD4 (Forkead box protein D4), INSL4, and INSL6 (insulin-like 4, 6 protein). Mutations or rearrangements of DMRT1 are the most likely cause of the 46,XY DSD phenotypes. However, in humans, there have been no mutations identified within the DMRT1 gene itself, jet.
Fibroblast growth factor 9
FGF9 encodes fibroblast growth factor 9 (FGF9), one of a number of growth factors that play a role in various developmental processes such as cell proliferation, cell survival, cell migration, and cell differentiation. Fgf9 is expressed in the bi-potential gonad immediately after the expression of Sry. Mice null for Fgf9 show male-to-female sex reversal along with impaired development of Sertoli cells. However, this is only evident on some genetic backgrounds, but not on others (Colvin et al. 2001; Schmahl et al. 2004). In the absence of Fgf9, Sox9 expression is not maintained, Sertoli cells fail to differentiate, testis development is aborted and the resulting somatic cells express genes characteristic for ovarian development and the female pathway. There is data supporting the concept that SRy and NR5A1 initiate a positive feedback loop by up-regulating SOX9, which in turn up-regulates FGF9, further increasing Sox9 expression (Kim et al. 2006). FGF9 mediates its molecular function through the membrane-bound receptor FGFR2 (fibroblast growth factor receptor 2). FGFR2 is integrated in the plasma membrane of progenitor Sertoli cells and is critical for Sertoli cell proliferation and differentiation in the developing testis. Although no mutations in FGFR2 have yet been reported in human DSD patients (Kim et al. 2007b), due to its crucial role in Sertoli cell proliferation and differentiation, mutations in this gene could account for some unexplained cases of DSD.
A new role for MAP kinases MAP3K1 and Map3k4 in gonad development
In a linkage study of a large family with DSD, a putative testis determining gene was mapped to a region on the long arm of chromosome 5 (Jawaheer et al. 2003). A follow-up study identified a mutation in the gene MAP3K1, which segregated with the phenotype in the family. Further mutations within the MAP3K1 gene were identified within a second family and two out of eleven sporadic cases analyzed. The mutations found in the first family and in the two sporadic cases were shown to alter the phosphorylation of MAP3K1 downstream targets, such as p38 and ERK1/2, and to enhance the binding of RHOA to the MAP3K1 complex. Map3k1 is expressed in the mouse gonad prior to, and following, sex determination but Map3k1 null mice do not show a gonadal phenotype. The spatiotemporal expression profile of Map3k1, together with the identification of mutations within the MAP3K1 gene in cases of human 46,XY DSD strongly implicates the mitogen-activated protein kinase (MAPK) pathway in normal human and mouse sex determination (Pearlman et al. 2010). However, a mutation in another member of the MAPK family, Map3k4, was recently shown to be associated with XY sex reversal in mice, providing further evidence, that mutations in genes of the MAPK signaling pathways may be associated with DSD (Bogani et al. 2009; Bashamboo et al. 2010).
Genes required for ovary development
While many genes have been identified as part of the testis developmental pathways, until recently little was known about the molecular mechanisms underlying ovarian development. Figure 1c gives an overview of the current knowledge of molecular mechanisms leading to ovarian development (see also Table 1 for a summary of the mouse and human phenotypes of genes that are described below).
Wingless-type MMTV integration site family, member 4
WNT4 encodes member 4 of the wingless-type MMTV integration site family, a family of locally acting signaling molecules that are known to play a role in a range of developmental processes. Wnt4 is expressed in the mesonephric mesenchyme and coelomic epithelium in mouse as early as E9.5. At E11, it is expressed in the mesonephros and the bi-potential gonad of both sexes but is then downregulated in the developing testis at around E11.5. The expression persists in the mesonephros as well as the developing ovary and the mesenchyme surrounding the Müllerian ducts (Stark et al. 1994; Vainio et al. 1999). WNT4 plays a role in Mullerian duct formation, oocyte development, and sex-specific patterning of the vasculature (Heikkila et al. 2005). Lack of Wnt4 in the mouse results in partial female-to-male sex reversal, suggesting that WNT4 acts to positively regulate ovary differentiation. The gonads of XX embryos appear to be masculinized, round shaped, unencapsulated, and associated with a fat body but do not form testis cords or express Sertoli cell markers. These mice lack Müllerian ducts and retain Wolffian ducts, have degenerating oocytes, and produce testosterone (Vainio et al. 1999). A human patient resembling this Wnt4 null mouse phenotype was shown to carry a mutation in WNT4 (Biason-Lauber et al. 2004). Ectopic over-expression of Wnt4 in mice resulted in disruption of testis-specific vasculature (the coelomic vessel still forms, but the structure and branching seem to be abnormal) and inhibition of testosterone synthesis in XY embryos (Jordan et al. 2003). As the coelomic vessel forms in XX embryos with WNT4 over-expression, it suggests either, that WNT4 is not the only factor required to suppress male-specific vasculature or that the testes express another factor that is able to compensate for WNT4-mediated repression. Yao et al. (2004) propose that WNT4 regulates vascular boundaries and the maintenance of germ cell survival in the ovary via follistatin (Fst) as both Wnt4 and Fst null ovaries develop the male-specific coelomic vessel and show a substantial apoptotic loss of germ cells at E16.5. Wnt4 −/− ovaries show a loss of Bmp2 and Fst expression between E12.5 and E14.5 and Fst −/− ovaries (Matzuk et al. 1995) have normal Bmp2 and Wnt4 expression within that time frame. These results suggest that WNT4 acts upstream of (and directly or indirectly activates) Bmp2 and Fst in the developing ovary (Yao et al. 2004). Wnt4 −/− XY embryos do not show complete sex reversal, suggesting that inactivation of additional genes are required to disrupt the male-specific pathways and promote female ovarian development (Kim et al. 2006; Kim and Capel 2006).
Studies of in vitro cultured XX gonads showed that addition of exogenous FGF9 induces Sox9 and blocks Wnt4 expression. In Wnt4 −/− XX gonads SOX9 and FGF9 were found to be transiently up-regulated between E11.5 and E12, indicating that WNT4 would usually act as repressor of the male pathway by suppressing Sox9 up-regulation. However, no mechanism has been proposed to explain how this interaction occurs (Kim et al. 2006).
Human patients with duplications of chromosome 1p, including WNT4, show a broad range of gonadal anomalies including complete sex reversal (Jordan et al. 2001).
The Wnt morphogens are known to signal through binding to integral membrane-bound co-receptors LRP5 and LRP6 (low-density lipoprotein receptor-related protein 5 and 6). These co-receptors heterodimerize with the integral membrane receptor Frizzled and thus transfer the signal into the cytoplasm of the cell. Within the cell, this leads to the dephosphorylation of cytoplasmic β-catenin (via the glycogen synthase kinase 3 beta/casein kinase 1 (GSK3-β/CK-1) complex), which results in the stabilization and accumulation of β-catenin in the cytoplasm. At a certain threshold, dephosphorylated β-catenin enters the nucleus and interacts with the TCF/LEF/BCL9 (transcription factor/lymphoid enhancer-binding factor/B-cell CLL/lymphoma 9 protein) complex of transcription factors. This results in the transcriptional activation of downstream target genes. In the absence of Wnt signaling molecules, β-catenin is phosphorylated and subjected to proteosomal degradation, which results in the repression of target genes (Bernard and Harley 2007). It seems likely that WNT4 uses this canonical β-catenin pathway in the developing gonad.
R-spondin 1
R-spondin 1 (RSPO1) encodes a secreted factor that can stabilize β-catenin as part of the canonical Wnt-signaling pathway and is expressed at high levels in mouse, as well as human gonads around the critical time of gonad development (mouse: (Parma et al. 2006; Chassot et al. 2008a; Smith et al. 2008), human: (Tomaselli et al. 2011)). Its role in ovarian development was first implicated when a homozygous single nucleotide insertion within the RSPO1 coding sequence was identified in four 46,XX testicular DSD patients from a consanguineous family. In addition, a homozygous exonic deletion was identified in an unrelated sporadic case of 46,XX testicular DSD (Parma et al. 2006). This study was the first to show that inactivation of a single gene can cause female-to-male sex reversal. A subsequent study identified a homozygous splice-site mutation in a 46,XX ovotesticular DSD patient (Tomaselli et al. 2008). In vivo studies on ovotesticular tissue from this patient showed no changes in CTNNB1 (β-catenin) mRNA levels, but β-catenin protein levels were decreased. In addition, WNT4 mRNA expression was significantly decreased compared to normal ovaries. These results suggest that RSPO1 acts either upstream of WNT4 or synergistically with WNT4 during early ovarian development (Tomaselli et al. 2011). The conserved expression pattern of RSPO1 in other vertebrates suggests that it is a critical gene in ovarian development. XX Rspo1 knockout mice have masculinised gonads, but do not show complete sex reversal (Tomizuka et al. 2008). This phenotype is similar to Wnt4 null mice, and provides further evidence, that these genes act within the same pathway to activate β-catenin. Wnt4, Fst, and Bmp2 were down-regulated in Rspo1 −/− XX ovaries at E13.5, suggesting that RSPO1 acts as a positive regulator of Wnt4 (Yao et al. 2004; Tomizuka et al. 2008; Chassot et al. 2008b; Trautmann et al. 2008). Chassot et al. (2008b) showed that in the absence of Rspo1, the canonical Wnt-signaling pathway was not activated, and there was no increase in Wnt4 expression. The phenotype of Rspo1 null ovaries could be rescued by stabilized β-catenin expression in ovarian somatic cells, suggesting that β-catenin acts downstream of Rspo1 to block testis development in XX gonads (Chassot et al. 2008b).
In 46,XX female individuals, both WNT4 and RSPO1 are known to promote ovarian development and repress testis development. In 46,XY DSD gonadal dysgenesis females with a duplication of chromosome 1p, that includes both WNT4 and RSPO1, testis development is suppressed and the ovarian pathway dominates. WNT4 as well as RSPO1 perform their actions in the somatic cell population of the ovary via β-catenin and the canonical Wnt-signaling pathway.
Conditional knockout of β-catenin in ovarian somatic cells (the putative precursors of granulosa cells) leads to ovarian defects similar to those of Rspo1 and Wnt4 null mice (Manuylov et al. 2008). Ectopic over-expression of a stabilized form of β-catenin in mouse somatic cells, leads to the disruption of testis development in XY embryos and causes male-to-female sex reversal (Maatouk et al. 2008). This suggests that β-catenin is a key ovarian and anti-testis signaling molecule. As a consequence, 46,XY DSD gonadal dysgenesis female patients, without any apparent mutations in known testis-determining genes, could be the result of stabilized β-catenin which then undermines the role of SRY in the developing testis by inhibiting its critical downstream target SOX9 (Maatouk et al. 2008).
Forkhead box L2
Forkhead box L2 (FOXL2) encodes the forkhead transcription factor 2, which is a member of the winged helix/forkhead transcription factors (Hannenhalli and Kaestner 2009). A role for FOXL2 in ovarian development was first suggested with the identification of a deletion near FOXL2 in the polled/intersex syndrome (PIS) goat (Pailhoux et al. 2001). In humans, mutations in FOXL2 are associated with blepharophimosis/ptosis/epicanthus inversus syndrome (BPES), which is characterized by eyelid defects with or without ovarian dysfunction, such as premature ovarian failure (POF) (Crisponi et al. 2001). In addition, BPES patients have been shown to carry a 7.4-kb deletion of a cis-regulatory region, which is thought to interrupt the FOXL2 promoter (D’Haene et al. 2009).
Foxl2 −/− mice develop normal testis during embryogenesis but show masculinization of the supporting cells in the postnatal gonad (Ottolenghi et al. 2005). Foxl2 null and Wnt4/Foxl2 double knockout ovaries have reduced Fst and Bmp2 expression at E13.5. In Wnt4 −/− ovaries, the expression of Fst is back to wild-type levels at birth (postnatal day 0; P0), whereas the expression of Fst remains decreased in both, the Foxl2 −/− and Wnt4 −/−, Foxl2 −/− double knockout ovaries. Bmp2 expression remains significantly reduced in all three mouse strains. In vitro studies show that FOXL2 and BMP2 up-regulate Fst expression. Exogenous BMP2 increased Fst expression in ex vivo studies on fetal mouse gonads, but this was counteracted by the BMP antagonist Noggin (Kashimada et al. 2011). Together this data suggest that FOXL2 cooperates with BMP2 to ensure correct expression of Fst in the developing ovary, whereas WNT4 is necessary for the initiation, but not the maintenance of Fst expression.
Foxl2 −/−, Wnt4 −/− double knockout XX animals show testis differentiation and the development of male germ cells (Ottolenghi et al. 2007). Surprisingly, conditional knockout of Foxl2 in the adult mouse ovary results in trans-differentiation into testis (Uhlenhaut et al. 2009). The ovarian structures become more testis-like, with tubular features and Sertoli-like cells. Gene expression was investigated using in situ hybridization and showed that some male-specific genes were up-regulated, including Sox9. These results indicate that Foxl2 has a role in maintaining the ovary even during adulthood, presumably by suppressing the pro-testis action of Sox9/Fgf9. There is experimental evidence that this role is carried out together with ESR1/ESR2 (estrogen receptor 1 and 2) by binding to sequences within the TESCO region and thus controlling Sox9 expression (Bagheri-Fam et al. 2010). This study may be clinically relevant in humans, as somatic mutations in FOXL2 could be the underlying cause of ovarian dysfunction in adult females.
As previously mentioned (see DMRT1 section), Matson et al. (2011) have recently shown that loss of Dmrt1 in mouse leads to the upregulation of Foxl2, theca cell formation, estrogen production, and eventually feminized germ cells.
Identifying novel genes in the sex determining pathway
We are currently still unable to determine the causative mutation in a relatively high percentage of DSD patients. This together with the recent discovery of several new genes in patients with previously unexplained DSD lend support to the idea that both, the genes involved in gonad development, as well as the etiology of DSD, are still incompletely understood. Mutations in regulatory elements of known DSD/gonad development genes, as well as novel gonad-determining genes, are likely to be responsible for a significant percentage of yet unexplained DSD cases.
Gene expression profiling, using microarrays
Over the years, several strategies were used to identify novel genes involved in gonad development. One approach was the analysis of differentially expressed genes using microarrays. Several studies performed these types of analyses using mouse (Beverdam and Koopman 2006) and human (Houmard et al. 2009) gonadal tissues. While the lists of genes that show sexually dimorphic expression patterns identified many of the known gonad genes (although rarely SRY) this approach has not lead to the discovery of any novel genes involved in sex determination and the etiology of DSD.
CNV arrays—genomic rearrangements associated with DSD
As previously mentioned, CNV arrays have been used over the last couple of years to screen for and identify copy number variations in patients affected by DSD. Throughout this review, we mentioned a number of cases where this approach lead to the identification of the causative mutation in DSD patients. Nevertheless, there are still various genomic rearrangements associated with human DSD phenotypes that have been reported but where no causative gene could be identified.
A duplication of 1p has been shown to be associated with 46,XY DSD gonadal dysgenesis, although the gene(s) causing the DSD phenotype are still unknown (Wieacker and Volleth 2007). Several deletions of terminal 10q have been associated with 46,XY DSD gonadal dysgenesis together with other somatic anomalies. The gene responsible for this DSD phenotype is still unknown, but EMX2 has been suggested to be a likely candidate (Ogata et al. 2000). Deletions and duplications of chromosome 22q11.2 have been identified in three cases of 46,XX testicular DSD (which were SRY-negative) (Aleck et al. 1999; Erickson et al. 2003; Seeherunvong et al. 2004). The causative gene(s) haven't been identified, but this region contains the MAPK1 gene, which appears to play an important role in proper testis development. Deletions on chromosomes 11p13 and 9p24 were identified using CGH arrays and were found to be associated with syndromic forms of 46,XY DSD (Le Caignec et al. 2007; Vinci et al. 2007). Other CGH arrays revealed a novel recurrent 15q24 microdeletion syndrome with the deletions being between 1.7 and 3.9 Mb in size. The syndrome is characterized, amongst other features, by genital anomalies in male, including micropenis and hypospadias (Sharp et al. 2007; Andrieux et al. 2009), but the causative genes have not been identified.
SRY-related HMG-box gene 3—an unexpected role in 46,XX DSD
By far, the most successful approach to the identification and functional analysis of gonad genes used a combination of human DSD patient studies and mouse models. The recently published example of SOX3 highlights this once more.
SOX3 is another member of the SOX gene family of transcription factors. SOX3 shows near sequence identity to SRY so it has been proposed that the Y-linked SRY evolved from the X-linked SOX3 gene (Foster et al. 1992; Graves 2001). This process occurred as the mammalian sex chromosomes evolved and differentiated. SOX3 is not expressed in the developing gonads in either sex, and loss-of-function mutations in SOX3 do not affect sex determination in humans or mice. However, a transgenic mouse line over-expressing Sox3 showed ectopic expression of this gene in the bi-potential gonad. XX embryos showed female-to-male sex reversal, and further analyses suggest that SOX3 induced testis differentiation by up-regulating Sox9 expression in these animals through a similar mechanism to SRY (Sutton et al. 2011). The same study identified three 46,XX testicular DSD patients with genomic rearrangements: two duplications including SOX3 and one deletion upstream of SOX3, in the putative SOX3 regulatory region. It was hypothesized that these rearrangements caused ectopic expression of SOX3 in the embryonic gonad, where it was able to substitute for the absence of SRY and drive testis development. Together, these findings suggest that SOX3 and SRY are functionally interchangeable in testis determination and lend further support to the hypothesis that SRY evolved from SOX3.
Conclusion and future directions of DSD research
Since the discovery of SRY in 1990, we have significantly increased our knowledge of genes and gene networks involved in gonad development and the etiology of DSD. Nevertheless, still unexplained DSD cases, as well as gaps in the network of testis and ovary development show us that there is still much to understand.
With the development of more powerful algorithms, comparative genomic hybridization (CGH)/SNP chip arrays have the potential to detect subtle rearrangements of genes involved in gonad determination and differentiation. These two arrays-based methods allow the detection of rearrangements in putative regulatory elements, which, if mutated, may cause DSD. The use of microarrays to detect CNVs has had some success in identifying causative mutations in unexplained cases of DSD. However, this technique is limited to the detection of large rearrangements and may miss smaller genomic rearrangements or point mutations. Familial cases of both 46,XY DSD and 46,XX DSD often show a broad range of phenotypes in affected individuals within the same family, including instances, where the underlying genetic mutation has been identified (Temel et al. 2007; Lourenco et al. 2009). This phenotypic variability may be explained by additional mutations in other genes up and downstream of the target gene, which may interact or influence its activity. This has been shown in a familial case of hypogonadotropic hypogonadism, where a compound heterozygous GNRHR (gonadotropin-releasing hormone receptor) and a heterozygous FGFR1 mutation were identified (Pitteloud et al. 2007). In addition, almost all cases of non-syndromic ovotesticular DSD remain unexplained although there have been considerable genetic analyses of candidate genes (Temel et al. 2007; McElreavey et al. 1992). Although mutations in the androgen receptor (AR) gene are the most common genetic cause of 46,XY DSD, pathogenic mutations in 46,XY under-androgenized patients who are suspected to have mutations in this gene are only found in about 50% of affected individuals (Audi et al. 2010). The analyses of presumably multifactoral DSD phenotypes, such as hypospadias or cryptorchidism, have rarely identified the genetic cause, although epidemiological studies give clear evidence of a major genetic component in these conditions (Fukami et al. 2006; Schnack et al. 2008; Kohler et al. 2009). Next generation sequencing (NGS) or massively parallel sequencing approaches (MPS), such as whole exome, single chromosome, or targeted sequencing of sets of candidate genes (together referred to as targeted sequencing from this point), as well as whole genome sequencing, should provide substantially higher resolution for mutation detection and should assist in resolving unexplained cases of DSD. Whole genome sequencing, which combines the advantages of all other methods, is a powerful tool to detect the range of genomic mutations, including SNPs and all size ranges of genomic re-arrangements and CNVs, in coding as well as non-coding and putative regulatory regions, in a non-biased manner. Currently, this method is still costly and requires immense bioinformatic analysis. However, as the cost of whole genome sequencing continues to decrease and as bioinformatic analysis improves, this will eventually become the method of choice for many researchers. In the interim, targeted sequencing approaches and whole exome sequencing offer a great opportunity to gain new insights into key genes required for gonad development with a lower level of bioinformatic complexity.
Despite the differences between human and mouse and the improvements in the techniques and bioinformatics, comparative genomics will still play a key role. Given the increasing resolution of the new techniques and the increasing power of bioinformatics, we are likely to identify an increasing number of new genes, which are mutated or rearranged in DSD patients. Comparative genetics and mouse models will be essential for establishing the biological function of mutations that do not lie within known DSD genes and crucial in verifying novel genes with a role in gonad development.
Abbreviations
- ACD:
-
Acampomelic campomelic dysplasia
- AMH:
-
Anti-Muellerian hormone
- AR:
-
Androgen receptor
- BMP2:
-
Bone morphogenic protein 2
- BPES:
-
Blepharophimosis/ptosis/epicantus inversus syndrome
- CAH:
-
Congenital adrenal hypoplasia
- CBLN4:
-
Cerebellin 4 precursor
- CBX2:
-
Chromobox homolog 2
- CHD:
-
Congenital heart defect
- CK-1:
-
Casein kinase 1
- CNV:
-
Copy number variation
- CTNNB1:
-
β-catenin
- CYP19A1:
-
Cytochrome P450 family XIX, subfamily, A, polypeptide 1 or aromatase
- DAX1:
-
Dosage sensitive sex reversal adrenal, hypoplasia critical region, X, chromosome gene 1
- DHH:
-
Desert hedgehog
- DMRT1 2, 3:
-
Doublesex and mab-3 related, transcription factor 1, 2, 3
- Dmy:
-
DM-domain gene on the Y chromosome
- Dpc:
-
Day post coitum
- DSD:
-
Disorders of sex development
- E:
-
Embryonic day
- EMX2:
-
Empty spiracles homeobox 2
- ems:
-
Empty spiracles
- ERK1:
-
Mitogen-activated protein kinase 3
- ERK2:
-
Mitogen-activated protein kinase 1
- FGF9:
-
Fibroblast growth factor 9
- FGFR1 2:
-
Fibroblast growth factor receptor 1, 2
- FOG2:
-
Zinc finger protein multitype 2
- FOXD4:
-
Forkhead box protein D4
- FOXL2:
-
Forkhead box L2
- FST:
-
Follistatin
- GATA4:
-
GATA binding protein 4
- GNRHR:
-
Gonadotropin-releasing hormone receptor
- GSK3-β:
-
Glycogen synthase kinase 3 β
- HMG:
-
High mobility group
- HSD3B2:
-
3 beta-hydroxysteroid dehydrogenase/Delta 5-4-isomerase type 2
- IGFs:
-
Insulin-like growth factors
- IHH:
-
Idiopathic hypogonadotrophic hypogonadism
- IHH:
-
Indian hedgehog
- INHA:
-
Inhibin α-subunit
- INSL4 6:
-
Insulin-like 4, 6 protein
- LHX9:
-
LIM homeobox 9
- LMX1B:
-
LIM homeobox transcription factor 1-β
- LRP5 6:
-
Low density lipoprotein receptor related, protein 5, 6
- M33:
-
Mouse Polycomb group member M33
- MAP3K1:
-
Mitogen-activated protein kinase kinase kinase 1
- Map3k4:
-
Mitogen-activated protein kinase kinase kinase 4
- MAPK:
-
Mitogen-activated protein kinase
- MPS:
-
Massively parallel sequencing
- NEIL2:
-
Endonuclease VIII-like 2
- NGS:
-
Next generation sequencing
- NR0B1:
-
Nuclear receptor subfamily 0 group B, member 1
- NR5A1:
-
Nuclear receptor subfamily 5 group A member 1
- Ods:
-
Odsex
- P:
-
Postnatal day
- P38:
-
Mitogen-activated protein kinases 11 12, 13 and 14
- PIS:
-
Polled/intersex syndrome
- PTCH1:
-
Patched 1
- PGDS:
-
Prostaglandin-D synthase
- RHOA:
-
Ras homolog gene family member A
- RSPO1:
-
R-spondin 1
- SF1:
-
Steroidogenic factor-1
- SHH:
-
Sonic hedgehog
- SOX3 8, 9, 10:
-
SRY-related HMG box 3, 8, 9, 10
- STAR:
-
Steroidogenic acute regulatory protein
- SRY:
-
Sex-determining region Y
- TCF/LEF/BCL9:
-
Transcription factor/lymphoid enhancer-binding factor/B cell, CLL/Lymphoma 9 protein
- TES:
-
Testis-specific enhancer of Sox9
- TESCO:
-
Testis enhancer sequence core element
- VNN:
-
Vanin-1
- WAGR syndrome:
-
Wilms' tumor aniridia, genitourinary, anomalies and mental retardation, syndrome
- WNT4:
-
Wingless-type MMTV integration site family, member 4
- WT1:
-
Wilms' tumor 1
- ZFPM2:
-
Zinc finger protein multitype 2
References
Achermann JC, Ito M, Hindmarsh PC, Jameson JL (1999) A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet 22:125–126
Aleck KA, Argueso L, Stone J, Hackel JG, Erickson RP (1999) True hermaphroditism with partial duplication of chromosome 22 and without SRY. Am J Med Genet 85:2–4
Andrieux J, Dubourg C, Rio M et al (2009) Genotype–phenotype correlation in four 15q24 deleted patients identified by array-CGH. Am J Med Genet A 149A:2813–2819
Arango NA, Lovell-Badge R, Behringer RR (1999) Targeted mutagenesis of the endogenous mouse Mis gene promoter: in vivo definition of genetic pathways of vertebrate sexual development. Cell 99:409–419
Armstrong JF, Pritchard-Jones K, Bickmore WA, Hastie ND, Bard JB (1993) The expression of the Wilms' tumour gene, WT1, in the developing mammalian embryo. Mech Dev 40:85–97
Audi L, Fernandez-Cancio M, Carrascosa A et al (2010) Novel (60%) and recurrent (40%) androgen receptor gene mutations in a series of 59 patients with a 46, XY disorder of sex development. J Clin Endocrinol Metab 95:1876–1888
Bagheri-Fam S, Sinclair AH, Koopman P, Harley VR (2010) Conserved regulatory modules in the Sox9 testis-specific enhancer predict roles for SOX, TCF/LEF, Forkhead, DMRT, and GATA proteins in vertebrate sex determination. Int J Biochem Cell Biol 42:472–477
Barbaro M, Oscarson M, Schoumans J et al (2007) Isolated 46, XY gonadal dysgenesis in two sisters caused by a Xp21.2 interstitial duplication containing the DAX1 gene. J Clin Endocrinol Metab 92:3305–3313
Barbaro M, Balsamo A, Anderlid BM et al (2009) Characterization of deletions at 9p affecting the candidate regions for sex reversal and deletion 9p syndrome by MLPA. Eur J Hum Genet 17:1439–1447
Barbaux S, Niaudet P, Gubler MC et al (1997) Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 17:467–470
Bardoni B, Zanaria E, Guioli S et al (1994) A dosage sensitive locus at chromosome Xp21 is involved in male to female sex reversal. Nat Genet 7:497–501
Barrionuevo F, Bagheri-Fam S, Klattig J et al (2006) Homozygous inactivation of Sox9 causes complete XY sex reversal in mice. Biol Reprod 74:195–201
Barrionuevo F, Georg I, Scherthan H et al (2009) Testis cord differentiation after the sex determination stage is independent of Sox9 but fails in the combined absence of Sox9 and Sox8. Dev Biol 327:301–312
Bashamboo A, Ledig S, Wieacker P, Achermann JC, McElreavey K (2010) New technologies for the identification of novel genetic markers of disorders of sex development (DSD). Sex Dev 4:213–224
Benko S, Gordon CT, Mallet D et al (2011) Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J Med Genet 48:825–830
Bernard P, Harley VR (2007) Wnt4 action in gonadal development and sex determination. Int J Biochem Cell Biol 39:31–43
Beverdam A, Koopman P (2006) Expression profiling of purified mouse gonadal somatic cells during the critical time window of sex determination reveals novel candidate genes for human sexual dysgenesis syndromes. Hum Mol Genet 15:417–431
Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ (2004) A WNT4 mutation associated with Mullerian-duct regression and virilization in a 46, XX woman. N Engl J Med 351:792–798
Biason-Lauber A, Konrad D, Meyer M, DeBeaufort C, Schoenle EJ (2009) Ovaries and female phenotype in a girl with 46, XY karyotype and mutations in the CBX2 gene. Am J Hum Genet 84:658–663
Birk OS, Casiano DE, Wassif CA et al (2000) The LIM homeobox gene Lhx9 is essential for mouse gonad formation. Nature 403:909–913
Bishop CE, Whitworth DJ, Qin Y et al (2000) A transgenic insertion upstream of sox9 is associated with dominant XX sex reversal in the mouse. Nat Genet 26:490–494
Bitgood MJ, Shen L, McMahon AP (1996) Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr Biol 6:298–304
Bogani D, Siggers P, Brixey R et al (2009) Loss of mitogen-activated protein kinase kinase kinase 4 (MAP3K4) reveals a requirement for MAPK signalling in mouse sex determination. PLoS Biol 7:e1000196
Bouma GJ, Washburn LL, Albrecht KH, Eicher EM (2007) Correct dosage of Fog2 and Gata4 transcription factors is critical for fetal testis development in mice. Proc Natl Acad Sci USA 104:14994–14999
Bowles J, Cooper L, Berkman J, Koopman P (1999) Sry requires a CAG repeat domain for male sex determination in Mus musculus. Nat Genet 22:405–408
Bowles J, Schepers G, Koopman P (2000) Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev Biol 227:239–255
Bradford ST, Hiramatsu R, Maddugoda MP et al (2009) The cerebellin 4 precursor gene is a direct target of SRY and SOX9 in mice. Biol Reprod 80:1178–1188
Bullejos M, Koopman P (2001) Spatially dynamic expression of Sry in mouse genital ridges. Dev Dyn 221:201–205
Burris TP, Guo W, Le T, McCabe ER (1995) Identification of a putative steroidogenic factor-1 response element in the DAX-1 promoter. Biochem Biophys Res Commun 214:576–581
Calvari V, Bertini V, De Grandi A et al (2000) A new submicroscopic deletion that refines the 9p region for sex reversal. Genomics 65:203–212
Cameron FJ, Sinclair AH (1997) Mutations in SRY and SOX9: testis-determining genes. Hum Mutat 9:388–395
Canto P, Soderlund D, Reyes E, Mendez JP (2004) Mutations in the desert hedgehog (DHH) gene in patients with 46, XY complete pure gonadal dysgenesis. J Clin Endocrinol Metab 89:4480–4483
Canto P, Vilchis F, Soderlund D, Reyes E, Mendez JP (2005) A heterozygous mutation in the desert hedgehog gene in patients with mixed gonadal dysgenesis. Mol Hum Reprod 11:833–836
Cantu JM, Hernandez A, Vaca G et al (1981) Trisomy 22q12 leads to qter: “aneusomie de recombinaison” of a pericentric inversion. Ann Genet 24:37–40
Capel B (1998) Sex in the 90s: SRY and the switch to the male pathway. Annu Rev Physiol 60:497–523
Capel B (2000) The battle of the sexes. Mech Dev 92:89–103
Chaboissier MC, Kobayashi A, Vidal VI et al (2004) Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development 131:1891–1901
Chassot AA, Gregoire EP, Magliano M, Lavery R, Chaboissier MC (2008a) Genetics of ovarian differentiation: Rspo1, a major player. Sex Dev 2:219–227
Chassot AA, Ranc F, Gregoire EP et al (2008b) Activation of beta-catenin signaling by Rspo1 controls differentiation of the mammalian ovary. Hum Mol Genet 17:1264–1277
Clark AM, Garland KK, Russell LD (2000) Desert hedgehog (Dhh) gene is required in the mouse testis for formation of adult-type Leydig cells and normal development of peritubular cells and seminiferous tubules. Biol Reprod 63:1825–1838
Colvin JS, Green RP, Schmahl J, Capel B, Ornitz DM (2001) Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell 104:875–889
Cory AT, Boyer A, Pilon N, Lussier JG, Silversides DW (2007) Presumptive pre-Sertoli cells express genes involved in cell proliferation and cell signalling during a critical window in early testis differentiation. Mol Reprod Dev 74:1491–1504
Cox JJ, Willatt L, Homfray T, Woods CG (2011) A SOX9 duplication and familial 46, XX developmental testicular disorder. N Engl J Med 364:91–93
Crispino JD, Lodish MB, Thurberg BL et al (2001) Proper coronary vascular development and heart morphogenesis depend on interaction of GATA-4 with FOG cofactors. Genes Dev 15:839–844
Crisponi L, Deiana M, Loi A et al (2001) The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet 27:159–166
Crocker M, Coghill SB, Cortinho R (1988) An unbalanced autosomal translocation (7;9) associated with feminization. Clin Genet 34:70–73
D’Haene B, Attanasio C, Beysen D et al (2009) Disease-causing 7.4 kb cis-regulatory deletion disrupting conserved non-coding sequences and their interaction with the FOXL2 promotor: implications for mutation screening. PLoS Genet 5:e1000522
Das DK, Sanghavi D, Gawde H, Idicula-Thomas S, Vasudevan L (2011) Novel homozygous mutations in desert hedgehog gene in patients with 46, XY complete gonadal dysgenesis and prediction of its structural and functional implications by computational methods. Eur J Med Genet 54:e529–e534
de Santa Barbara P, Mejean C, Moniot B et al (2001) Steroidogenic factor-1 contributes to the cyclic-adenosine monophosphate down-regulation of human SRY gene expression. Biol Reprod 64:775–783
Erickson RP, Skinner S, Jacquet H et al (2003) Does chromosome 22 have anything to do with sex determination: further studies on a 46, XX,22q11.2 del male. Am J Med Genet A 123A:64–67
Evans T, Felsenfeld G (1989) The erythroid-specific transcription factor Eryf1: a new finger protein. Cell 58:877–885
Finelli P, Pincelli AI, Russo S et al (2007) Disruption of friend of GATA 2 gene (FOG-2) by a de novo t(8;10) chromosomal translocation is associated with heart defects and gonadal dysgenesis. Clin Genet 71:195–204
Foster JW, Brennan FE, Hampikian GK et al (1992) Evolution of sex determination and the Y chromosome: SRY-related sequences in marsupials. Nature 359:531–533
Foster JW, Dominguez-Steglich MA, Guioli S et al (1994) Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 372:525–530
Fukami M, Wada Y, Miyabayashi K et al (2006) CXorf6 is a causative gene for hypospadias. Nat Genet 38:1369–1371
Garg V, Kathiriya IS, Barnes R et al (2003) GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424:443–447
Georg I, Bagheri-Fam S, Knower KC et al (2010) Mutations of the SRY-responsive enhancer of SOX9 are uncommon in XY gonadal dysgenesis. Sex Dev 4:321–325
Giese K, Cox J, Grosschedl R (1992) The HMG domain of lymphoid enhancer factor 1 bends DNA and facilitates assembly of functional nucleoprotein structures. Cell 69:185–195
Gordon CT, Tan TY, Benko S et al (2009) Long-range regulation at the SOX9 locus in development and disease. J Med Genet 46:649–656
Graves JA (2001) From brain determination to testis determination: evolution of the mammalian sex-determining gene. Reprod Fertil Dev 13:665–672
Grutzner F, Rens W, Tsend-Ayush E et al (2004) In the platypus a meiotic chain of ten sex chromosomes shares genes with the bird Z and mammal X chromosomes. Nature 432:913–917
Haber DA, Buckler AJ, Glaser T et al (1990) An internal deletion within an 11p13 zinc finger gene contributes to the development of Wilms' tumor. Cell 61:1257–1269
Hammes A, Guo JK, Lutsch G et al (2001) Two splice variants of the Wilms' tumor 1 gene have distinct functions during sex determination and nephron formation. Cell 106:319–329
Hanley NA, Hagan DM, Clement-Jones M et al (2000) SRY, SOX9, and DAX1 expression patterns during human sex determination and gonadal development. Mech Dev 91:403–407
Hannenhalli S, Kaestner KH (2009) The evolution of Fox genes and their role in development and disease. Nat Rev Genet 10:233–240
Heikinheimo M, Ermolaeva M, Bielinska M et al (1997) Expression and hormonal regulation of transcription factors GATA-4 and GATA-6 in the mouse ovary. Endocrinology 138:3505–3514
Heikkila M, Prunskaite R, Naillat F et al (2005) The partial female to male sex reversal in Wnt-4-deficient females involves induced expression of testosterone biosynthetic genes and testosterone production, and depends on androgen action. Endocrinology 146:4016–4023
Hirayama-Yamada K, Kamisago M, Akimoto K et al (2005) Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A 135:47–52
Hossain A, Saunders GF (2001) The human sex-determining gene SRY is a direct target of WT1. J Biol Chem 276:16817–16823
Houmard B, Small C, Yang L et al (2009) Global gene expression in the human fetal testis and ovary. Biol Reprod 81:438–443
Huang B, Wang S, Ning Y, Lamb AN, Bartley J (1999) Autosomal XX sex reversal caused by duplication of SOX9. Am J Med Genet 87:349–353
Hughes IA, Houk C, Ahmed SF, Lee PA (2006) Consensus statement on management of intersex disorders. J Pediatr Urol 2:148–162
Ingham PW (1998) Transducing hedgehog: the story so far. EMBO J 17:3505–3511
Jawaheer D, Juo SH, Le Caignec C et al (2003) Mapping a gene for 46, XY gonadal dysgenesis by linkage analysis. Clin Genet 63:530–535
Jordan BK, Mohammed M, Ching ST et al (2001) Up-regulation of WNT-4 signaling and dosage-sensitive sex reversal in humans. Am J Hum Genet 68:1102–1109
Jordan BK, Shen JH, Olaso R, Ingraham HA, Vilain E (2003) Wnt4 overexpression disrupts normal testicular vasculature and inhibits testosterone synthesis by repressing steroidogenic factor 1/beta-catenin synergy. Proc Natl Acad Sci USA 100:10866–10871
Kashimada K, Pelosi E, Chen H et al (2011) FOXL2 and BMP2 act cooperatively to regulate follistatin gene expression during ovarian development. Endocrinology 152:272–280
Katoh-Fukui Y, Tsuchiya R, Shiroishi T et al (1998) Male-to-female sex reversal in M33 mutant mice. Nature 393:688–692
Katoh-Fukui Y, Owaki A, Toyama Y et al (2005) Mouse Polycomb M33 is required for splenic vascular and adrenal gland formation through regulating Ad4BP/SF1 expression. Blood 106:1612–1620
Kim Y, Capel B (2006) Balancing the bipotential gonad between alternative organ fates: a new perspective on an old problem. Dev Dyn 235:2292–2300
Kim Y, Kobayashi A, Sekido R et al (2006) Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination. PLoS Biol 4:e187
Kim S, Bardwell VJ, Zarkower D (2007a) Cell type-autonomous and non-autonomous requirements for Dmrt1 in postnatal testis differentiation. Dev Biol 307:314–327
Kim Y, Bingham N, Sekido R et al (2007b) Fibroblast growth factor receptor 2 regulates proliferation and Sertoli differentiation during male sex determination. Proc Natl Acad Sci USA 104:16558–16563
Kohler B, Lin L, Mazen I et al (2009) The spectrum of phenotypes associated with mutations in steroidogenic factor 1 (SF-1, NR5A1, Ad4BP) includes severe penoscrotal hypospadias in 46, XY males without adrenal insufficiency. Eur J Endocrinol 161:237–242
Koopman P (2005) Sex determination: a tale of two Sox genes. Trends Genet 21:367–370
Koopman P, Munsterberg A, Capel B, Vivian N, Lovell-Badge R (1990) Expression of a candidate sex-determining gene during mouse testis differentiation. Nature 348:450–452
Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R (1991) Male development of chromosomally female mice transgenic for Sry. Nature 351:117–121
Kreidberg JA, Sariola H, Loring JM et al (1993) WT-1 is required for early kidney development. Cell 74:679–691
Kuo CT, Morrisey EE, Anandappa R et al (1997) GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev 11:1048–1060
Le Caignec C, Delnatte C, Vermeesch JR et al (2007) Complete sex reversal in a WAGR syndrome patient. Am J Med Genet A 143A:2692–2695
Lecointre C, Pichon O, Hamel A et al (2009) Familial acampomelic form of campomelic dysplasia caused by a 960 kb deletion upstream of SOX9. Am J Med Genet A 149A:1183–1189
Lee DG, Han DH, Park KH, Baek M (2011) A novel WT1 gene mutation in a patient with Wilms' tumor and 46, XY gonadal dysgenesis. Eur J Pediatr 170(8):1079-82
Lourenco D, Brauner R, Lin L et al (2009) Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med 360:1200–1210
Lourenco D, Brauner R, Rybczynska M et al (2011) Loss-of-function mutation in GATA4 causes anomalies of human testicular development. Proc Natl Acad Sci USA 108:1597–1602
Lovell-Badge R (1992) The role of Sry in mammalian sex determination. CIBA Found Symp 165:162–179, discussion 179–182
Ludbrook LM, Harley VR (2004) Sex determination: a ‘window’ of DAX1 activity. Trends Endocrinol Metab 15:116–121
Luo X, Ikeda Y, Parker KL (1994) A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 77:481–490
Lutfalla G, Roest Crollius H, Brunet FG, Laudet V, Robinson-Rechavi M (2003) Inventing a sex-specific gene: a conserved role of DMRT1 in teleost fishes plus a recent duplication in the medaka Oryzias latipes resulted in DMY. J Mol Evol 57(Suppl 1):S148–S153
Maatouk DM, DiNapoli L, Alvers A et al (2008) Stabilization of beta-catenin in XY gonads causes male-to-female sex-reversal. Hum Mol Genet 17:2949–2955
Manuylov NL, Smagulova FO, Leach L, Tevosian SG (2008) Ovarian development in mice requires the GATA4-FOG2 transcription complex. Development 135:3731–3743
Matson CK, Murphy MW, Sarver AL et al (2011) DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 476:101–104
Matsuda M, Nagahama Y, Shinomiya A et al (2002) DMY is a Y-specific DM-domain gene required for male development in the medaka fish. Nature 417:559–563
Matzuk MM, Lu N, Vogel H et al (1995) Multiple defects and perinatal death in mice deficient in follistatin. Nature 374:360–363
McElreavey K, Rappaport R, Vilain E et al (1992) A minority of 46, XX true hermaphrodites are positive for the Y-DNA sequence including SRY. Hum Genet 90:121–125
Miyamoto Y, Taniguchi H, Hamel F, Silversides DW, Viger RS (2008) A GATA4/WT1 cooperation regulates transcription of genes required for mammalian sex determination and differentiation. BMC Mol Biol 9:44
Molkentin JD (2000) The zinc finger-containing transcription factors GATA-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem 275:38949–38952
Molkentin JD, Lin Q, Duncan SA, Olson EN (1997) Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev 11:1061–1072
Moniot B, Berta P, Scherer G, Sudbeck P, Poulat F (2000) Male specific expression suggests role of DMRT1 in human sex determination. Mech Dev 91:323–325
Morais da Silva S, Hacker A, Harley V et al (1996) Sox9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nat Genet 14:62–68
Nanda I, Shan Z, Schartl M et al (1999) 300 million years of conserved synteny between chicken Z and human chromosome 9. Nat Genet 21:258–259
Nanda I, Kondo M, Hornung U et al (2002) A duplicated copy of DMRT1 in the sex-determining region of the Y chromosome of the medaka, Oryzias latipes. Proc Natl Acad Sci USA 99:11778–11783
Nemer G, Fadlalah F, Usta J et al (2006) A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat 27:293–294
Nicholl RM, Grimsley L, Butler L et al (1994) Trisomy 22 and intersex. Arch Dis Child Fetal Neonatal Ed 71:F57–F58
Nishida H, Miyagawa S, Vieux-Rochas M et al (2008) Positive regulation of steroidogenic acute regulatory protein gene expression through the interaction between Dlx and GATA-4 for testicular steroidogenesis. Endocrinology 149:2090–2097
O’Bryan MK, Takada S, Kennedy CL et al (2008) Sox8 is a critical regulator of adult Sertoli cell function and male fertility. Dev Biol 316:359–370
Ogata T, Muroya K, Sasagawa I et al (2000) Genetic evidence for a novel gene(s) involved in urogenital development on 10q26. Kidney Int 58:2281–2290
Ottolenghi C, Moreira-Filho C, Mendonca BB et al (2001) Absence of mutations involving the LIM homeobox domain gene LHX9 in 46, XY gonadal agenesis and dysgenesis. J Clin Endocrinol Metab 86:2465–2469
Ottolenghi C, Omari S, Garcia-Ortiz JE et al (2005) Foxl2 is required for commitment to ovary differentiation. Hum Mol Genet 14:2053–2062
Ottolenghi C, Pelosi E, Tran J et al (2007) Loss of Wnt4 and Foxl2 leads to female-to-male sex reversal extending to germ cells. Hum Mol Genet 16:2795–2804
Pailhoux E, Vigier B, Chaffaux S et al (2001) A 11.7-kb deletion triggers intersexuality and polledness in goats. Nat Genet 29:453–458
Parma P, Radi O, Vidal V et al (2006) R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet 38:1304–1309
Pearlman A, Loke J, Le Caignec C et al (2010) Mutations in MAP3K1 cause 46, XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. Am J Hum Genet 87:898–904
Pelletier J, Bruening W, Li FP et al (1991) WT1 mutations contribute to abnormal genital system development and hereditary Wilms' tumour. Nature 353:431–434
Pierucci-Alves F, Clark AM, Russell LD (2001) A developmental study of the Desert hedgehog-null mouse testis. Biol Reprod 65:1392–1402
Pitteloud N, Quinton R, Pearce S et al (2007) Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest 117:457–463
Poirier C, Qin Y, Adams CP et al (2004) A complex interaction of imprinted and maternal-effect genes modifies sex determination in odd sex (Ods) mice. Genetics 168:1557–1562
Polanco JC, Wilhelm D, Davidson TL, Knight D, Koopman P (2010) Sox10 gain-of-function causes XX sex reversal in mice: implications for human 22q-linked disorders of sex development. Hum Mol Genet 19:506–516
Pop R, Conz C, Lindenberg KS et al (2004) Screening of the 1 Mb SOX9 5′ control region by array CGH identifies a large deletion in a case of campomelic dysplasia with XY sex reversal. J Med Genet 41:e47
Porter FD (2008) Smith–Lemli–Opitz syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet 16:535–541
Poulat F, Girard F, Chevron MP et al (1995) Nuclear localization of the testis determining gene product SRY. J Cell Biol 128:737–748
Raymond CS, Shamu CE, Shen MM et al (1998) Evidence for evolutionary conservation of sex-determining genes. Nature 391:691–695
Raymond CS, Parker ED, Kettlewell JR et al (1999) A region of human chromosome 9p required for testis development contains two genes related to known sexual regulators. Hum Mol Genet 8:989–996
Raymond CS, Murphy MW, O'Sullivan MG, Bardwell VJ, Zarkower D (2000) Dmrt1, a gene related to worm and fly sexual regulators, is required for mammalian testis differentiation. Genes Dev 14:2587–2595
Refai O, Friedman A, Terry L et al (2010) De novo 12;17 translocation upstream of SOX9 resulting in 46, XX testicular disorder of sex development. Am J Med Genet A 152A:422–426
Sadovsky Y, Dorn C (2000) Function of steroidogenic factor 1 during development and differentiation of the reproductive system. Rev Reprod 5:136–142
Schepers GE, Teasdale RD, Koopman P (2002) Twenty pairs of sox: extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev Cell 3:167–170
Schepers G, Wilson M, Wilhelm D, Koopman P (2003) SOX8 is expressed during testis differentiation in mice and synergizes with SF1 to activate the Amh promoter in vitro. J Biol Chem 278:28101–28108
Schlaubitz S, Yatsenko SA, Smith LD et al (2007) Ovotestes and XY sex reversal in a female with an interstitial 9q33.3–q34.1 deletion encompassing NR5A1 and LMX1B causing features of Genitopatellar syndrome. Am J Med Genet A 143A:1071–1081
Schmahl J, Kim Y, Colvin JS, Ornitz DM, Capel B (2004) Fgf9 induces proliferation and nuclear localization of FGFR2 in Sertoli precursors during male sex determination. Development 131:3627–3636
Schnack TH, Zdravkovic S, Myrup C et al (2008) Familial aggregation of hypospadias: a cohort study. Am J Epidemiol 167:251–256
Seeherunvong T, Perera EM, Bao Y et al (2004) 46, XX sex reversal with partial duplication of chromosome arm 22q. Am J Med Genet A 127A:149–151
Sekido R, Lovell-Badge R (2008) Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 453:930–934
Sekido R, Bar I, Narvaez V, Penny G and Lovell-Badge R (2004) SOX9 is up-regulated by the transient expression of SRY specifically in Sertoli cell precursors. Dev Biol 274:271–279
Sharp AJ, Selzer RR, Veltman JA et al (2007) Characterization of a recurrent 15q24 microdeletion syndrome. Hum Mol Genet 16:567–572
Shinoda K, Lei H, Yoshii H et al (1995) Developmental defects of the ventromedial hypothalamic nucleus and pituitary gonadotroph in the Ftz-F1 disrupted mice. Dev Dyn 204:22–29
Siggers P, Smith L, Greenfield A (2002) Sexually dimorphic expression of Gata-2 during mouse gonad development. Mech Dev 111:159–162
Sinclair AH, Berta P, Palmer MS et al (1990) A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346:240–244
Smith CA, McClive PJ, Western PS, Reed KJ, Sinclair AH (1999) Conservation of a sex-determining gene. Nature 402:601–602
Smith CA, Shoemaker CM, Roeszler KN et al (2008) Cloning and expression of R-Spondin1 in different vertebrates suggests a conserved role in ovarian development. BMC Dev Biol 8:72
Smith CA, Roeszler KN, Ohnesorg T et al (2009) The avian Z-linked gene DMRT1 is required for male sex determination in the chicken. Nature 461:267–271
Smyk M, Berg JS, Pursley A et al (2007) Male-to-female sex reversal associated with an approximately 250 kb deletion upstream of NR0B1 (DAX1). Hum Genet 122:63–70
Sock E, Schmidt K, Hermanns-Borgmeyer I, Bosl MR, Wegner M (2001) Idiopathic weight reduction in mice deficient in the high-mobility-group transcription factor Sox8. Mol Cell Biol 21:6951–6959
Stark K, Vainio S, Vassileva G, McMahon AP (1994) Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 372:679–683
Sultan C, Biason-Lauber A, Philibert P (2009) Mayer–Rokitansky–Kuster–Hauser syndrome: recent clinical and genetic findings. Gynecol Endocrinol 25:8–11
Sutton E, Hughes J, White S et al (2011) Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest 121:328–341
Swain A, Lovell-Badge R (1999) Mammalian sex determination: a molecular drama. Genes Dev 13:755–767
Taylor HS, Block K, Bick DP, Sherins RJ, Layman LC (1999) Mutation analysis of the EMX2 gene in Kallmann's syndrome. Fertil Steril 72:910–914
Temel SG, Gulten T, Yakut T et al (2007) Extended pedigree with multiple cases of XX sex reversal in the absence of SRY and of a mutation at the SOX9 locus. Sex Dev 1:24–34
Tevosian SG, Albrecht KH, Crispino JD et al (2002) Gonadal differentiation, sex determination and normal Sry expression in mice require direct interaction between transcription partners GATA4 and FOG2. Development 129:4627–4634
Tomaselli S, Megiorni F, De Bernardo C et al (2008) Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum Mutat 29:220–226
Tomaselli S, Megiorni F, Lin L et al (2011) Human RSPO1/R-spondin1 is expressed during early ovary development and augments beta-catenin signaling. PLoS One 6:e16366
Tomita-Mitchell A, Maslen CL, Morris CD, Garg V, Goldmuntz E (2007) GATA4 sequence variants in patients with congenital heart disease. J Med Genet 44:779–783
Tomizuka K, Horikoshi K, Kitada R et al (2008) R-spondin1 plays an essential role in ovarian development through positively regulating Wnt-4 signaling. Hum Mol Genet 17:1278–1291
Trautmann E, Guerquin MJ, Duquenne C et al (2008) Retinoic acid prevents germ cell mitotic arrest in mouse fetal testes. Cell Cycle 7:656–664
Tremblay JJ, Robert NM, Viger RS (2001) Modulation of endogenous GATA-4 activity reveals its dual contribution to Mullerian inhibiting substance gene transcription in Sertoli cells. Mol Endocrinol 15:1636–1650
Uhlenhaut NH, Jakob S, Anlag K et al (2009) Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell 139:1130–1142
Umehara F, Tate G, Itoh K et al (2000) A novel mutation of desert hedgehog in a patient with 46, XY partial gonadal dysgenesis accompanied by minifascicular neuropathy. Am J Hum Genet 67:1302–1305
Vainio S, Heikkila M, Kispert A, Chin N, McMahon AP (1999) Female development in mammals is regulated by Wnt-4 signalling. Nature 397:405–409
Val P, Swain A (2010) Gene dosage effects and transcriptional regulation of early mammalian adrenal cortex development. Mol Cell Endocrinol 323:105–114
van Silfhout A, Boot AM, Dijkhuizen T et al (2009) A unique 970 kb microdeletion in 9q33.3, including the NR5A1 gene in a 46, XY female. Eur J Med Genet 52:157–160
Veitia RA, Nunes M, Quintana-Murci L et al (1998) Swyer syndrome and 46, XY partial gonadal dysgenesis associated with 9p deletions in the absence of monosomy-9p syndrome. Am J Hum Genet 63:901–905
Vetro A, Ciccone R, Giorda R et al (2011) XX males SRY negative: a confirmed cause of infertility. J Med Genet 48:710–712
Vidal VP, Chaboissier MC, de Rooij DG, Schedl A (2001) Sox9 induces testis development in XX transgenic mice. Nat Genet 28:216–217
Viger RS, Mertineit C, Trasler JM, Nemer M (1998) Transcription factor GATA-4 is expressed in a sexually dimorphic pattern during mouse gonadal development and is a potent activator of the Mullerian inhibiting substance promoter. Development 125:2665–2675
Viger RS, Guittot SM, Anttonen M, Wilson DB, Heikinheimo M (2008) Role of the GATA family of transcription factors in endocrine development, function, and disease. Mol Endocrinol 22:781–798
Vinci G, Chantot-Bastaraud S, El Houate B et al (2007) Association of deletion 9p, 46, XY gonadal dysgenesis and autistic spectrum disorder. Mol Hum Reprod 13:685–689
Wagner T, Wirth J, Meyer J et al (1994) Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 79:1111–1120
Wainwright EN, Wilhelm D (2010) The game plan: cellular and molecular mechanisms of mammalian testis development. Curr Top Dev Biol 90:231–262
White S, Ohnesorg T, Notini A et al (2011) Copy number variation in patients with disorders of sex development due to 46, XY gonadal dysgenesis. PLoS One 6:1–10
Wieacker P, Volleth M (2007) WNT4 and RSPO1 are not involved in a case of male-to-female sex reversal with partial duplication of 1p. Sex Dev 1:111–113
Wilhelm D, Englert C (2002) The Wilms tumor suppressor WT1 regulates early gonad development by activation of Sf1. Genes Dev 16:1839–1851
Wilhelm D, Martinson F, Bradford S et al (2005) Sertoli cell differentiation is induced both cell-autonomously and through prostaglandin signaling during mammalian sex determination. Dev Biol 287:111–124
Wilhelm D, Hiramatsu R, Mizusaki H et al (2007) SOX9 regulates prostaglandin D synthase gene transcription in vivo to ensure testis development. J Biol Chem 282:10553–10560
Wilson MJ, Jeyasuria P, Parker KL, Koopman P (2005) The transcription factors steroidogenic factor-1 and SOX9 regulate expression of Vanin-1 during mouse testis development. J Biol Chem 280:5917–5923
Yao HH, Capel B (2002) Disruption of testis cords by cyclopamine or forskolin reveals independent cellular pathways in testis organogenesis. Dev Biol 246:356–365
Yao HH, Whoriskey W, Capel B (2002) Desert hedgehog/patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev 16:1433–1440
Yao HH, Matzuk MM, Jorgez CJ et al (2004) Follistatin operates downstream of Wnt4 in mammalian ovary organogenesis. Dev Dyn 230:210–215
Yoshida M, Suda Y, Matsuo I et al (1997) Emx1 and Emx2 functions in development of dorsal telencephalon. Development 124:101–111
Acknowledgements
This work was supported by an NHMRC Program grant to AS and the Victorian Government's Operational Infrastructure Support Program.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editors: Tariq Ezaz and Jennifer Graves.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Eggers, S., Sinclair, A. Mammalian sex determination—insights from humans and mice. Chromosome Res 20, 215–238 (2012). https://doi.org/10.1007/s10577-012-9274-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-012-9274-3