
Abstract
Y-linked Dmy (also called dmrt1bY) in the teleost fish medaka, W-linked Dm-W in the African clawed frog (Xenopus laevis), and Z-linked Dmrt1 in the chicken are all sex chromosome-linked Dmrt1 homologues required for sex determination. Dmy and Dm-W both are Dmrt1 palalogues evolved through Dmrt1 duplication, while chicken Dmrt1 is a Z-linked orthologue. The eutherian sex-determining gene, Sry, evolved from an allelic gene, Sox3. Here we analyzed the exon–intron structures of the Dmrt1 homologues of several vertebrate species through information from databases and by determining the transcription initiation sites in medaka, chicken, Xenopus, and mouse. Interestingly, medaka Dmrt1 and Dmy and Xenopus Dm-W and Dmrt1 have a noncoding-type first exon, while mouse and chicken Dmrt1 do not. We next compared the 5′-flanking sequences of the Dmrt1 noncoding and coding exons 1 of several vertebrate species and found conservation of the presumptive binding sites for some transcription factors. Importantly, based on the phylogenetic trees for Dmrt1 and Sox3 homologues, it was implied that the sex-determining gene Dmy, Dm-W, and Sry have a higher substitution rate than thier prototype genes. Finally, we discuss the evolutionary relationships between vertebrate sex chromosomes and the sex-determining genes Dmy/Dm-W and Sry, which evolved by neofunctionalization of Dmrt1 and Sox3, respectively, for sex determining function. We propose a coevolution model of sex determining gene and sex chromosome, in which undifferentiated sex chromosomes easily allow replacement of a sex-determining gene with another new one, while specialized sex chromosomes are restricted a particular sex-determining gene.
Similar content being viewed by others


Introduction
Both genotypic and environmental sex-determining systems exist in vertebrates (Graves 2008). In the former, heterogametic sex chromosomes determine the fate of sex, male (XY) or female (ZW). In the XX/XY-type sex-determining system, the Y-linked Sry genes of most eutherian mammals and the Dmy (also known as dmrt1bY) gene of the teleost fish medaka (Oryzias latipes) function as sex-determining genes that trigger testis formation (Sinclair et al. 1990; Koopman et al. 1991; Matsuda et al. 2002, 2007; Nanda et al. 2002). In contrast, the molecular mechanisms of ZZ/ZW-type systems are poorly understood, although our recent studies have proved that a W-linked gene Dm-W is a female sex-determining gene in the African clawed frog (Xenopus laevis) (Yoshimoto et al. 2008, 2010; Okada et al. 2009; Yoshimoto and Ito 2011). Importantly, a Dmrt1 duplication caused the emergence of Xenopus Dm-W (Yoshimoto et al. 2008), as well as medaka Dmy (Matsuda et al. 2002; Nanda et al. 2002). Our findings indicated that DMRT1 and its paralogous protein DM-W could have mutually opposite roles in sex determination, supporting a novel ZZ/ZW-type system model in which DM-W dominantly orients female development by antagonizing DMRT1 (Yoshimoto et al. 2010). Smith et al. (2009) also have recently reported that the two copies of Z-linked Dmrt1 gene are necessary for male sex determination in the chicken (Gallus gallus domesticus). The avian Dmrt1 gene may have been located in the Z chromosome to induce male development by maintaining its two-fold gene dosage in ZZ males (Nanda et al. 2000; Smith et al. 2009). The Y-linked Dmy in medaka and the W-linked Dm-W in X. laevis emerged as a Dmrt1 positive and dominant-negative type paralogue during species divergence, respectively (Kondo et al. 2004; Bewick et al. 2011), suggesting that both the sex-determining genes of the XX/XY and ZZ/ZW systems were generated by neofunctionalization of Dmrt1 (Yoshimoto et al. 2010). These findings support the idea that a DMRT1-driven male-determining system is involved in non-mammalian vertebrate species (Yoshimoto et al. 2010; Yoshimoto and Ito 2011). In contrast, the male sex-determining gene Sry evolved from Sox3 during evolution of eutherian mammals by neofunctionalization and established an SRY/SOX9-driven male-determining system.
Here, to clarify the molecular evolution of the sex-determining genes Dmy, Dm-W, and Sry, and their prototype genes Dmrt1 and Sox3 in vertebrates (Matsuda et al. 2002; Yoshimoto et al. 2008; Foster and Graves 1994), we compared the gene structures, presumptive promoter regions, and/or substitution rates among Dmrt1 and Sox3 family members in various species. Finally, we discuss the relationships between sex-determining genes and sex chromosomes during vertebrate evolution.
Materials and methods
Animal care and use
The Institutional Animal Care and Use Committee of Kitasato University approved all experimental procedures involving O. latipes, X. laevis, G. gallus, and Mus musculus.
RNA ligase-mediated rapid amplification of cDNA 5′ ends (RLM-5′RACE)
Total RNA was isolated using an RNeasy mini kit (Qiagen) from the gonads of M. musculus (C57BL/6), G. gallus, X. laevis, and O. latipes (Carbio). The Dmrt1 orthologue transcription initiation sites were determined using RNA obtained with the FirstChoice RLM-RACE kit (Ambion). Primers used for PCR were those supplied with the kit, and Dmrt1-specific primers were as follows: M. musculus Dmrt1, 5′-AATCAGGCTGCACTTCTTGC -3′ and 5′-ACTTCTTGCTTCCAGAACCC-3′; G. gallus Dmrt1, 5′-CACTTCTTGCACTGGCAGTC-3′ and 5′-ACATGCAGAACCGCTTGTGC-3′; X. laevis Dmrt1α, 5′-ATAACCCGTTGTCTCTCTGC-3′ and 5′-TCCATGATTTCTGCATCGGG-3′; O. latipes Dmrt1, 5′-TTCCAGCGGCAGAAGCGCTTG-3′ and 5′-GGCCTTTCAGCGGAGACACG-3′. The former primer set was used for first-round PCR, the latter for the second round. Products of the second round were inserted into pBluescript-KS (+) (Agilent Technologies) and sequenced.
Comparative analysis of genomic sequences
Comparisons of the 5′-flanking regions of Dmrt1 orthologues and paralogues were performed with mVISTA using the alignment program AVID (http://genome.lbl.gov/vista/index.shtml), which is suitable for globally aligning DNA sequences with its accuracy and ability to detect weak homologies. The Dmrt1 5′-flanking sequences of eight species (Homo sapiens, M. musculus, Canis lupus familiaris, Monodelphis domestica, Ornithorhynchus anatinus, G. gallus, Anolis carolinensis, Xenopus (Silurana) tropicalis) were obtained from the UCSC Genome Browser (http://genome.brc.mcw.edu). The 5′-flanking sequences of X. laevis Dm-W, O. latipes (Carbio) Dmrt1, and Dmy were obtained from GenBank (AB365520, AP006154, and AP006152, restrictively; http://www.ncbi.nlm.nih.gov/genbank/). About 3 kbp 5′-flanking region of X. laevis Dmrt1α was isolated and sequenced, which was deposited in the GenBank/EBI Data Bank under accession number AB678700. We compared the 5′-flanking regions from the transcription start sites of M. musculus Dmrt1, G. gallus Dmrt1, X. tropicalis Dmrt1, O. latipes (Carbio) Dmrt1, or O. latipes (Carbio) Dmy, and Dmrt1 sequences of H. sapiens, C. familiaris, M. domestica, O. anatinus, or A. carolinensis to the 3′ end of the next upstream gene, kank1. About 3 kbp sequences upstream of the transcription start sites of X. laevis Dmrt1α and Dm-W were also compared.
Construction of molecular phylogenetic trees and substitution rate calculations
Phylogenetic analyses were performed using integrated tool MEGA 5 (Tamura et al. 2011). The nucleotide sequences used for the analyses were obtained from the GenBank/EBI Data Bank as follows. O. latipes (HNI) Dmrt1, AY157712; O. latipes (HNI) Dmy, AY12924; O. latipes (Carbio) Dmrt1, AF319994; O. latipes (Carbio) Dmy, AY129240; O. latipes (YZ) Dmrt1, AY442916 and AY524417; O. latipes (YZ) Dmy, AY448017; Oryzias marmoratus Dmrt1, AY521023; X. laevis Dmrt1β, AB252635; X. laevis Dm-W, NM001114842; Xenopus. andrei Dmrt1β1, HQ220773; X. andrei Dmrt1β2, HQ220774; X. andrei Dm-W, HQ220853; Xenopus itombwensis Dmrt1β1, HQ220748; X. itombwensis Dmrt1β2, HQ220749; X. itombwensis Dm-W, HQ220850; Bufo marinus Dmrt1, FJ697175; H. sapiens SOX3, NM005634; H. sapiens SRY, NM003140; Pan troglodytes Sox3, AC149044; P. troglodytes Sry, NM001008988; Nomascus leucogenys Sox3, XM003272598; N. leucogenys Sry, HM757941; Macaca mulatta Sox3, NM001193752; M. mulatta Sry, NM001032836; Ornithorhynchus anatinus Sox3, XP001511549. DNA sequences were aligned using the MUSCLE (Edgar 2004) and gaps (insertions/deletions) were removed. Phylogenetic trees for the DNA binding (DM or HMG) domain regions, the non-DNA binding (non-DM or non-HMG) domain regions, and their combined regions were constructed using a maximum likelihood method after selecting the substitution model that best fitted the data using MODELTEST (Bayesian information criterion). Nucleotide numbers used: medaka DM domain, 111 bp; medaka non-DM domain, 672 bp; Xenopus DM domain, 105 bp; Xenopus non-DM region, 35 bp; Sox3 or Sry HMG domain, 216 bp; Sox3 non-HMG region, 1124 bp; and Sry non-HMG region, 225 bp. The bootstrap consensus tree inferred from 500 replicates was generated. Branches corresponding to partitions reproduced in less than 60% bootstrap replicates were collapsed. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site (indicated next to the branches). The substitution rates of sex-determining genes and their paralogues from each divergence were calculated using the phylogenetic tree branch lengths. Tajima's nonparametric relative-rate test (Tajima 1993) was performed for testing the molecular evolutionary clock hypothesis.
Results
Xenopus and medaka Dmrt1 homologues possess a noncoding first exon
We previously showed that both the Xenopus tropicalis (also called Silurana tropicalis) Dmrt1 and X. laevis Dm-W genes have a noncoding exon 1 (Yoshimoto et al. 2006, 2008), as does medaka Dmy (Matsuda et al. 2002). To confirm the existence of the noncoding exon 1 and analyze the 5′-flanking sequences containing the promoter regions in the X. laevis Dmrt1α and medaka Dmrt1 genes, we determined their transcription initiation sites by performing rapid amplification of the 5′ end using X. laevis and medaka testis RNAs and by cloning the products into a plasmid (see “Materials and methods”). X. laevis is an allotetraploid species (Hughes and Hughes 1993) and has two Dmrt1 genes, Dmrt1α and β, while Xenopus (Silurana) tropicalis is a diploid (Evans 2008). Because we were not able to obtain the genomic sequence corresponding to the Dmrt1β gene, we did not confirm the existence of the noncoding exon 1 of the β gene. The comparison between the obtained sequences and the genomic sequence revealed that both X. laevis Dmrt1α and medaka Dmrt1 had a noncoding exon 1, consisting of 140 and 115 bases, respectively (Fig. 1). These findings suggest that authentic Dmrt1 homologues in teleost fish and amphibians have a noncoding exon 1.

Exon–intron structures of Dmrt1 orthologues and its paralogous sex-determining genes Dmy and Dm-W in vertebrates. The number shows the size (bp) of each exon. Noncoding exon 1 is shown as a blue box; other noncoding and coding regions in exons are shown as white and gray boxes, respectively. The locations of DM domains, male-specific motifs, and P/S (proline/serine)-rich regions are indicated by orange, green, and purple boxes, respectively. Medaka, Oryzias latipes; Frog (Xl), Xenopus laevis; Frog (Xt), Xenopus (Silurana) tropicalis; Chicken, Gallus gallus; Platypus, Ornithorhynchus anatinus; Opossum, Monodelphis domestica; Mouse, Mus musculus; Human, Homo sapiens
The Dmrt1 noncoding exon 1 degenerated during vertebrate evolution
We next examined whether or not the exon–intron structures of Dmrt1 orthologue and paralogue were conserved during vertebrate evolution, using the genomic and/or EST databases of teleost fish medaka, amphibian Xenopus, chicken, and mammalian species (human and mouse). We also predicted the Dmrt1 structures of monotreme platypus and marsupial opossum by comparative analysis between their genomic databases. The exon–intron structures, including the splicing sites, are well conserved among various vertebrate species except for the noncoding exons (Fig. 1). Interestingly, the comparative analysis did not detect a Dmrt1 noncoding exon 1 in mouse, human, or chicken. To confirm the absence of the noncoding exon 1, we then determined the Dmrt1 transcription initiation sites using mouse and chicken testis RNAs. The transcription initiation sites of the mouse and chicken Dmrt1 genes were located at 236 and 54 bases, respectively, upstream of the ATG translation initiation codon, indicating that each Dmrt1 gene has a coding first exon. These findings suggest that a Dmrt1 noncoding exon 1 has degenerated during amniote or homoiothermal evolution.
Comparative analysis of the 5′-flanking sequences among vertebrate Dmrt1 homologues
To understand the conservation and molecular evolution of the 5′-flanking sequences of Dmrt1 orthologues during vertebrate evolution and of the neofuctionalization-type sex-determining genes Dmy and Dm-W after Dmrt1 duplication, we conducted a comparative analysis of the 5′-flanking sequences of the Dmrt1 homologues in several vertebrate species using a comparative genomics tool, mVISTA (see “Materials and methods”). The analysis detected no significant homologous regions among mouse, chicken, clawed frog, and medaka fish over approximately 5 kb of the Dmrt1 5′-flanking sequences. However, in the more closely related species, such as human/mouse Dmrt1 (Fig. 2a), and X. tropicalis/X. laevis Dmrt1 (Fig. 2b), some homologous regions were found within 500 bp of the transcription start site. Since about 2.5 kb repeated sequence is inserted into the medaka Dmy promoter region (Herpin et al. 2010), we also searched the upstream region of the repeated sequence. As expected, two homologous regions were identified in the region −500 to −75 of medaka Dmrt1 and −3,120 to −2,386 of medaka Dmy (Fig. 2c). Curiously, we could not detect any sequence homology between X. laevis Dm-W and X. tropicalis or X. laevis Dmrt1. This may be because Dm-W emerged earlier than Dmy and much more base substitutions were accumulated—Dm-W and Dmy diverged from Dmrt1 13–64 million years ago (Bewick et al. 2011) and about 10 million years ago (Kondo et al. 2004), respectively. In any case, it will be necessary to obtain the gene structure and sequence information of X. laevis Dmrt1β, which is infered to be an ancestral paralogue of Dm-W (Bewick et al. 2011).

Comparisons of the 5′-flanking regions among several vertebrate Dmrt1 homologues using mVISTA. Graphs were constructed using the AVID alignment program. Numbers with minus sign correspond to bp upstream of the transcription start site (+1). a Comparison of the first 500 bp of the mouse or chicken Dmrt1 promoter sequence, located upstream of the coding exon 1, with that of human DMRT1. b Comparison of the first 500 bp of the frog (Xl) Dmrt1α, frog (Xl) Dm-W, medaka Dmrt1, or medaka Dmy promoter sequence, located upstream of the noncoding exon 1, with that of frog (Xt) Dmrt1. c Comparison of the medaka Dmy 5′-flanking region with the first 500 bp of medaka Dmrt1. The 8-kb 5′-flanking region of the transcription initiation site of medaka Dmy was used, and the homologous regions are shown. d Comparison of the 2-kb region between the 3′-flanking region of kank1 and the 5′-flanking region of Dmrt1, a region conserved in several vertebrate species. The graph was constructed in comparison to the human 2-kb sequence. The locations of the conserved regions are shown in Supplementary Table 1. Dog, Canis lupus familiaris; Lizard, Anolis carolinensis
To understand basic mechanisms for transcription of Dmrt1 orthologues, we searched for DNA-binding motifs of transcription factors in the 5′-flanking 500 bases upstream of the transcription start sites of the Dmrt1 homologues, using the program TFSEARCH 1.3 (http://www.cbrc.jp/research/db/TFSEARCH.html). We could find a few motifs common to the human and mouse Dmrt1 promoter regions upstream of the coding exon 1, except for the binding elements for AML-1a, C/EBP, and GATA2 within the homologous regions detected by mVISTA (Supplementary Fig. 1A). In the promoter regions upstream of the noncoding exon 1, we could not find any common motifs arranged in order among the Xenopus and medaka Dmrt1 homologues. However, there was a conserved array of DNA-binding motifs, that is, Sox5/Nkx-2.5/AML-1a/Sox5/HNF-1/GATA-1/CdxA/YY1, in the homologous regions corresponding to the region −500 to −250 of medka Dmrt1 and the −3,120 to −2,862 of medaka Dmy (Supplementary Fig. 1B).
The medaka and Xenopus Dmrt1 genes have a noncoding exon 1. Therefore, we predicted there would be a homologous region between the upstream regions of the noncoding exons 1 of Xenopus/medaka Dmrt1 and the far upstream regions of the coding exons 1 of mouse/human and chicken Dmrt1. We searched the regions from the transcription start sites of Xenopus and medaka Dmrt1 to the 3′ end of the next upstream gene, kank1 but did not find any sequene homology. Besides, a Blat search (http://genome.ucsc.edu/cgi-bin/hgBlat) using the wider region between the Dmrt1 and kank 1 genes revealed a significantly homologous region, consisting of about 600 bases, among the various species of mammals, chicken, and lizard (Supplementary Fig. 2). This region on the human genome is located at about 40 kb upstream of Dmrt1 (Supplementary Table 1), which corresponds to about 60 kb downstream of kank1. Figure 2d shows the results of mVISTA analysis on this region. There was a high sequence conservation among the regions of eutherian human, mouse and dog, marsupial opossum, and monotreme platypus. Moreover, the human region showed a significant homology with those of chicken and lizard, but not of frog. From mammals to lizards, the region included common DNA-binding motifs, such as Cdxa, GATA-X, Pbx-1, AP-1, and Nkx-2.5 (Supplementary Fig. 2), which might regulate the transcription of Dmrt1 and/or kank1. Lei and Heckert (2002, 2004) reported that transcription factors Sp1 and Egr1/Gata4 regulate transcription of the rat Dmrt1 gene in the testes by binding to about 100 b and 3 kb upstream of the gene, respectively. Our search in the upstream regions of vertebrate Dmrt1 genes identified no consensus sequences for Sp1 and Egr1/Gata 4 binding except for rats/mice and rats, respectively.
The sex-determining genes Dmy/Dm-W and Sry show a higher substitution rate than their prototype genes, Dmrt1 and Sox3, respectively
To elucidate the molecular evolution of the sex-determining genes, we first examined the substitution rates of the sex-determining gene Dmy and its prototype gene Dmrt1 from three groups of medaka (O. latipes HNI, Carbio, and YZ), and Dm-W and Dmrt1β from three species of Xenopus (X. laevis, X. andrei, and X. itombwensis). The substitution rates of Dmy and Dm-W were higher than those of Dmrt1 and Dmrt1β, respectively (Fig. 3). Particularly, the DNA-binding DM domains of Dmy and Dm-W showed a higher substitution rate, about 46 and 7 times, respectively (Fig. 3). To examine whether substitution rates vary significantly between the sex-determining genes and their prototype genes, Tajima's relative rate test was performed on the DNA sequences corresponding to Fig. 3. The Tajima's test showed that the molecular clock hypothesis was rejected (p < 0.05) between each region of medaka Dmrt1 and Dmy (Supplementary Table 3) and between the DM domain region or combined region of Xenopus Dmrt1 and Dm-W except for X. itombwensis (Supplementary Table 4). These results indicated that the sex-determining genes Dmy and Dm-W significantly have a higher substitution rate than their prototype genes. The substitution rates of the DM domains of medaka and Xenopus Dmrt1 were lower than those of their non-DM domain regions (Fig. 3). This was expected because the DNA-binding domain is known to be functionally conservative. Interestingly, the substitution rate of the DM domain, on the contrary, was higher than that of the non-DM domain region in Dmy and was equal to in Dm-W.

Comparisons of substitution rates among Dmy, Dm-W, and their prototype gene Dmrt1. The phylogenetic trees were constructed from three groups of medaka and O. marmoratus (Om) as an outgroup (a) or three species of Xenopus and Bufo marinus (Bm) as an outgroup (b), using the maximum likelihood method based on the Tamura 3-parameter model with 4 discrete gamma distribution categories (a) or Kimura 2-parameter model (b). The three trees of each panel were derived from the DM domain region (upper), the non-DM domain region (middle), and their combined region (lower). The number shows the branch length, which was defined as the number of nucleotide substitutions per site for the branch. The substitution rates of Dmy or Dm-W and their prototype gene Dmrt1 (right side of each panel) were calculated using the branch lengths from the position of their common ancestor (white block) to the branch tip corresponding to each gene (gray block). Bootstrap percentage values of 500 replications are shown in bold above the node. Dmy and Dm-W diverged from the their prototype genes 10 and 13–64 million years ago, respectively (Kondo et al. 2004; Bewick et al. 2011). *An average of the substitution rates of sex-determining genes or thier prototype gene. Ol, O. latipes; Xl, X. laevis; Xa, X. andrei; Xi, X. itombwensis
We next examined substitution rates of the mammalian sex-determining gene Sry and its prototype gene Sox3 from four species of primates—H. sapiens, P. troglodytes, N. leucogenys, and M. mulatta (Fig. 4a). Because there is little homology in the non-HMG domains between Sox3 and Sry, we calculated the substitution rates of only the HMG domain to compare the two genes (Fig. 4a). As expected, the rate of the DNA-binding HMG domain of Sry was about seven times higher than that of Sox3. Tajima's test was also performed, indicating that the molecular clock hypothesis was rejected between the HMG regions of Sry and Sox3 (p < 0.05) (Supplementary Table 5). These results coincided with the relationships between Dmy/Dm-W and their prototype gene Dmrt1. We next compared substitution rates between the HMG domain and non-HMG domain regions by constructing phylogenetic trees of Sox3 or Sry (Fig. 4b, c). The substitution rate of the Sox3 HMG domain was lower than that of the non-HMG domain (Fig. 4b), as was seen for the DNA-binding DM domain of medaka and Xenopus Dmrt1. Interestingly, the substitution rate of the HMG domain of Sry was also lower than that of its non-HMG domain (Fig. 4c), which was not the case for Dmy or Dm-W.

Comparisons of substitution rates among a sex-determining gene Sry and its prototype gene Sox3. The phylogenetic trees were constructed from the HMG domain regions of Sox3 and Sry (a) in four species of primates and Ornithorhynchus anatinus (Oa) as an outgroup, Sox3 (b) or Sry (c) in three species of primates and Macaca mulatta (Mm) as an outgroup, using the maximum likelihood method based on the Kimura 2-parameter model (a), Hasegawa-Kishino-Yano model (b), or the Kimura 2-parameter model (c). The three trees in (b) and (c) were derived from the HMG domain region (upper), the non-HMG domain region (middle), and their combined region (lower). Calculations of the substitution rates were performed as described in Fig. 3. Sry diverged from the prototype genes 148–166 million years ago (Marques-Bonet et al. 2009). A common ancestor of Homo. sapiens, Pan. troglodytes, and Nomascus leucogenys diverged from M. mulatta 18 million years ago (Marques-Bonet et al. 2009). Hs, H. sapiens; Pt, Pan troglodytes; Nl, Nomascus leucogenys
Discussion
Degeneration of a Dmrt1 noncoding exon 1 during vertebrate evolution
Here, we performed a comparative analysis of Dmrt1 homologue genomic and cDNA sequences in medaka, Xenopus, chicken, and mouse, with determination of the transcription initiation sites (Fig. 1) and showed that a Dmrt1 noncoding exon 1 exists in the fish and frog, but not in chicken or mouse. This suggests that non-coding exon 1 degenerated during vertebrate evolution. Why did it degenerate? It may have been due to the need for a promoter change to modify the transcription machinery for the Dmrt1 mRNA expression. In mice, DMRT1 is expressed in primordial germ cells (PGCs) and somatic cells of XX- and XY-indifferent gonads (Lei et al. 2007). In chicken and Xenopus, Dmrt1 is expressed in somatic cells of ZZ- and ZW-indifferent gonads (Oréal et al. 2002; Yoshimoto et al. 2010). In medaka, Dmy and Dmrt1 are expressed in PGC- and spermatogonium-supporting cells, respectively, of XY gonads (Kobayashi et al. 2004). Dmrt1 is not essential for fetal testis and ovary development in mice (Raymond et al. 2000) but is necessary for male and female germ cell development (Matson et al. 2010; Krentz et al. 2011) and for postnatal testicular development (Matson et al. 2011). In contrast, Dmrt1 is required for male determination in chicken (Smith et al. 2009) and has the potential to induce primary male development in Xenopus (Yoshimoto et al. 2010). Promoter reporter analyses of a transgenic mouse carrying about 9-kb upstream of the rat Dmrt1 coding exon 1 and of transgenic Xenopus carrying about 3-kb upstream of the X. tropicalis Dmrt1 noncoding exon 1 revealed that this region is necessary for the transgene expression in Sertoli cells and male germ cells (Lei et al. 2009) and in indifferent gonads (Yoshimoto et al. 2006), respectively. Totally, these findings do not show any differences in the promoter activity and regulation of Dmrt1 between the animals with and without non-coding exon 1.
We propose one hypothesis that some important regulatory sequences upstream of the Dmrt1 noncoding exon 1 required for sex determination and differentiation in poikilothermic vertebrate species were abolished during homeotherm evolution (Fig. 2; Supplemental Fig. 1). It is possible that these regulatory sequences are involved in temperature sensitivity because Dmrt1 expression is related to the temperature-dependent sex determination and sex reversal in some poikilothermic reptile, amphibian, and teleost species (Kettlewell et al. 2000; Murdock and Wibbels 2003; Sakata et al. 2006; Hattori et al. 2007; Anand et al. 2008; Graves 2008). It is important to clarify whether or not reptiles have a noncoding exon 1 within the Dmrt1 gene.
Dmy, Dm-W, and Sry as neofunctionalization-type sex-determining genes
In general, all of the new genes might arise from redundant copies of the preexisting genes (Ohno 1970). In the neofunctionalization model of gene duplication, one copy retains the original function, and the other evolves a new function (Lynch et al. 2001). The vertebrate sex-determining genes Sry and Dmy/Dm-W may have evolved from Sox3 and Dmrt1, respectively, as a neofunctionalization-type gene for sex determination. Neofunctionalization-type genes have higher substitution rates than those of their prototype genes (Fig. 3; Fig. 4a; Supplemental Tables 3–5). This coincides with the results of amino acid sequence comparisons among vertebrate DMRT1 family proteins (Supplementary Table 2). In this context, the chicken Z-linked Dmrt1 does not appear to be a neofunctionalization-type gene (data not shown; Supplementary Table 2), although avian Z-linked Dmrt1 is a strong candidate for the male-determining gene.
The substitution rates of the transcription factor genes Dmrt1 and Sox3 indicated that their DM and HMG domains are relatively conserved, compared to other regions, during species divergence (Fig. 3; Fig. 4b); this indicates the functional importance of these sequence-specific DNA-binding domains. In contrast, the Dmrt1 and Sox3-derived sex-determining genes were different in that point; the rate of the DNA-binding domain was significantly higher than that of the non-DNA binding region in Dmy, was equal to in Dm-W, and lower in Sry (Figs. 3, 4c). The difference in substitution rates between Dmy and Dm-W could be due to the restricted small numbers of Dm-W sequences used for the analysis. Zhang (2004) reported that the Dmy DM domain is likely to be under positive Darwinian selection. Full sequencing of the Dm-W orthologues in several Xenopus species would answer whether or not there is a common evolutionary mechanism of DM domain in the Dmrt1-derived sex-determining genes. On the other hand, the contrasting results between Dmy and Sry might have been caused by differences in the functional importance of the non-DNA binding regions. The results in the present study indicate that the HMG-domain sequence of Sry is much more conserved than the other region during species divergence. In fact, it is proved that Sry could be replaced with Sox3 for the male determination in the transgenic studies (Sutton et al. 2011), indicating that the targent element of Sry is still conserved after divergence from Sox3. In contrast, the higher substitution rate of Dmy DM-domain than the other region suggests that the target elements are modified after duplication. Further transgenic experiments with a replacement of Dmy with Dmrt1 would give an answer to the intriguing quesiton. In the future, it will be important to analyze the molecular evolution of the Dmrt1- and Sox3-derived sex-determining genes, from the view of the differences in the neofuntionalization-type emergence process; Dmy/Dm-W and Sry evolved through individualized autosomal Dmrt1 duplication and allelic Sox3 mutation, respectively (Fig. 5). This molecular process may be closely related to the coevolution of the sex-determining genes and sex chromosomes.

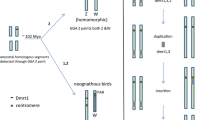
Proposed model for evolutionary relationships between the appearance of sex-determining genes (SDGs) and sex chromosomes in vertebrates. First, a candidate SDG emerges or evolves on one chromosome of a pair of autosomes by insertion or mutation. Then, the candidate gene may be established as an SDG during species divergence with few morphological changes in the two chromosomes, in cases like that of the heterogametic XY or ZW sex chromosomes in the teleost fish medaka (O. latipes) carrying the Y-linked SDG Dmy, or the African clawed frog (X. laevis) carrying the W-linked SDG Dm-W, respectively. If the new SDG emerges as a stronger regulator for sex determination, the original SDG or its candidate might degenerate into a psuedogene, as in the case of sex determination in O. luzonensis, which is closely related to the medaka species. In contrast, if an SDG strongly contributes to the stability of a sex-determining system during species divergence, differentiation of the sex chromosomes might be allowed, leading to specialization of the heterogametic sex chromosomes and to stabilization of the SDG. This might be the case for the heterogametic XY sex chromosomes in eutherian mammals carrying the Y-linked SDG, Sry
A hypothesis—undifferentiated sex chromosome state allows a sex-determining gene to change
Closely related species to medaka fish (O. latipes), which has Dmy as a sex-determining gene on the Y chromosome, are O. curvinotus, O. luzonensis, and O. mekongensis. These four species (including medaka) all have 48 chromosomes with a genetic XX/XY-type sex-determining system. O. curvinotus has Dmy, which is located on the orthologous Y chromosome (chromosome 1 of O. latipes, called latipes linkage group 1 (LG1)). Interestingly, the Dmy gene degenerats into a pseudogene in O. luzonensis (Kondo et al. 2004), and, there is no Dmy-orthologous gene in O. mekongensis, suggesting that the sex-determining gene Dmy arose from a duplicated copy of the autosomal Dmrt1 gene after divergence of the three species from O. mekongensis, approximately 10 million years ago (Kondo et al. 2004). Importantly, the Y chromosomes in O. luzonensis and O. mekongensis are not LG1, but LG12 and LG2, respectively (Tanaka et al. 2007; unpublished data in Takehana et al. 2008), suggesting that a new sex-determining gene arose in each of the two species. In addition, no sex-chromosomal heteromorphism was observed in the genus Oryzias species, as is seen in O. latipes and O. luzonensis (Takehana et al. 2007; Tanaka et al. 2007). The sex-reversed XX males and XY females in these species are completely fertile, suggesting that there is no functional differentiation between the X and Y chromosomes except for the male determining role of Dmy in the Y chromosoome (Takehana et al. 2007; Tanaka et al. 2007). This idea is supported by the fertility of the transgenic medaka sex-reversed XX males carrying the Dmy expression vector and the XY females with spontaneous Dmy gene mutations (Matsuda et al. 2002, 2007). Consequently, it is plausible that a sex-determining gene is not stabilized during species diversification under the genomic condition in which sex chromosomes are undifferentiated (Fig. 4).
We recently performed a FISH analysis of Dm-W in X. laevis and showed that the Dm-W-harboring chromosome 3 is the W sex chromosome, and its homologous partner is the Z sex chromosome (Yoshimoto et al. 2008), although conventional chromosomal staining did not identify any morphological differences between the Z and W chromosomes. In addition, the sex chromosomes are indistiguishable in the females and males of several other species examined in the genus Xenopus, including diploid and polyploid (tetraploid, octaploid, and tetraoctaploid) species (Tymowska and Fischberg 1973). It is interesting that most Xenopus species may not have developed differentiated sex chromosomes during and after the species divergence mediated through polyploidization. More recently, Bewick et al. (2011) have identified Dm-W orthologues in at least seven species of the genus Xenopus—four tetraploid species including X. laevis and X. largeni, and three octaploid species including X. itombwensis—but that many other Xenopus species lack a Dm-W orthologue. They also suggested that Dm-W might be degenerated in closely related Xenopus species to the ones bearing Dm-W, as in the case of the teleost fish O. luzonensis. They concluded that Dm-W arose from a partial Dmrt1β duplication in the Xenopus genus after diverged from its sister genus Silurana, which has a diploid genome, but before the divergence of X. leavis and X. clivii, that is, 13–64 million years ago. Therefore, there should be other kinds of sex-determining genes in Xenopus species lacking Dm-W gene. Importantly, X. laevis ZW transgenic individuals carrying the Dm-W knockdown vector develop testes (Yoshimoto et al. 2010); among them, one ZW individual formed sperm. In addition, X. laevis ZZ animals that had undergone male-to-female sex reversal were fertile (Hayes et al. 2010). Taken together, these findings suggest that there might be no functional differentiation between the Z and W chromosomes except for the sex-determining gene Dm-W. This is similar to the relationship between the X and Y chromosomes in the four closely related medaka species of the genus Oryzias described above. Thus, the heterogametic sex chromosome may be only a vehicle for the sex-determining gene in the two independent genera Oryzias and Xenopus.
In summary, there is likely some relationship between the undifferentiated state of sex chromosome and the change of sex-determining gene in the cases of both the Oryzias fish and the Xenopus frog. Therefore, we hypothesize that an undifferentiated state of sex chromosomes allows change of a sex-determining gene and provokes evolution of a neofunctionalization-type sex-determining gene, regardless of the heterogametic sex (Fig. 5).
Is sex chromosome specialization involved in sex-determining gene stabilization?
Compared to poikilotherm vertebrate evolution, male heterogametic (XX/XY) and female heterogametic (ZZ/ZW) sex-determining systems are almost perfectly conserved during mammalian and avian evolution, respectively, with some exceptions, such as the Ryukyu spiny rats and mole voles (Kobayashi et al. 2007; Matthey 1933; Fregda 1983). A similar phenomenan of the loss of a Y chromosome appeared to occur independently during species diversity of the spiny rats and mole vole (Just et al. 2007). In poikilotherm animals, flexibility of gene expression for sex determination and sex-reversal might be required in response to the environmental change, and the sex chromosomes are morphologically homomorphic in both sexes in many fishes, amphibians, and reptiles. On the other hand, it is possible that a homeothermic condition, as in birds and mammals, makes it possible to control the gene expression superior to the ourter environment and requires no flexibility of the genetic systems, and thus might allow sex chromosomes to become highly differentiated between sexes. Once the sex chromosomes were highly differentiated, some functional differentiation might be accelerated between the female and male sex chromosomes except for the role of sex determination. As a result, the sex-determining gene becomes stabilized on the sex chromosome (Fig. 5). This could be one reason why Sry is highly conserved as a sex-determining gene in most species of eutherian mammals. Based on this scenario, one same sex-determining gene might be common to most avian species—the avian Z-linked Dmrt1 gene is plausible to be a male-determining gene. Likewise, as for female determination, it is possible that a particular W-linked female determining gene is common to many avian species.
In the future, it will be interesting to clarify molecular mechanisms of coevolution between sex-determining genes and sex chromosomes in various vertebrate species including the spiny rats and mole voles mentioned above, the Japanese wrinkled frog Rana rugosa, which underwent change of heterogametic sex from XY male and ZW female, and some species of reptiles and fishes, which have temperature sex determination (TSD) and/or genetic sex determination (GSD). The identification of sex-determining genes in these species would lead to molecular understanding of the coevolution and supply information to discuss on our model proposed here (Fig. 5) with reference to TSD, GSD-TSD transitions (Quinn et al. 2007; Grossen et al. 2011), and ZW-XY transitions (Miura 2007; Quinn et al. 2011).
Abbreviations
- DM:
-
Doublesex and mab-3
- Dmrt1:
-
Doublesex and mab-3 related transcription factor 1
- Dmy:
-
Doublesex and mab-3 related gene Y—the sex-determining gene on the medaka Y chromosome
- Dm-W:
-
Doublesex and mab-3 related gene W—the sex-determining gene on the Xenopus W chromosome
- FISH:
-
Fluorescence in situ hybridization
- HMG:
-
High mobility group
- PGC:
-
Primordial germ cell
- Sox3:
-
Sry (sex-determining region Y)-box 3
- Sry:
-
Sex-determining region Y—the sex-determining gene on the eutherian Y chromosome
References
Anand A, Patel M, Lalremruata A et al (2008) Multiple alternative splicing of Dmrt1 during gonadogenesis in Indian mugger, a species exhibiting temperature-dependent sex determination. Gene 425:56–63
Bewick AJ, Anderson DW, Evans BJ (2011) Evolution of the closely related, sex-related genes DM-W and DMRT1 in African clawed frogs (Xenopus). Evolution 65:698–712
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113
Evans BJ (2008) Genome evolution and speciation genetics of allopolyploid clawed frogs (Xenopus and Silurana). Frontiers Biosci 13:4687–4706
Foster JW, Graves JAM (1994) An SRY-related sequence on the marsupial X chromosome: implications for the evolution of the mammalian testis-determining gene. Proc Natl Acad Sci USA 91:1927–1931
Fregda K (1983) Aberrant sex chromosome mechanisms in mammals. Evolutionary aspects. Differentiation 23(Suppl):S23–S30
Graves JAM (2008) Weird animal genomes and the evolution of vertebrate sex and sex chromosomes. Annu Rev Genet 42:565–586
Grossen C, Neuenschwander S, Perrin N (2011) Temperature-dependent turnovers in sex-determination mechanisms: a quantitative model. Evolution 65:64–78
Hattori RS, Gould RJ, Fujioka T et al (2007) Temperature-dependent sex determination in Hd-rR medaka Oryzias latipes: gender sensitivity, thermal threshold, critical period, and DMRT1 expression profile. Sex Dev 1:138–146
Hayes TB, Khoury V, Narayan A et al (2010) Atrazine induces complete feminization and chemical castration in male African clawed frogs (Xenopus laevis). Proc Natl Acad Sci USA 107:4612–4617
Herpin A, Braasch I, Kraeussling M et al (2010) Transcriptional rewiring of the sex determining dmrt1 gene duplicate by transposable elements. PLoS Genet 6:e1000844
Hughes MK, Hughes AL (1993) Evolution of duplicate genes in a tetraploid animal, Xenopus laevis. Mol Biol Evol 10:1360–1369
Just W, Baumstark A, Süss A et al (2007) Ellobius lutescens: sex determination and sex chromosome. Sex Dev 1:211–221
Kettlewell JR, Raymond CS, Zarkower D (2000) Temperature-dependent expression of turtle Dmrt1 prior to sexual differentiation. Genesis 26:174–178
Kobayashi T, Matsuda M, Kajiura-Kobayashi H et al (2004) Two DM Domain Genes, DMY and DMRT1, involved in testicular differentiation and development in the Medaka, Oryzias latipes. Dev Dyn 231:518–526
Kobayashi T, Yamada F, Hashimoto T, Abe S, Matsuda Y, Kuroiwa A (2007) Exceptional minute sex-specific region in the X0 mammal, Ryukyu spiny rat. Chromosome Res 15:175–187
Kondo M, Nanda I, Hornung U, Schmid M, Schartl M (2004) Evolutionary origin of the medaka Y chromosome. Curr Biol 21:1664–1669
Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R (1991) Male development of chromosomally female mice transgenic for Sry. Nature 351:117–121
Krentz AD, Murphy MW, Sarver AL, Griswold MD, Bardwell VJ, Zarkower D (2011) DMRT1 promotes oogenesis by transcriptional activation of Stra8 in the mammalian fetal ovary. Dev Biol 356:63–70
Lei N, Heckert LL (2002) Sp1 and Egr1 regulate transcription of the Dmrt1 gene in Sertoli cells. Biol Reprod 66:675–684
Lei N, Heckert LL (2004) Gata4 regulates testis expression of Dmrt1. Mol Cell Biol 24:377–388
Lei N, Hornbaker KI, Rice DA, Karpova T, Agbor VA, Heckert LL (2007) Sex-specific differences in mouse DMRT1 expression are both cell type- and stage-dependent during gonad development. Biol Reprod 77:466–475
Lei N, Karpova T, Hornbaker KI, Rice DA, Heckert LL (2009) Distinct transcriptional mechanisms direct expression of the rat Dmrt1 promoter in Sertoli cells and germ cells of transgenic mice. Biol Reprod 81:118–125
Lynch M, O'Hely M, Walsh B, Force A (2001) The probability of preservation of a newly arisen gene duplicate. Genetics 159:1789–1804
Marques-Bonet T, Ryder OA, Eichler EE (2009) Sequencing primate genomes: what have we learned? Annu Rev Genomics Hum Genet 10:355–386
Matson CK, Murphy MW, Griswold MD, Yoshida S, Bardwell VJ, Zarkower D (2010) The mammalian doublesex homolog DMRT1 is a transcriptional gatekeeper that controls the mitosis versus meiosis decision in male germ cells. Dev Cell 19:612–624
Matson CK, Murphy MW, Sarver AL, Griswold MD, Bardwell VJ, Zarkower D (2011) DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 476:101–104
Matsuda M, Nagahama Y, Shinomiya A et al (2002) DMY is a Y-specific DM-domain gene required for male development in the medaka fish. Nature 417:559–563
Matsuda M, Shinomiya A, Kinoshita M et al (2007) DMY gene induces male development in genetically female (XX) medaka fish. Proc Natl Acad Sci USA 104:3865–3870
Matthey R (1933) La formule chromosomique et le probléme de la détermination sexuelle chez Ellobius letescens (Thomas) (Rodentia-Muridae-Microtinae). Arch Julius Klaus—Stift Vererb Forsch 28:65–73
Miura L (2007) An evolutionary witness: the frog Rana rugosa underwent change of heterogametic sex from XY male to ZW female. Sex Dev 1:323–331
Murdock C, Wibbels T (2003) Expression of Dmrt1 in a turtle with temperature-dependent sex determination. Cytogenet Genome Res 101:302–308
Nanda I, Zend-Ajusch E, Shan Z et al (2000) Conserved synteny between the chicken Z sex chromosome and human chromosome 9 includes the male regulatory gene DMRT1: a comparative (re)view on avian sex determination. Cytogenet Cell Genet 89:67–78
Nanda I, Kondo M, Hornung U et al (2002) A duplicated copy of DMRT1 in the sex-determining region of the Y chromosome of the medaka, Oryzias latipes. Proc Natl Acad Sci USA 99:11778–11783
Ohno S (1970) Evolution by gene duplication. Springer, Heidelberg
Okada E, Yoshimoto S, Ikeda N et al (2009) Xenopus W-linked DM-W induces Foxl2 and Cyp19 expression during ovary formation. Sex Dev 3:38–42
Oréal E, Mazaud S, Picard JY, Magre S, Carré-Eusèbe D (2002) Different patterns of anti-Müllerian hormone expression, as related to DMRT1, SF-1, WT1, GATA-4, Wnt-4, and Lhx9 expression, in the chick differentiating gonads. Dev Dyn 225:221–232
Quinn AE, Georges A, Sarre SD, Guarino F, Ezaz T, Graves JA (2007) Temperature sex reversal implies sex gene dosage in a reptile. Science 316:411
Quinn AE, Sarre SD, Ezaz T, Marshall Graves JA, Georges A (2011) Evolutionary transitions between mechanisms of sex determination in vertebrates. Biol Lett 7:443–448
Raymond CS, Murphy MW, O'Sullivan MG, Bardwell VJ, Zarkower D (2000) Dmrt1, a gene related to worm and fly sexual regulators, is required for mammalian testis differentiation. Genes Dev 14:2587–2595
Sakata N, Miyazaki K, Wakahara M (2006) Up-regulation of P450arom and down-regulation of Dmrt-1 genes in the temperature-dependent sex reversal from genetic males to phenotypic females in a salamander. Dev Genes Evol 216:224–228
Sinclair AH, Berta P, Palmer MS et al (1990) A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346:240–244
Smith CA, Roeszler KN, Ohnesorg T et al (2009) The avian Z-linked gene DMRT1 is required for male sex determination in the chicken. Nature 461:267–271
Sutton E, Hughes J, White S et al (2011) Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest 121:328–341
Tajima F (1993) Simple methods for testing molecular clock hypothesis. Genetics 135:599–607
Takehana Y, Demiyah D, Naruse K, Hamaguchi S, Sakaizumi M (2007) Evolution of different Y chromosomes in two medaka species, Oryzias dancena and O. latipes. Genetics 175:1335–1340
Takehana Y, Hamaguchi S, Sakaizumi M (2008) Different origins of ZZ/ZW sex chromosomes in closely related medaka fishes, Oryzias javanicus and O. hubbsi. Chromosome Res 16:801–811
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tanaka K, Takehana Y, Naruse K, Hamaguchi S, Sakaizumi M (2007) Evidence for different origins of sex chromosomes in closely related Oryzias fishes: substitution of the master sex-determining gene. Genetics 177:2075–2081
Tymowska J, Fischberg M (1973) Chromosome complements of the genus Xenopus. Chromosoma 44:335–342
Yoshimoto S, Ito M (2011) A ZZ/ZW-type sex determination in Xenopus laevis. FEBS 278:1020–1026
Yoshimoto S, Okada E, Oishi T et al (2006) Expression and promoter analysis of Xenopus DMRT1 and functional characterization of the transactivation property of its protein. Dev Growth Differ 48:597–603
Yoshimoto S, Okada E, Umemoto H et al (2008) A W-linked DM-domain gene, DM-W, participates in primary ovary development in Xenopus laevis. Proc Natl Acad Sci USA 105:2469–2474
Yoshimoto S, Ikeda N, Izutsu Y, Shiba T, Takamatsu N, Ito M (2010) Opposite roles of DMRT1 and its W-linked paralogue, DM-W, in sexual dimorphism of Xenopus laevis: implications of a ZZ/ZW-type sex-determining system. Development 137:2519–2526
Zhang J (2004) Evolution of DMY, a newly emergent male sex-determination gene of medaka fish. Genetics 166:1887–1895
Acknowledgements
We thank Dr. Hiroki Oota for helpful comments and suggestions. We are grateful to the reviewers and editors for their important comments and suggestions and critical reading of the manuscript. This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education Culture, Sports, Science, and Technology (M. I.).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editors: Tariq Ezaz and Jennifer Graves.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Table 1
Location of the conserved regions from the first exon of mammalian, avian, and reptile Dmrt1 (DOC 60 kb)
Supplemental Table 2
Amino acid sequence identity (%) of the DM domains among several vertebrate Dmrt1 orthologues, Xenopus Dm-W, and medaka Dmy (DOC 69 kb)
Supplemental Table 3
Tajima's relative rate test for sex-determining genes Dmy and its prototype gene Dmrt1 (DOC 63 kb)
Supplemental Table 4
Tajima's relative rate test for sex-determining genes Dm-W and its prototype gene Dmrt1 (DOC 71 kb)
Supplemental Table 5
Tajima's relative rate test for sex-determining genes Sry and its prototype gene Sox3 (DOC 59 kb)
Supplementary Fig. 1
Putative transcription factors and their binding cis-elements within the 5′-flanking sequences of Dmrt1 homologues. Binding sites for transcription factors were analyzed using the program TFSEARCH 1.3 (http://www.cbrc.jp/research/db/TFSEARCH.html). Black regions correspond to conserved regions in the mVISTA graph of Fig. 2. (A) Region 500-bp upstream of the coding exon 1 of human, mouse, and chicken Dmrt1. (B) Region 500-bp upstream of the noncoding exon 1 of frog Dmrt1 and Dm-W, and medaka Dmrt1 and Dmy. The regions −3,120 to −2,862 and −2,484 to −2,386 of medaka Dmy, which are homologous to the regions −500 to −250 and −150 to −75 of medaka Dmrt1, restrictively (Fig. 2c), is also shown. Human, H. sapiens; Mouse, M. musculus; Chicken, G. gallus; Frog (Xt), Xenopus tropicalis; Frog (Xl), X. laevis; Medaka, O. latipes (JPEG 1,698 kb)
Supplementary Fig. 2
Alignment of an evolutionarily conserved region and its potential binding sites for transcription factors among several amniote species. Nucleotides conserved among more than five species are shaded. Binding sites for transcription factors were analyzed as in Figure S1. The putative binding sites are indicated below the nucleotide sequences. Location of the conserved regions is shown in Supplementary Table 1 (JPEG 1,553 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mawaribuchi, S., Yoshimoto, S., Ohashi, S. et al. Molecular evolution of vertebrate sex-determining genes. Chromosome Res 20, 139–151 (2012). https://doi.org/10.1007/s10577-011-9265-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-011-9265-9