
Abstract
Previously we found that terfenadine, an H1 histamine receptor antagonist, acts as a potent apoptosis inducer in melanoma cells through modulation of Ca2+ homeostasis. In this report, focusing our attention on the apoptotic mechanisms activated by terfenadine, we show that this drug can potentially activate distinct intrinsic signaling pathways depending on culture conditions. Serum-deprived conditions enhance the cytotoxic effect of terfenadine and caspase-4 and -2 are activated upstream of caspase-9. Moreover, although we found an increase in ROS levels, the apoptosis was ROS independent. Conversely, terfenadine treatment in complete medium induced ROS-dependent apoptosis. Caspase-4, -2, and -9 were simultaneously activated and p73 and Noxa induction were involved. ROS inhibition prevented p73 and Noxa expression but not p53 and p21 expression, suggesting a role for Noxa in p53-independent apoptosis in melanoma cells. Finally, we found that terfenadine induced autophagy, that can promote apoptosis. These findings demonstrate the great potential of terfenadine to kill melanoma cells through different cellular signaling pathways and could contribute to define new therapeutic strategies in melanoma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Histamine is a biogenic amine that mediates numerous physiological and pathological processes through the activation of specific histamine receptors (H1, H2, H3, and H4) [1]. Although many in vitro and in vivo studies on the modulatory roles of histamine in tumor development and metastasis have been reported, the effect of histamine in the progression of some types of tumors remains controversial. In this respect, we have previously found that histamine stimulates melanoma cell growth and that H1 histamine receptor antagonists induce apoptosis in a variety of human melanoma cell lines [2, 3]. Other reports also prove the antitumoral activity of H1 histamine receptor antagonists in human leukemia, myeloma [4, 5], colon, and liver cancer cells [6].
In one exploration of the molecular pathways involved in the apoptosis induced by H1 antihistaminics, A375 cells were treated with terfenadine (TEF), a highly potent H1 histamine receptor antagonist [7]. We found that TEF induces DNA damage and caspase-2-dependent apoptosis [2]. Moreover, these effects are the consequence of a cytosolic Ca2+ increase induced by TEF treatment in a dose-dependent manner [3].
The endoplasmic reticulum (ER) is one of the most important intracellular Ca2+ storage organelles [8]. It has been established that Ca2+ plays a vital executioner role in the regulation of apoptosis. If the Ca2+ concentration in the cytoplasm increases, there is a resultant increase in nuclear Ca2+. In the nucleus, Ca2+ modulates nucleases involved in apoptosis [9]. Ca2+ also regulates another family of proteins involved in the control of apoptosis, the calpains. Calpains have been implicated in activation of ER-localized caspase-4 in human cells [10]. Ca2+ release from ER can also activate apoptosis through a mitochondrial-dependent pathway. ER Ca2+ release triggers rapid accumulation of Ca2+ by mitochondria and mitochondrial membrane permeabilization [11, 12], resulting in the release of pro-apoptotic factors from mitochondria such as cytochrome c and caspase-9 activation [13]. Moreover, the accumulation of Ca2+ in the mitochondrial matrix can stimulate oxidative phosphorylation and enhance reactive oxygen species (ROS) generation. Oxidative stress markedly sensitizes mitochondria toward mitochondrial membrane permeabilization induction and can also cause damage to nucleic acids and proteins [14].
In response to DNA damage, p53 becomes stabilized by phosphorylation and induces cell-cycle arrest through the accumulation of the cyclin-dependent kinase inhibitor p21. If p53 accumulates above a particular threshold, it can induce apoptosis by induction of pro-apoptotic members of the Bcl-2 family [15]. In addition, it has been found that cells can trigger p53-independent DNA-damage-induced apoptosis through transcriptional upregulation of the p53 homolog p73. p73-induced apoptosis is mediated by induction of Bax, Puma, and Noxa [16].
Multiple stress signals, such as ROS and rapid increases in intracellular Ca2+, can trigger both apoptosis and macroautophagy. In physiological conditions, macroautophagy (referred to here as autophagy) is the major pathway that eukaryotic cells use to degrade and recycle long-lived proteins and aging cytoplasmic components. However, in the presence of an apoptotic stimulus, autophagy has been described as both an alternative route to cell death (termed autophagic or type II cell death) and an adaptation mechanism to environmental stress. Moreover, autophagy and apoptosis are cross-linked at multiple levels, although their functional interrelationship is still being worked out [17–20].
In the present work, we have observed that the presence of serum in the medium of treatment decreases the intensity of the death stimulus produced by TEF on A375 melanoma cells. Thus, we have postulated that the environmental conditions in which cells receive TEF treatment may influence engagement of apoptotic mechanisms. The major aims of this study were to identify the apoptotic pathways activated by TEF and to analyze ROS involvement in TEF-induced apoptosis and autophagy on melanoma cells in different culture conditions.
Materials and methods
Chemicals and reagents
TEF, α-tocopherol (vitamin E), cyclic pifithrin-alpha, BAPTA-AM and 3-methyladenine were purchased from Sigma-Aldrich Quimica, S.A. (Madrid, Spain). Caspase-2 inhibitor was from Calbiochem (Darmstadt, Germany). Caspase-4 and caspase-9 inhibitors were from PromoCell GmbH (Heidelberg, Germany).
Acrylamide and bis-acrylamide solutions and Precision Plus Protein Standards were obtained from Bio-Rad Laboratories (Hercules, CA, USA). Bicinchoninic acid solution and electrophoresis reagents were all obtained from Sigma-Aldrich Quimica, S.A. (Madrid, Spain).
Polyclonal rabbit anti-cleaved caspase-3 antibody and Atg7 rabbit monoclonal antibody were obtained from Cell Signaling Technology (Danvers, MA, USA). Mouse monoclonal to histone H2A.X (phospho S139), mouse monoclonal antibody anti-p21, mouse monoclonal antibody anti-Noxa, and rabbit polyclonal antibody to LC3B were from Abcam (Cambridge, UK). Mouse monoclonal antibody PhosphoDetect™ anti-ATM (pSer1981), rabbit polyclonal antibody PhosphoDetect™ anti-p53 (pSer15), and mouse monoclonal antibody anti-p73 were obtained from Calbiochem (Darmstadt, Germany). A mouse anti-cytochrome c antibody was purchased from BD Pharmingen (San Diego, CA, USA) and the anti-actin antibody was from Sigma-Aldrich Co. (St Louis, MO, USA). HRP-F(ab’)2 goat anti-mouse IgG (H + L) and HRP-F(ab’)2 goat anti-rabbit IgG (H + L) secondary antibodies were from Zymed Laboratories (South San Francisco, CA, USA), and Alexa Fluor® goat anti-rabbit and Alexa Fluor® goat anti-mouse secondary antibodies were from Molecular Probes (Europe BV, Leiden, The Netherlands).
Cell culture
Human melanoma cell lines A375, HT144 and Hs294T (obtained from American Type Culture Collection, Rockville, MD, USA) were routinely cultured in 75 cm2 flasks and grown in a monolayer in Dulbecco’s Modified Eagle’s Medium (Sigma-Aldrich Quimica, S.A., Madrid, Spain), supplemented with 10% (v/v) heat-inactivated FCS (Biochrom AG, Berlin, Germany), 2 mM l-glutamine and antibiotic solution (100 μg ml−1 streptomycin and 100 IU ml−1 penicillin) (Sigma-Aldrich Quimica, S.A., Madrid, Spain) at 37°C in a 5% CO2–95% air–water saturated atmosphere. Trypsinization was performed at harvesting. The cell lines were examined during culture with an inverted microscope (Olympus CK-2, Olympus, Melville, NY, USA). Cells in the exponential phase of growth were used in the experiments. Stock solutions of TEF (50 mM) were prepared in dimethyl sulfoxide (Sigma-Aldrich Quimica, S.A., Madrid, Spain). The final treatment concentration of dimethyl sulfoxide was not >0.05% (v/v) dimethyl sulfoxide/culture medium. Control groups were either untreated or treated with the corresponding doses of dimethyl sulfoxide alone.
Cell viability assay
Cells were seeded into flat-bottomed 96-well microtiter plates at a density of 1 × 105 cells ml−1 and allowed to attach to the wells overnight. Subsequently, cells were treated, and controls without treatment were also included in each experiment. Cell viability was determined by means of a colorimetric XTT viability assay kit (Roche Molecular Biochemicals, Indianapolis, IN, USA) in accordance with the instructions of the supplier. Absorbance at 490 nm was measured using a microplate reader (ELx800, Bio-Tek Instruments, Winooski, VT), and cell viability was calculated in relation to untreated control cells as follows: (experimental absorbance/untreated control absorbance) × 100. Twelve replicate wells were analyzed for each test, and each assay was repeated 3–4 times.
Nuclear staining and immunofluorescence
Melanoma cells were seeded at a density of 1 × 105 cells ml−1 overnight on sterile glass coverslips in 24-well plates before being treated. Treated and untreated cells on the coverslips were washed twice with phosphate-buffered saline (PBS) and fixed and permeabilized with 70% methanol at −20°C overnight. Blocking of non-specific binding of antibodies was performed by incubation of cells in PBS buffer containing 1% BSA at room temperature (RT) for 60 min. Incubations with primary and secondary antibodies, diluted in PBS buffer, were performed at 4°C overnight and at RT for 60 min, respectively. Cells were incubated with 20 μg ml−1 propidium iodide (Sigma-Aldrich Quimica, S.A., Madrid, Spain) or with 1 μg ml−1 4′,6-diamidino-2-phenylindole (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 15 min at RT to stain the nucleus. Between all steps, cells were washed for 3 × 10 min with PBS at RT. Coverslips with stained cells were mounted using Fluoromount G (Southern Biotech, Hatfield, PA, USA). Digital images were obtained with a fluorescent microscope (Olympus BX51, Olympus, Melville, NY, USA) or by sequential acquisition. Images were obtained by sequential acquisition with a confocal microscope (Olympus FV500, Olympus, Melville, NY, USA).
Transmission electron microscopy
Treated and untreated A375 cells were harvested and washed in 0.1 M phosphate buffer, pH 7.3, sedimented at 1,500 rpm, and immediately fixed with 2.5% glutaraldehyde (Sigma-Aldrich Quimica, S.A., Madrid, Spain) in the same buffer for 1 h. Subsequently, they were post-fixed with 1% OsO4 in cacodylate buffer, then alcohol-dehydrated and embedded in Epon as previously reported [21]. Thin sections were collected on nickel grids and stained with uranyl acetate for observation under a Philips EM 208 electron microscope (Philips Electronic Instruments, Eindhoven, Netherlands).
Flow cytometry
To measure intracellular ROS, treated and untreated cells were loaded with 10 μM carboxi-H2DCFDA probe (Molecular Probes; Europe BV, Leiden, The Netherlands) during the last 30 min of treatment. Then, cells were harvested by trypsinization and washed twice with PBS before being analyzed by flow cytometry. Flow cytometric analysis was performed on at least 10,000 cells using a Coulter Epics Elite ESP (EPICS Division Coulter Corp.), and results were analyzed using the WinMDI 2.8 program.
Mitochondrial superoxide detection
MitoSOX™ Red mitochondrial superoxide indicator (Molecular Probes, Europe BV, Leiden, The Netherlands) is a fluorogenic dye for highly selective detection of the superoxide in the mitochondria in living cells. MitoSOX™ Red reagent is live-cell permeant and is rapidly and selectively targeted to the mitochondria. Once in the mitochondria, MitoSOX™ Red reagent is oxidized by superoxide and exhibits red fluorescence. Cells were seeded into 96-well microplates at a density of 1 × 105 cells ml−1 and allowed to attach to the wells overnight. Subsequently, cells were treated, and controls without treatment were also included in each experiment. Cells were loaded with 5 μM MitoSox and incubated for 10 min at 37°C, protected from light. Then, cells were washed with warm buffer and mitochondrial superoxide was measured using a fluorimeter/microplate reader (Fluoroskan Ascent, Labsystems) at excitation/emission of approximately 510/580 nm.
Caspase activity assays
The activity of caspases-2, -4, -9, and -3 was measured using commercially available Promokine colorimetric assay kits (PromoCell GmbH, Heidelberg, Germany). Briefly, samples of untreated and treated cells were washed with cold PBS and lysed on ice in 50 μl of cold lysis buffer. Cell lysates were centrifuged at 10,000×g for 1 min to precipitate cellular debris. The protein content of each sample was quantified by means of the bicinchoninic acid method. One hundred micrograms of protein extracts were incubated with 100 μl of kit products, and assays were performed in triplicate in 96-well plates on the basis of the manufacturer’s protocol using a microplate reader (ELx800, Bio-Tek Instruments, Winooski, VT) at 405 nm. Comparison of the absorbance from apoptotic samples with that of an untreated control allowed determination of fold increase in caspase activity.
Superoxide dismutase (SOD) and catalase (CAT) activity assays
The activity of SOD was measured using the Superoxide Dismutase (SOD) Activity Assay Kit, from BioVision (Mountain View, CA). This kit utilizes WST-1 that produces a water-soluble formazan dye upon reduction with superoxide anion. The rate of the reduction with a superoxide anion is linearly related to the xanthine oxidase (XO) activity, and is inhibited by SOD. Therefore, the inhibition activity of SOD can be determined by a colorimetric method. Samples of untreated and treated cells were lysed in ice cold 0.1 M Tris/HCl, pH 7.4 containing 0.5% Triton X-100, 5 mM β-ME, 0.1 mg/ml PMSF. Cell lysate was centrifuged the crude tissue at 14,000×g for 5 min at 4°C. The supernatant contained total SOD activity from cytosolic and mitochondria. The assay was performed in triplicate using 96-well microplates, according to the manufacturer’s instruction. The absorbance was read at 450 nm using a microplate reader (ELx800, Bio-Tek Instruments, Winooski, VT) and the activity was calculated as the inhibition rate of XO.
CAT was measured using the Catalase Activity Assay Kit (BioVision). In the assay, catalase first reacts with H2O2 to produce water and oxygen. The unconverted H2O2 reacts with OxiRed™, probe to produce a product, which can be measured by a colorimetric method. Cell lysates were centrifuged at 10,000×g for 15 min at 4°C and the supernatants were collected for the assay. The assay was performed in triplicate using 96-well microplates, according to the manufacturer’s instruction. The absorbance was read at 570 nm using a microplate reader (ELx800, Bio-Tek Instruments, Winooski, VT) and the activity was calculated in nmol/min/ml.
SDS–polyacrylamide gel electrophoresis and immunoblotting
Treated and untreated melanoma cells were harvested by trypsinization. Washed in cold PBS and lysed in RIPA lysis buffer (80 mM Tris·HCl pH 8, 150 mM NaCl, 1% NP 40, 0.5% sodium deoxycholate, 0.1% SDS) containing Protease Inhibitor Cocktail (Sigma-Aldrich Quimica, S.A., Madrid, Spain) for 15 min on ice. Lysates were then cleared by centrifugation at 10,000 g, for 5 min. Total protein concentration was determined using the bicinchoninic acid assay. Equal amounts of total proteins from each sample were resolved by electrophoresis on an SDS–polyacrylamide gel and then transferred onto a nitrocellulose membrane (Whatman GmbH, Dassel, Germany). The blots were blocked with PBS containing 5% non-fat milk and 0.1% Tween-20 for 1 h and then probed overnight with an appropriate dilution of the primary antibody. After washing, membranes were incubated for 1 h with horseradish peroxidase-linked secondary antibody. Finally, proteins were visualized by enhanced chemiluminescence using the SuperSignal® West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL, USA).
Transfection of Atg7 small interference RNA (siRNA)
A375 and HT144 melanoma cells were transfected by means of the SignalSilence Atg7 siRNA I (Cell Signalling Technology, Inc., Danvers, MA, USA) using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) as transfection agent, according to the manufacturer’s instructions. The Fluorescein Conjugate SignalSilence Control siRNA (Cell Signalling Technology, Inc., Danvers, MA, USA) was used for optimization experiments, assessment of transfection efficiency and as a negative siRNA control.
Cells (104) were seeded in 96-well microplates and then 100 nM Atg7 siRNA per well were mixed with Lipofectamine RNAiMAX agent in OPTI-MEM I medium (Invitrogen, Carlsbad, CA, USA) and added to the cells. Seventy-two hours after transfection, cell lysates were obtained to verify the Atg7 protein knockdown by Western Blotting using an Atg7 monoclonal antibody. In parallel experiments, 48 h after transfection, cells were treated with 10 μM TEF for several hours and cell viability was determined by XTT assay. Four separate experiments were performed and the gene knockdown was higher than 70% with less than 5% of cell death.
Statistical analysis
The level of statistical significance between sample means was determined using the Student t-test; P < 0.05 was considered to be statistically significant, and P < 0.01 was considered to be statistically very significant.
Results
Terfenadine induces distinct apoptotic pathways in A375 human melanoma cells
We began by studying the ability of TEF to induce apoptosis in various melanoma cell lines treated in complete medium (containing 10% FCS). We assessed the effect of different concentrations of TEF using the XTT assay (Fig. 1a) and we saw that the cytotoxic effect of TEF was dose-dependent in all tested cell lines. The half maximal inhibitory concentration value (IC50) after 24 h of TEF treatment in complete medium was 10.4 μM for A375 cells, 9.9 μM for Hs294T cells and 9.6 for HT144 cells. Then, we analyzed the effect of TEF treatment on A375 cells in medium deprived of serum (Fig. 1b). In this case, the effect of TEF was also dose-dependent. The IC50 after 8 h of TEF treatment in the absence of serum was 6.8 μM for A375 cells. These results indicate that TEF induces dose-dependent cytotoxicity in both culture conditions.

Dose and time-dependent apoptosis induced by TEF on A375 melanoma cells in different culture conditions. a The dose-dependent effect of TEF on A375, HT144 and Hs294T cells. Cells were treated with different concentrations of TEF in complete medium (supplemented with 10% FCS) and cell viability was assessed by means of an XTT viability assay after the indicated times. b The dose-dependent effect of TEF/SS on A375 cells. Cells were treated with different concentrations of TEF in serum starvation (SS) conditions and cell viability was assessed by means of an XTT viability assay after the indicated times. c The time-dependent effect of TEF on A375 cells. Cells were treated with 10 μM TEF in complete medium or in absence of FCS for the indicated times, and cell viability was assessed by means of an XTT viability assay. d Activated caspase-3 detection. Control cells, cells treated with 10 μM TEF/SS for 6 h and cells treated with 10 μM TEF for 24 h were stained for cleaved caspase-3 (red). Then, cell nuclei (blue) were counterstained with 4′,6-diamidino-2-phenylindole for 10 min, and the cells were analyzed under a fluorescence microscope. Bar 10 μm
As shown in Fig. 1c, the cytotoxic effect of TEF was time-dependent either in serum-free or in complete medium. Moreover, starvation (SS) enhanced the cytotoxic effect of TEF. While 10 μM TEF/SS required only about 8 h to induce practically total cell death, 10 μM TEF required about 32–40 h to induce the same level of cytotoxicity. We studied the expression of cleaved caspase-3 by fluorescence microscopy, and we found that 10 μM TEF/SS and 10 μM TEF treatment for 6 and 24 h, respectively, induced caspase-3 activation and morphological nuclear changes that are characteristic of apoptosis (Fig. 1d), indicating that TEF induces cell death by apoptosis in human melanoma cells either in complete medium or in medium deprived of serum.
To study if SS was involved in the molecular mechanisms of apoptosis induced by TEF, a time-course analysis of activation of the initiator caspases-2, -4, and -9 and the executioner caspase-3 was performed in melanoma cells treated with TEF (Fig. 2). We found that TEF/SS activated caspases-2, -4, and -3 after 2 and 3 h of treatment, but there was no significant activation of caspase-9 at that time point. The activation of caspase-9 was observed only after 4 h of treatment, when the activity of caspases-2, -4, and -3 had increased dramatically (Fig. 2a). These results indicate that caspase-9 is activated downstream of caspase-3.

Effect of serum starvation on TEF-induced caspase pattern activation. Cells were treated with 10 μM TEF/SS (a) and with 10 μM TEF in complete medium (b) for the indicated periods of time. Total cell lysates were prepared according to the manufacturer’s protocol. Assays were performed in triplicate in 96-well plates using a microplate reader. The activity of caspases was calculated as fold increase relative to control cells without TEF treatment. Earlier time points are not shown in a and b because the levels are no different to control ones. Control cells were cultured in the absence of FCS for 4 h in a and in complete medium in b. c Effect of caspase-2 inhibitor (z-VDVAD-FMK), caspase-4 inhibitor (z-LEVD-FMK), and caspase-9 inhibitor (z-LEHD-FMK) on cell death induced by 10 μM TEF. Cells were pre-treated with caspase inhibitors when indicated for 1 h and then challenged with 10 μM TEF for 24 h. Then, cell viability was assessed using the XTT assay. Data represent mean ± SD of three determinations from three separate experiments. *P values < 0.05; **P values < 0.01
In contrast, the addition of TEF in complete medium was found to activate all of the initiator caspases-2, -4, and -9 after 8 h of treatment. The activation levels of the studied caspases reached a maximal level at 28 h (Fig. 2b). These results suggest that TEF activates three distinct apoptotic pathways. To confirm the role of initiator caspases in TEF-induced apoptosis, we pre-treated A375 cells with specific inhibitors for each caspase (-2, -4, and -9) before the addition of TEF. We found that each specific caspase inhibitor partially blocked the effect of TEF at 28 h of treatment (Fig. 2c).
Terfenadine induces ROS generation
To test if TEF treatment could lead to ROS generation, we measured intracellular ROS levels by flow cytometry after labeling cells with carboxi-H2DCFDA, a specific ROS-detecting fluorescent dye. We began by investigating the effect of SS on ROS generation and observed that the absence of serum in culture medium for 4 h was enough to produce a significant increase in ROS levels. Moreover, we found that TEF/SS also produced a low increase in ROS levels after 4 h of treatment, compared to control cells cultured in serum-free medium for 4 h (Fig. 3a). We further analyzed ROS generation by TEF in complete medium and found a significant increase of ROS levels at 30 min. Then, ROS generation decreased to control levels at 8 h, but an important increase followed at 16 h and reached a maximal level at 28 h of treatment (Fig. 3b). Taken together, these results indicate that TEF induces an intracellular ROS increase. In addition, pre-treatment with BAPTA-AM, a calcium chelator, prevented ROS production (Fig. 3c), indicating that the observed rise of ROS generation is dependent on intracellular calcium concentration increase. Moreover, we observed a mitochondrial superoxide increase after TEF treatment using MitoSox, a mitochondrial superoxide indicator (Fig. 3d). Overall, these results indicate that TEF treatment induces mitochondrial and cytosolic ROS levels increase. Furthermore, pre-treatment with the antioxidant α-tocopherol (vitamin E) for 1 h before the addition of TEF effectively blocked ROS induction in TEF-treated cells, either in serum-free medium or in complete medium (Fig. 3e).

TEF induces ROS production in A375 melanoma cells. Flow cytometry analysis of intracellular ROS using a carboxi-H2DCFDA probe. A375 cells were treated with 10 μM TEF in the absence (a) or in the presence (b) of FCS for the indicated periods of time. ROS levels are expressed as fold increase relative to control cells cultured in complete medium. c ROS induction by TEF is prevented by BAPTA-AM. A375 melanoma cells were treated with 10 μM TEF for the indicated times in the presence of 10 μM BAPTA-AM. Control cells were cultured in complete medium. ROS levels are expressed as fold increase relative to control cells cultured in complete medium. d Analysis of mitochondrial superoxide using MitoSOX. A375 cells were treated with 10 μM TEF for the indicated periods of time. Mitochondrial superoxide levels were measured by reading absorbance at 548 nm. Control cells were cultured in complete medium. e ROS induction by TEF is blocked by vitamin E (vit E). A375 melanoma cells were treated with 10 μM TEF/SS or 10 μM TEF for 30 min, either in the presence or in the absence of 100 μM vit E. Control cells were cultured in complete medium. Data represent mean values ± SD of three determinations from three separate experiments. *P values < 0.05; **P values < 0.01
In order to investigate the role of the endogenous antioxidant defense system in TEF treatment, we studied the activation of SOD and CAT, which are in charge of the superoxide and hydrogen peroxide remove, respectively [14]. We saw that TEF did not induce SOD activity. Indeed, SOD activity decreased at the time of 8 and 24 h of treatment (Fig. 4a). However, CAT activity was notably increased at 30 min and later on at 8 and 24 h (Fig. 4b). These results indicate that the observed ROS levels increase is not due to a failure in the antioxidant defense system.

Analysis of SOD and CAT activation in A375 melanoma cells. Cells were treated with 10 μM TEF for the indicated times. After treatment, total cell lysates were prepared according to the manufacturer’s protocol and assays were performed in triplicate in 96-well plates. The activity of SOD (a) and CAT (b) was measured by reading absorbance at 450 and 570 nm respectively using a microplate reader
Involvement of ROS generation in terfenadine-induced DNA damage and apoptosis
To analyze the role of ROS in TEF-induced apoptosis, we assessed cell viability of TEF-treated A375 cells in the presence of vitamin E. Vitamin E did not protect cells from cytotoxicity induced by TEF/SS, but it almost completely blocked the cytotoxic effect of TEF in cells treated in complete medium (Fig. 5a). The histone H2A.X becomes phosphorylated in response to DNA double-strand breaks, and we further studied the effect of ROS on DNA damage by analyzing phosphorylated H2A.X expression by Western blot. We found that both TEF/SS and TEF induced a significant increase of phosphorylated H2A.X. In addition, inhibition of ROS with vitamin E significantly reduced phosphorilated H2A.X in A375 cells treated with TEF in complete medium but not in cells treated with TEF/SS (Fig. 5b). These results indicate that oxidative stress is an upstream effector of terfenadine-induced DNA damage and apoptosis under normal conditions but not in cells treated in serum-free medium. Moreover, vitamin E prevented citotoxicity (Fig. 5c), caspase-3 cleavage and phosphorylated H2A.X expression (Fig. 5d) in HT144 melanoma cells treated with TEF in complete medium.

Involvement of ROS generation in cytotoxicity and DNA damage induced by terfenadine. a, b Effects of vitamin E (vit E) on cell death and DNA damage induced by TEF in A375 melanoma cells. a Cells were treated with 10 μM TEF/SS or 10 μM TEF for the indicated times; either in the presence or in the absence of 100 μM vit E. Cell viability was assessed using the XTT viability assay. Data represent mean ± SD of three determinations from three separate experiments. NS P values > 0.5; **P values < 0.01. b Western blot analysis of H2A.X (pSer 139) normalized with actin. 50 μg of total cell extracts were separated by SDS–polyacrylamide gel electrophoresis and analyzed by immunoblotting. A375 cells were treated with 10 μM TEF/SS or with 10 μM TEF either in the presence or in the absence of 100 μM vit E for the indicated periods of time. c, d Effects of vitamin E (vit E) on cell death and DNA damage induced by TEF in HT144 melanoma cells. c Cells were treated with 10 μM TEF for 28 h; either in the presence or in the absence of 100 μM vit E. Cell viability was assessed using the XTT viability assay. Data represent mean ± SD of three determinations from three separate experiments. **P values < 0.01. d Western blot analysis of cleaved caspase-3 and H2A.X (pSer 139) normalized with actin. 50 μg of total cell extracts were separated by SDS–polyacrylamide gel electrophoresis and analyzed by immunoblotting. HT144 cells were treated with 10 μM TEF either in the presence or in the absence of 100 μM vit E for 28 h
Involvement of ROS generation in the p53/p73 pathway induced by terfenadine
p53/p73 pathway were analyzed by Western Blot and we found that p53, p73 and p21 levels decreased in TEF/SS treated A375 cells but no alteration of Noxa levels was observed (Fig. 6a). Conversely, an increase of p53, p73, p21 and Noxa protein levels was observed in cells treated with TEF in complete medium (Fig. 6c), suggesting that under these conditions, the p53/p73 pathway could be involved inn TEF-induced apoptosis.

Involvement of ROS generation in the p53/p73 pathway induced by TEF. Western blot analysis of p53 (pSer 15), p73, p21, and Noxa normalized with actin. 50 μg of total cell extracts were loaded per lane. A375 cells were treated with 10 μM TEF/SS in a and b or with 10 μM TEF in c and d, either in the presence b, d or in the absence a, c of 100 μM vitamin E (vit E) for the indicated times. e HT144 cells were treated with 10 μM TEF in the presence or in the absence of Vit E for 28 h
We further analyzed the effect of ROS in p53/p73 pathway activation. As expected, the presence of vitamin E did not alter the expression of tested proteins in TEF/SS-treated cells (Fig. 6b). However, when A375 cultures were treated in complete medium, vitamin E significantly reduced TEF-induced p73 accumulation and Noxa expression but did not block p53 and p21 induction (Fig. 6d). Similar results were obtained with HT144 cells treated with TEF in complete medium, in the absence or presence of Vit E (Fig. 6e).
p73 localization assay by confocal microscopy demonstrated that 16 h of TEF treatment induced p73 translocation from cytoplasm to the nucleus in A375 cells (Fig. 7a), which reflects its activation. Moreover, vitamin E partially prevents this translocation, indicating ROS dependence. We also studied cytochrome c localization by confocal microscopy. As shown in Fig. 7b, TEF treatment for 16 h induced cytochrome c release from mitochondria to cytosol, as is reflected by the transition from a punctate distribution in the cytoplasm in control cells to diffuse localization in the cytoplasm in treated cells. Vitamin E also partially prevented cytochrome c release from the mitochondria to the cytosol. Overall, these results indicate that p73 and Noxa induction by TEF—but not p53 and p21 induction—depend on ROS generation and that the mitochondrial apoptotic pathway is involved in TEF-induced apoptosis.

Terfenadine induces ROS-dependent p73 nuclear translocation and cytochrome c release. a p73 localization analysis. A375 control cells and 10 μM TEF-treated cells for 16 h, either in the presence or in the absence of 100 μM vitamin E (vit E) were labeled for p73 (green), and then nuclei were counterstained with 4′,6-diamidino-2-phenylindole (blue). b Cytochrome c localization analysis. Control and 10 μM TEF-treated cells for 16 h, either in the presence or in the absence of 100 μM vit E were labeled for cytochrome c (green), and then cells were incubated with 4′,6-diamidino-2-phenylindole to stain the nucleus (blue). The cells were viewed and analyzed with a confocal microscope. Bars 10 μm
Terfenadine induces p53-independent citotoxicity
Since vitamin E prevents cytotoxicity induced by TEF, but not p53 expression increase, we assessed the effect of cyclic pifithrin-alpha (PFT-α), a pharmacological inhibitor of p53 transcriptional activity, on cell viability of cells treated with TEF during 24 h in complete medium. We found that PFT-α did not prevent the cytotoxic effect of TEF either in A375 (Fig. 8a) or in HT144 (Fig. 8b). These results indicate that TEF induces apoptosis independently of p53 transcriptional activity in melanoma cells.

Effect of p53 inhibition on cell death induced by TEF. A375 (a) and HT144 melanoma cells (b) were incubated with 30 μM pifithrin-alpha (PFT-α) during 1 h before 10 μM TEF addition for 28 h in complete medium. Then, cell viability was assessed using the XTT viability assay. Data represent mean ± SD of three determinations from three separate experiments. NS P values > 0.5
Terfenadine induces autophagy by ROS-dependent and -independent mechanisms
Because reticular stress and ROS production have been linked with autophagy, we investigated autophagy induction by TEF treatment. Electron microscopy examination of cells treated with TEF for 8 h revealed a massive vacuolization of the cytoplasm (Fig. 9a). Autophagic vacuoles of both double and multiple membranes (Fig. 9b) and at various stages (Fig. 9c) can be observed. Moreover, Western blot analysis revealed that both TEF and TEF/SS could rapidly elicit accumulation of LC3B-II, a lipidated form of LC3B, which is localized on the autophagic vacuoles (Fig. 10a). Indeed, confocal microscopic analysis indicated formation of the LC3B punctate appearance in cells treated with TEF for 8 h and with TEF/SS for 4 h, which would represent the autophagic vacuoles (Fig. 10b).

Terfenadine induces ultrastructural features of autophagy. A375 cells were treated with 10 μM TEF for 8 h and processed for electron microscopy. a The induction of autophagy is manifested by massive vacuolization of the cytoplasm. Arrows indicate autophagic vacuoles different in size. b Note the double membrane or the multimembrane structure of the autophagic vacuoles. c In the same cell, immature autophagic vacuoles characterized by an electron density equivalent to the cytoplasm coexist with late vesicles, in which catabolic processes have been already started (characterized by an increased electron density). Bars 1 μm

ROS involvement in TEF-induced autophagy. a Western blot analysis of LC3B normalized with actin. 50 μg of total cell extracts were loaded per lane. Cells were treated with 10 μM TEF or with 10 μM TEF/SS for the indicated times. b LC3B localization analysis. Control cells cultured in complete medium and cells treated with 10 μM TEF for 8 h were stained for LC3B (red), and then cells were incubated with 4′,6-diamidino-2-phenylindole to stain the nuclei (blue). The cells were viewed and analyzed with a confocal microscope. Bar 10 μm. c Western blot analysis of LC3B normalized with actin. 50 μg of total cell extracts were loaded per lane. Cells were treated with 10 μM TEF or with 10 μM TEF/SS in the presence of 100 μM vitamin E (vit E) for the indicated times
We next examined how ROS production could affect autophagy triggered by TEF and found that vitamin E did not block LC3B-II accumulation induced by TEF/SS but strongly inhibited LC3B-I to LC3B-II conversion when cells were treated in the presence of serum (Fig. 10c). These results indicate that autophagy induced by TEF can be independent or dependent of ROS generation.
Finally, we down-regulated Atg 7 by means of siRNA transfection to investigate how autophagy inhibition could affect TEF-induced cell death. Transfection optimization was performed using a fluorescein conjugate control siRNA, that doesn’t lead to the specific degradation of any cellular message and also serves as a negative control. Figure 11a shows the high efficiency of transfection: Fluorescent cells have incorporated the fluorescein conjugate control siRNA. Using the same culture conditions, Atg7 siRNA was transfected to the cells and 72 h after transfection cell lysates were obtained to verify the Atg7 protein knockdown by Western Blotting (Fig. 11b). The viability experiments were performed by XTT assay after 48 h of transfection. As can be seen in Fig. 11c, Atg 7 down-regulation partially prevented cell death induced by TEF at initial times of treatment in both A375 and HT144 cells but not when TEF had finally killed most of the cells. These results indicate that autophagy acts by promoting cell death in TEF-treated melanoma cells.

Effect of Atg 7 downregulation in TEF-induced cytotoxicity. a Assessment of transfection efficiency using SignalSilence Control siRNA (Fluorescein Conjugate), that also serves as a negative control. Cells were viewed under a confocal microscope. b Down-regulation of Atg 7 protein following transfection with specific Atg 7 siRNA. 50 μg of total cell extracts of transfected and non transfected cells were loaded per lane, separated by electrophoresis and analyzed by immunoblotting. c Atg 7 down-regulation partially prevented TEF-induced cytotoxicity. Cell viability was determined after 48 h of siRNA transfection. *P values < 0.05
Discussion
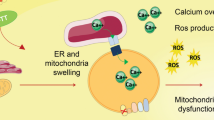
Most of the currently available anticancer drugs are often non-selective antiproliferative agents that preferentially kill dividing cells [22]. Thus, the search for new anticancer agents or therapeutic strategies without these side effects is pressing. In this context, we previously demonstrated that TEF induces dose-dependent apoptosis in a variety of human melanoma cell lines but not in normal melanocytes [2]. Depletion of ER Ca2+ stores and Ca2+ influx from the extracellular medium mediate TEF-induced apoptosis [3]. In the present work, we have shown that TEF can activate distinct apoptotic signaling pathways and that SS increases TEF-induced cytotoxicity in A375 human melanoma cells. Importantly, we have also observed that both TEF treatment and SS induce ROS generation; however, only the apoptosis induced by TEF in the presence of serum was ROS-dependent. TEF treatment in serum-free medium induced ROS-independent apoptosis because the inhibition of ROS production by vitamin E did not protect against the apoptosis. RE seems to form a largely interconnected tubular network that is in close contact with the mitochondria. Upon ER Ca2+ depletion, the mitochondrial surface, which is situated in close proximity to the ER, is exposed to high concentration of calcium. High concentrations of Ca2+ appear to have several negative effects on mitochondrial function. At the heart of understanding how Ca2+ can be both a physiological and a pathological effector of mitochondrial function is the issue of how Ca2+ modulates mitochondrial ROS generation. The majority of ROS are products of mitochondrial respiration. Stimulation of the respiratory chain activity by Ca2+ would enhance ROS output by making the whole mitochondrion work faster and consume more O2. Indeed, mitochondrial ROS generation correlates well with metabolic rate [23]. Here we have demonstrated that ROS induction by TEF is dependent on intracellular calcium concentration increase and that there is a production of ROS into the mitochondria in response to TEF treatment.
Other authors have also described that starvation provided protection to normal cells, but not to a variety of cancer cells treated with chemotherapy drugs that are implicated in the generation of ROS and DNA damage [23]. In addition, some results show that although most tumor cells exhibit an antioxidant phenotype, human melanoma cells have depressed antioxidant capacity compared to normal melanocytes [24]. We have seen that the mitochondrial antioxidant enzyme SOD was not activated by TEF, but CAT was activated in response to TEF treatment, indicating that the antioxidant system is not completely depressed. Our observation that caspase-4 was activated by TEF was not surprising because caspase-4 is activated upon ER Ca2+ release [10]. This result was validated by inhibition of caspase-4 activity. In addition, we have also shown that TEF can potentially activate distinct intrinsic apoptotic signaling pathways that are summarized in Fig. 11. The relevance of these signaling pathways and the precise mechanism of activation appear to differ depending on the intensity of the death stimulus or the presence of survival factors in the medium of TEF treatment.
A severe apoptotic stimulus produced by TEF in medium deprived of serum quickly leads to Ca2+-dependent DNA damage and activation of caspase-2 as the predominant mechanism of apoptosis induction, involving the mitochondrial apoptotic pathway [2, 3]. Caspase-2, which is activated by an autoproteolytic mechanism in response to DNA damage, can directly interact with the mitochondria and trigger mitochondrial membrane permeabilization and cytochrome c release [25, 26]. In this case, we have seen that caspase-9 is activated downstream of caspase-2 and -4. The mechanism of caspase-9 activation can result from cytochrome c release. Another possibility is that caspase-9 becomes activated by caspase-4 activity because it has been shown that, following its activation at the ER, caspase-4 can directly cleave pro-caspase-9 in a mitochondrial-independent manner [27, 28].
p53/p73 pathway induction in cells treated with TEF in medium deprived of serum was studied. We observed a decrease in p53, p73 and p21 and no change in Noxa protein levels, indicating that the p53/p73 pathway is not involved in the activation of the mitochondrial apoptotic pathway. A decrease in p53, p73 and p21 protein levels can result from the failure of proper protein folding caused by disruption of ER Ca2+ homeostasis [29]. We have also demonstrated that although ROS levels increase, the high intensity of the ER Ca2+ release seems sufficient to activate distinct apoptotic pathways without the contribution of ROS as an apoptotic signaling amplification mechanism.
By contrast, treatment with TEF in complete culture medium triggers ROS-mediated apoptosis and DNA damage. This treatment induces a lower intensity of Ca2+ release that requires ROS production by mitochondria to enhance the apoptotic stimulus. In this cellular context, we have observed a simultaneous activation of caspases-2, -4, and -9, suggesting activation of at least three distinct apoptotic mechanisms. We also found an accumulation of p53, p73, p21, and Noxa. These results indicate that the p53/p73 pathway can contribute to mitochondrial membrane permeabilization induction, cytochrome c release, and caspase-9 activation. Intriguingly, ROS inhibition by vitamin E partially prevented p73 and Noxa expression but not p53 and p21 induction, suggesting that Noxa expression and apoptosis are regulated independently of p53. Recently, other authors have described the role of Noxa in p53-independent apoptosis in melanoma cells [30–32]. On the other hand, overexpression of E2F1, which stimulates transcription of the p73 gene, has been shown to induce apoptosis in melanoma cells [33] and sensitize melanoma cells to apoptosis [34]. Moreover, Tuve and colleagues have demonstrated that a transcriptionally active form of p73 (TA-p73β) increases chemosensitivity of human malignant melanomas to the standard therapeutic agents adriamycin and cisplatin [35]. Importantly, we have shown that TEF induces p73 accumulation, p73 nuclear translocation, and Noxa expression, apparently in a p53-independent manner. We have hypothesized that p73 might have a role in Noxa induction and apoptosis. Taken together, these results suggest that agents such as TEF that induce the expression of p73 and Noxa might be clinically useful in the treatment of melanoma.
Finally, we have shown that TEF induces autophagy by electron microscopy, LC3 dot formation, and LC3 conversion from LC3-I to LC3-II. Again, the intensity of the death stimulus determines the mechanism responsible for autophagy activation. We observed that a severe apoptotic stimulus induced autophagy independently of ROS generation, whereas autophagy induced by a more moderate stimulus was dependent on ROS production. In the presence of an apoptotic stimulus, autophagy has been described as both an alternative route to cell death and an adaptation mechanism to environmental stress. In order to investigate the role of autophagy in cell death induced by TEF, we down-regulated the Atg 7 protein to block the autophagic process. The molecular machinery of autophagy was largely discovered in yeast and referred to as autophagy-related (Atg) genes. Formation of the autophagosome involves an ubiquitin-like conjugation system in which Atg12 is covalently bound to Atg5 and targeted to autophagosome vesicles. This conjugation reaction is mediated by the ubiquitin-E1-like enzyme Atg7 and the E2-like enzyme Atg10 [17–20]. Our results demonstrate that the inhibition of the autophagic protein Atg 7 leads to a decreased cell death rate in TEF-treated melanoma cells, indicating that autophagy acts by promoting death mechanism in cells challenged with TEF. However, whether autophagy induction in dying cells actually causes death, or whether it simply occurs as a process alongside it, remains controversial. It is becoming clear that apoptosis induction is often associated with increased autophagy and that they are interconnected, but the reciprocal influence of the two pathways has not been completely understood [36].
In summary, this study shows the great potential of TEF to kill A375 human melanoma cells in different culture conditions through distinct apoptotic pathways that emerge from Ca2+ homeostasis disruption (summarized in Fig. 12). Serum withdrawal enhances the intensity of the death stimulus induced by TEF. Caspase-4, which becomes activated upon ER stress, and caspase-2, activated in response to DNA damage, are the predominant apoptotic mechanisms. By contrast, TEF treatment in complete medium induces apoptosis through ROS generation, involving caspase-4, caspase-2, caspase-9, p73, and Noxa induction. Moreover, autophagy also promotes cell death under these culture conditions. Based on these results, we suggest that TEF alone or in combination with other antineoplasics could define a new therapeutic strategy for melanoma. Further in vivo studies could provide evidence regarding the antitumoral potential of this drug.

Schematic representation of the signaling apoptotic pathways induced by TEF. TEF rapidly induces ER calcium release through PLC-IP3 pathway [3], leading to caspase-4 activation and to intracellular calcium increase. Accumulation of calcium in the nucleus (N) causes DNA damage; and in the mitochondria (M) it induces ROS production. An increase of ROS levels can cause DNA damage in the nucleus and cytochrome c release from mitochondria, leading to caspase-9 activation. Caspase-9 can also be directly activated by caspase-4. Caspase-2 and the p53/p73 signaling pathway become activated in response to DNA damage. Caspase-2 can directly cause cytochrome c release from mitochondria. p53 can induce cell cycle arrest through p21 induction and p73 could be responsible of Noxa induction and subsequent cytochrome c release and caspase-9 activation. The initiator caspases-2, -4 and -9 cause executioner caspases activation and apoptosis
References
Parsons ME, Ganellin CR (2006) Histamine and its receptors. Br J Pharmacol 147(Suppl 1):S127–S135
Jangi SM, Díaz-Pérez JL, Ochoa-Lizarralde B, Martín-Ruiz I, Asumendi A, Pérez-Yarza G et al (2006) H1 histamine receptor antagonists induce genotoxic and caspase-2-dependent apoptosis in human melanoma cells. Carcinogenesis 27:1787–1796
Jangi SM, Ruiz-Larrea MB, Nicolau-Galmés F, Andollo N, Arroyo-Berdugo Y, Ortega-Martínez I et al (2008) Terfenadine-induced apoptosis in human melanoma cells is mediated through Ca2+ homeostasis modulation and tyrosine kinase activity, independently of H1 histamine receptors. Carcinogenesis 29:500–509
Jangi SM, Asumendi A, Arlucea J et al (2004) Apoptosis of human T-cell acute lymphoblastic leukemia cells by diphenhydramine, an H1 histamine receptor antagonist. Oncol Res 14:363–372
Mao X, Liang SB, Hurren R, Gronda M, Chow S, Xu GW et al (2008) Cyproheptadine displays preclinical activity in myeloma and leukemia. Blood 112:760–769
Liu JD, Wang YJ, Chen CH, Yu CF, Chen LC, Lin JK et al (2003) Molecular mechanisms of G0/G1 cell-cycle arrest and apoptosis induced by terfenadine in human cancer cells. Mol Carcinog 37:39–50
Simons FE (2004) Advances in H1-antihistamines. N Engl J Med 351:2203–2217
Groenendyk J, Michalak M (2005) Endoplasmic reticulum quality control and apoptosis. Acta Biochim Pol 52:381–395
Ermak G, Davies KJ (2002) Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol 38(10):713–721
Hitomi J, Katayama T, Eguchi Y et al (2004) Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aß-induced cell death. J Cell Biol 165:347–356
Kass GE, Orrenius S (1999) Calcium signaling and cytotoxicity. Environ Health Perspect 107(Suppl 1):25–35
Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305:626–629
Nicholson DW, Thornberry NA (2003) Apoptosis. Life and death decisions. Science 299:214–215
Orrenius S, Gogvadze V, Zhivotovsky B (2007) Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol 47:143–183
Schmitt E, Paquet C, Beauchemin M, Bertrand R (2007) DNA-damage response network at the crossroads of cell-cycle checkpoints, cellular senescence and apoptosis. J Zhejiang Univ Sci B 8:377–397
Roos WP, Kaina B (2006) DNA damage-induced cell death by apoptosis. Trends Mol Med 12:440–450
Ferraro E, Cecconi F (2007) Autophagic and apoptotic response to stress signals in mammalian cells. Arch Biochem Biophys 462:210–219
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007) Self-eating and self-killing: cross talk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8:741–752
Moretti L, Cha YI, Niermann KJ, Lu B (2007) Switch between apoptosis and autophagy: radiation-induced endoplasmic reticulum stress? Cell Cycle 6:793–798
Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G (2008) To die or not to die: that is the autophagic question. Curr Mol Med 8:78–91
Falcieri E, Gobbi P, Cataldi A, Zamai L, Faenza I, Vitale M (1994) Nuclear pores in apoptotic cell. Histochem J 26:754–763
Singh Y, Palombo M, Sinko PJ (2008) Recent trends in targeted anticancer prodrug and conjugate design. Curr Med Chem 15:1802–1826
Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G et al (2008) Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci USA 105:8215–8220
Meyskens FL Jr, Farmer P, Fruehauf JP (2001) Redox regulation in human melanocytes and melanoma. Pigment Cell Res 14:148–154
Robertson JD, Gogvadze V, Kropotov A, Vakifahmetoglu H, Zhivotovsky B, Orrenius S (2004) Processed caspase-2 can induce mitochondria-mediated apoptosis independently of its enzymatic activity. EMBO Rep 5:643–648
Zhivotovsky B, Orrenius S (2005) Caspase-2 function in response to DNA damage. Biochem Biophys Res Commun 331:859–867
Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y (2002) An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem 277:34287–34294
Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G et al (2002) Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem 277:21836–21842
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274
Qin JZ, Stennett L, Bacon P, Bodner B, Hendrix MJ, Seftor RE et al (2004) p53-independent NOXA induction overcomes apoptotic resistance of malignant melanomas. Mol Cancer Ther 3:895–902
Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V et al (2005) Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res 65:6282–6293
Armstrong JL, Veal GJ, Redfern CP, Lovat PE (2007) Role of Noxa in p53-independent fenretinide-induced apoptosis of neuroectodermal tumours. Apoptosis 12:613–622
Dong YB, Yang HL, Elliott MJ et al (1999) Adenovirus-mediated E2F-1 gene transfer efficiently induces apoptosis in melanoma cells. Cancer 86:2021–2033
Dong YB, Yang HL, Elliott MJ, McMasters KM (2002) Adenovirus-mediated E2F-1 gene transfer sensitizes melanoma cells to apoptosis induced by topoisomerase II inhibitors. Cancer Res 62:1776–1783
Tuve S, Racek T, Niemetz A, Schultz J, Soengas MS, Pützer BM (2006) Adenovirus-mediated TA-p73beta gene transfer increases chemosensitivity of human malignant melanomas. Apoptosis 11:235–243
Fimia GM, Piacentini M (2010) Regulation of autophagy in mammals and its interplay with apoptosis. Cell Mol Life Sci 67:1581–1588
Acknowledgments
This study was supported by grants from the Heath Department of the Government of the Basque Country (Exp: 2006111036) and the University of the Basque Country (GIC-IT-423-07). Microscopic photographs were taken in the General Service of Analytical and High Resolution Microscopy of the University of the Basque Country.
Conflict of interest
None declared.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Nicolau-Galmés, F., Asumendi, A., Alonso-Tejerina, E. et al. Terfenadine induces apoptosis and autophagy in melanoma cells through ROS-dependent and -independent mechanisms. Apoptosis 16, 1253–1267 (2011). https://doi.org/10.1007/s10495-011-0640-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-011-0640-y