
Abstract
The kidney plays a crucial role in the maintenance of the body calcium (Ca2+) balance. Ca2+ is an essential ion in all organisms and participates in a large variety of structural and functional processes. In mammals, active tubular Ca2+ reabsorption is restricted to the distal part of the nephron, i.e., the late distal convoluted (DCT2) and the connecting tubules (CNT), where approximately 10–15% of the total Ca2+ is reabsorbed. This active transcellular transport is hallmarked by the transient receptor potential vanilloid 5 (TRPV5) epithelial Ca2+ channel, regulated by an array of events, and mediated by hormones, including 1,25-dihydroxyvitamin D3, parathyroid hormone, and estrogen. Novel molecular mechanisms have been identified, such as the direct regulatory effects of klotho and tissue kallikrein on the abundance of TRPV5 at the apical membrane. The newly discovered mechanisms could provide potential pharmacological targets in the therapy of renal Ca2+ wasting. This review discusses the three basic molecular steps of active Ca2+ reabsorption in the DCT/CNT segments of the nephron, including apical entry, cytoplasmic transport, and basolateral extrusion of Ca2+. In addition, an overview of the recently identified mechanisms governing this active Ca2+ transport through the DCT2/CNT epithelial cells will be presented.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Calcium (Ca2+) is essential for the physiological functioning of all living cells. In humans, 99% of total body Ca2+ resides in the skeleton. The remaining 1% is distributed in soft tissues and extracellular fluid, which is the prime target of the Ca2+ homeostatic systems. Three tightly controlled mechanisms, including bone resorption and formation, intestinal absorption, and renal reabsorption, maintain Ca2+ homeostasis. In the kidney, approximately 45% of the plasma Ca2+, present in free ionized form, filters through the glomerulus and enters the proximal tubule segment of the nephron, where ∼65% of the filtered Ca2+ is passively reabsorbed [56, 62]. In the thick ascending loop of Henle (TAL), an additional 20% is reabsorbed through this passive paracellular pathway, mediated by the tight junction protein claudin-16 [3, 43]. In these segments, Ca2+ reabsorption is not specifically regulated and depends on gradients established by NaCl and water reabsorption [61]. The final regulation of Ca2+ excretion, according to physiological needs, appears to occur primarily in two segments of the distal part of the nephron, namely in the late part of the distal convoluted tubule (known as DCT2) and the connecting tubule (CNT; Table 1) [16].
Morphologic characteristics of the CNT
The above discussed distal segments of the nephron exhibit distinct morphological, as well as functional features. In the superficial cortex, the DCT/CNT region is short and flows directly into the cortical collecting duct (CCD). Midcortical and juxtamedullar nephrons, on the other hand, have longer DCT/CNTs merging with other CNT segments into arcades before transitioning to the CCD [56]. DCTs consist of two short segments, DCT1 and DCT2, both comprising a uniform population of principal cells, whereas the CNT contains both principal and two types of intercalated cells [3, 16, 43, 56]. Furthermore, the principal cells in the CNT have less cell–cell contacts and mitochondria, and their apical membrane contains fewer projections than DCT cells. Unlike the polygonal-shaped CNT cells, intercalated cells appear to be round with an apical membrane densely adorned with microprojections [43, 56]. The proton secreting α-intercalated cells have extensive apical microvilli with abundant expression of the H+/K+ exchanger and vacuolar H+-ATPase and numerous subapically localized small mitochondria, whereas the bicarbonate secreting β-intercalated cells have fewer apical microvilli, and their mitochondria tend to accumulate basolaterally, where the proton pump is also located [56]. Although the ratio of α- and β-intercalated cells vary depending on the actual physiological state, α-intercalated cells are more common in the CNT.
In addition to the ubiquitously expressed Na+/K+-ATPase, the Na+/Ca2+ exchanger (NCX1) and the plasma membrane ATPase type 1b (PMCA1b) have been found along the DCT2/CNT region, whereas the apically localized thiazide-sensitive Na+/Cl− co-transporter (NCC) and the transient receptor potential melastin subtype 6 are present in the DCT (see Xi et al. in this issue). The DCT2 region also shares additional similarities with the CNT segment, as both segments express the transient receptor potential vanilloid type 5 (TRPV5) and the Ca2+-binding protein calbindin-D28K. The tight junctions in these segments are impermeable for Ca2+, and transcellular Ca2+ transport occurs against an electrochemical gradient, supporting that Ca2+ reabsorption in the DCT2/CNT is mediated by active transepithelial transport.
Transepithelial Ca2+ reabsorption
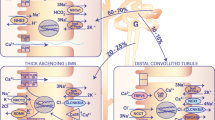
Transepithelial transport of Ca2+ is a three-step process. It initiates with influx of Ca2+ across the apical membrane mediated by TRPV5 [29]. Subsequently, entered Ca2+ is sequestered by the specialized intracellular carrier protein calbindin-D28K and diffuses to the basolateral membrane (Fig. 1). Finally, transporter proteins, such as NCX1 and PMCA1b, extrude Ca2+ from the epithelial cell into the circulation (Fig. 1). The identification and characterization of TRPV5 as the gatekeeper of renal epithelial Ca2+ transport [30] gave new momentum to the understanding of the molecular mechanisms underlying the process of active Ca2+ reabsorption.

Model of transcellular Ca2 reabsorption in DCT2 and CNT. The renal distal tubule in the nephron comprises anatomically discrete segments, including the thick ascending limb of the loop of Henle (TAL) and the distal convoluted tubule (DCT) that ends in the connecting tubule (CNT). The late part of the DCT (DCT2) and CNT play an important role in fine-tuning renal excretion of Ca2+. The epithelial Ca2+ channel (TRPV5) is primarily expressed apically in these segments and co-localizes with calbindin-D28K (28K), Na+/Ca2+ exchanger (NCX1), and the plasma membrane ATPase (PMCA1b). Upon entry via TRPV5, Ca2+ is buffered by 28K and diffuses to the basolateral membrane, where it is released and extruded by a concerted action of NCX1 and PMCA1b. In addition, the basolateral membrane exposes a parathyroid hormone receptor (PTHR) and the Na+/K+-ATPase consisting of the α-, β- and γ-subunit. PTHR activation by PTH stimulates TRPV5 activity, and entered Ca2+ can subsequently control the expression level of the Ca2+ transporters. At the apical membrane, there is a bradykinin receptor (BK2) that is activated by urinary tissue kallikrein (TK) to activate TRPV5-mediated Ca2+ influx. In the cell, entered Ca2+ acts as a negative feedback on channel activity, and 28K plays a regulatory role by association with TRPV5 under low intracellular Ca2+ concentrations. Extracellular urinary klotho directly stimulates TRPV5 at the apical membrane by modification of the N-glycan, whereas intracellular klotho enhances Na+/K+-ATPase surface expression that in turn activates NCX1-mediated Ca2+ efflux
TRPV5—the apical gate
TRPV5, also known as the epithelial Ca2+ channel, is a member of the TRP channel superfamily [29]. This channel comprises large and flexible intracellular amino- and carboxyl-terminal tails flanking six transmembrane segments (TM) and an additional hydrophobic stretch between TM5 and TM6, predicted to be the pore-forming region. The amino-terminal tail contains six ankyrin repeats [19, 54] that are important structural elements for both channel assembly and protein–protein interactions [13, 19]. Furthermore, the first extracellular loop between TM1 and TM2 contains an evolutionary conserved asparagine (N358) crucial for complex-glycosylation and in turn for regulating channel activity [11, 13, 33]. The carboxyl-terminal tail harbors three potential protein kinase C (PKC) sites, which suggests an important role for phosphorylation in channel activity. Moreover, in cultured mammalian cell systems, as well as in oocytes, TRPV5 is assembled into large homotetramers in order to acquire the active conformation state [26, 33]. Facing each other, the hydrophobic stretches between TM5 and TM6 in each subunit are postulated to form the aqueous pore centered at the fourfold symmetry axis.
Detailed electrophysiological studies have compellingly demonstrated the constitutive activity of TRPV5 at low intracellular Ca2+ concentrations and physiological membrane potentials [75]. The current–voltage relationship of TRPV5 shows strong inward rectification [29, 74, 75]. Another important functional feature of TRPV5 is the high Ca2+ selectivity, making this epithelial Ca2+ channel the most Ca2+-selective member in the TRP superfamily [75]. Finally, the generation of a TRPV5 deficient mouse strain (TRPV5−/−) provided compelling evidence for the physiological function of this channel. Active Ca2+ reabsorption in DCT2 and CNT is severely impaired in these animals as TRPV5−/− mice waste approximately six- to tenfold more Ca2+ than their wild-type littermates, which is in line with the postulated gatekeeper function of TRPV5 in active Ca2+ reabsorption [31].
Shortly after the identification of TRPV5, a homologous channel, sharing 75% amino acid identity with TRPV5, was cloned from intestine and named TRPV6 [53]. Although there are some functional differences between these channels, TRPV6 exhibits the same Ca2+ selectivity and current–voltage relationship [32, 47, 78]. Moreover, this latter channel has been implicated in intestinal Ca2+ absorption. Disturbances in the Ca2+ homeostasis were also reported in mice lacking TRPV6 (TRPV6−/−) as these animals display reduced intestinal Ca2+ absorption and low bone mineral density [1]. Although it has been shown that TRPV6 is moderately expressed in adult kidney [59] and the TRPV6−/− mice have increased urinary Ca2+ excretion, the exact role of TRPV6 in the kidney is not yet fully understood.
Calbindin-D28K—the intracellular shuttle
The principal cells of the DCT2/CNT segments are continuously challenged by a substantial Ca2+ influx through TRPV5, yet the cells manage to maintain a low intracellular Ca2+ concentration ([Ca2+]i). Maintaining the free [Ca2+]i at the basal level is essential for several reasons. High levels of free Ca2+ in the cytoplasm are known to induce apoptosis and protein precipitation. In addition, Ca2+ has an essential signaling function for many processes in the cell. More importantly, an increased [Ca2+]i has been shown to inhibit the activity of TRPV5 [75]. Based on the currently available data, three different models have been postulated for transepithelial Ca2+ transfer from the apical to the basolateral membrane. The first model is based on a passive diffusion tunneling through the endoplasmatic reticulum, vesicular transport along the microtubules involving lysosomes, and finally, facilitated diffusion. According to the second model, Ca2+-transporting cells utilize lysosomes to sequester Ca2+ and facilitate its movement to the basolateral membrane [35]. The apical Ca2+ influx through TRPV5 initiates the disruption of the actin cytoskeleton and the formation of Ca2+-enriched endocytic vesicles, which are transported along the microtubules and some fuse with lysosomes [46]. In a third model, the intracellular diffusion of Ca2+ is facilitated by the vitamin D3-dependent Ca2+-binding protein, calbindin-D28K, in the principal cells of DCT2 and CNT segments [7, 20]. Together with parvalbumin, calmodulin, and troponin C, calbindins are Ca2+-binding proteins that form a family of proteins with Ca2+ affinity [15]. Calbindin-D28K has three pairs of EF-hands that are the structural basis of the high Ca2+ affinity binding capacity [6]. Moreover, in the kidney, the expression of calbindin-D28K is restricted to DCT2, CNT, and CCD regions. It has also recently been shown that calbindin-D28K translocates to the TRPV5-containing plasma membranes upon a decrease in intracellular [Ca2+] and directly associates with this channel [39]. Due to the relatively slow Ca2+-binding kinetics of calbindin-D28K, hormone-induced Ca2+ signaling can also occur independently of the transcellular Ca2+ transport rate [37]. Bound to calbindin-D28K, Ca2+ is shuttled to the basolateral membrane, where Ca2+ is discharged into the blood flow by the basolateral Ca2+ extrusion systems. Finally, some studies reported that calbindin-D28K −/− mice fed a high Ca2+ diet have impaired renal Ca2+ handling as they excrete more Ca2+ in their urine than the wild-type control littermates [60], whereas other studies did not observe a difference that is probably due to the compensatory increase of renal calbindin-D9K expression [22]. These data suggest that calbindin-D28K facilitates the intracellular diffusion of Ca2+ in DCT2 and CNT.
NCX1 and PMCA1b—the basolateral extrusion system
The energy-consuming step of transcellular Ca2+ transport lies in the Ca2+ efflux process. In this step, intracellular Ca2+ is transported across the basolateral membrane against its electrochemical gradient, and the ions are extruded back to the blood flow. Two transporters have been implicated in this mechanism, PMCA1b and NCX1. Plasma membrane ATPases are high-affinity Ca2+ efflux pumps that maintain the resting Ca2+ concentration in virtually all cells [4]. The highest Ca2+-ATPase activity in kidney was reported in the DCT segment. However, earlier studies have suggested that the capacity of this PMCA pump in CNT seems to be insufficient to keep pace with the absorptive flux of Ca2+ because it can transport only ∼30% of the total Ca2+ efflux [2, 69]. In contrast to PMCA1b, the Na+/Ca2+ exchanger has been shown to be a prerequisite for transepithelial Ca2+ transport [2, 69]. NCX1 exchanges Ca2+ and Na+ generally in a 1:3 stoichiometric ratio. Moreover, NCX1 is a widely expressed protein as it can be found in several tissues, including the heart, brain, and skeletal muscle [45]. In the kidney, the expression of NCX1 is restricted to the distal part of the nephron, particularly the CNT segment, where it predominantly localizes along the basolateral membrane [3, 28, 43] and accounts for the remaining 70% of the Ca2+ efflux [2, 69].
Regulatory mechanisms of Ca2+ transport in DCT2/CNT
The aforementioned transporters comprise the machinery transporting Ca2+ from pro-urine to the blood in the DCT2/CNT. Several factors have been shown to contribute to the regulation of the Ca2+ transporting capacity of these particular nephron segments, which can be classified into four categories: (1) the control of the overall abundance of the transporter proteins by calciotropic hormones, (2) the rate of intracellular trafficking of the plasma membrane transporters, (3) alteration of activity of the transport proteins in the membrane by intracellular factors, and finally, (4) tuning apical Ca2+ influx by extracellular (luminal) factors.
Controlling the abundance of Ca2+ transporters
PTH
Parathyroid hormone (PTH) is an essential component of the Ca2+ homeostasis. The secretion of PTH from the parathyroid gland is triggered by changes in blood Ca2+ levels sensed by the parathyroid Ca2+-sensing receptor [8]. PTH receptors have been detected in DCT2/CNT, enabling the direct control of active Ca2+ reabsorption by PTH [57].
PTH-mediated regulation of the Ca2+ transporters was studied in parathyroidectomized rats [68]. Parathyroidectomy reduced the expression of TRPV5, calbindin-D28K, and NCX1. This decline in expression of Ca2+ transporters resulted in decreased active Ca2+ reabsorption and the development of hypocalcemia [68]. After PTH supplementation, the expression of Ca2+ transporters, as well as increased plasma Ca2+ concentration were normalized in these parathyroidectomized rats. In addition, the regulation by PTH was investigated in primary cultures of rabbit CNT cells. In these Ca2+-transporting cells, PTH resulted in an elevated expression of the Ca2+ transport proteins TRPV5, calbindin-D28K, NCX1, and PMCA1b. Taken together, these results indicate that PTH stimulates renal Ca2+ handling by co-regulating the expression of the Ca2+ transport proteins [68]. In addition, experiments in these primary CNT cell cultures supported a gatekeeper role of TRPV5 since a blockade of the apical Ca2+ influx by ruthenium red prevented the PTH-induced upregulation of the other Ca2+ transporters.
Vitamin D
The vitamin D endocrine system plays a pivotal role in Ca2+ homeostasis. In the recent years, it has become clear that the active form of vitamin D (1,25-dihydroxyvitamin D3, or abbreviated 1,25(OH)2D3) is a potent regulator of the Ca2+ transport proteins. Several groups have shown transcriptional regulation of TRPV5, calbindin-D28K, and NCX1 by 1,25(OH)2D3, whereas a 1,25(OH)2D3-sensitivity for PMCA1b is not consistently reported. Studies in vitamin-D-deficient knockout models showed an impressive downregulation of renal TRPV5, calbindin-D28K, and NCX1 mRNA that could be normalized by 1,25(OH)2D3 supplementation, whereas PMCA1b was not significantly affected [27]. On the other hand, several studies indicated that PMCA1b is positively regulated by 1,25(OH)2D3 in the intestine to increase Ca2+ absorption. Northern blot analysis indicated that repletion of vitamin-D-deficient chickens with vitamin D increases PMCA mRNAs in the duodenum, jejunum, ileum, and colon [9]. Because these studies and the role of vitamin D in Ca2+ homeostasis have been reviewed extensively [29, 64, 70], detailed information is not included in this review.
Estrogen
Although estrogen is generally not considered as a calciotropic hormone, it is widely accepted that it plays a role in renal Ca2+ handling. In rats, estradiol has been suggested to enhance the expression of TRPV5, NCX1, PMCA1b, and calbindin-D28K [67]. In line with these observations, a recent study with aromatase deficient mice lacking the aromatase enzyme (aromatase−/−) and, therefore, estrogen deficient, also showed decreased expression of these transporters and concomitant renal Ca2+ wasting [52]. Additionally, estradiol treatment of these animals normalized the urinary Ca2+ excretion and gene expression. In agreement with these observations, increased renal Ca2+ wasting, as well as renal stone formation in women after menopause, is a well-known phenomenon [25], suggesting that estrogens may significantly contribute to the regulation of the transepithelial Ca2+ transport in the DCT2/CNT.
Controlling the intracellular trafficking
S100A10/annexin-2
S100A10 (also known as annexin-2 light chain) is an auxiliary protein for TRPV5 [72]. With two Ca2+-insensitive EF-hands, S100A10 is predominantly present as a heterotetrameric complex with annexin-2, which has been implicated in many cellular processes, including endocytosis and exocytosis [21]. An important regulatory role has been proposed for the S100A10–annexin-2 heteromer in TRPV5 functioning [72]. The binding of annexin-2 to TRPV5 through S100A10 was shown to facilitate the translocation of TRPV5 toward the plasma membrane. This association of S100A10 and TRPV5 takes place through a short conserved peptide sequence, located in the carboxyl-terminus of TRPV5. Moreover, co-expression of S100A10, annexin-2, and TRPV5 has been observed in DCT2/CNT [72]. Taken together, these findings show that the S100A10–annexin-2 complex is a significant component for trafficking of TRPV5 toward the plasma membrane.
Rab11a
The small GTPase Rab11a has also been identified as a novel TRPV5- associated protein [71]. Rab11a is one of the key regulatory proteins that controls the recycling of endosomes [10, 76]. Rab11a was found to co-localize with TRPV5 in the DCT2/CNT. Here, both TRPV5 and Rab11a are present in subapical vesicular structures [71]. In addition, a direct protein–protein interaction was observed between Rab11a and TRPV5, suggesting that TRPV5 channels, present on the apical plasma membrane, recycle from the intracellular (recycling) endosomes in a Rab11-dependent manner.
Clathrin and caveolin
Van de Graaf and coworkers observed that the extraction of TRPV5 from the cell surface takes place in a constitutive clathrin-dependent manner [73]. In addition, the same authors showed that following its internalization, TRPV5 is not immediately targeted to protein degradation. Instead, by entering a Ca2+-dependent recycling pathway, TRPV5 remained stable in the subapical endosomal fraction [73]. Another recent study by Huang et al. suggested a caveolin-1-mediated internalization of TRPV5, which was inhibited by PKC [12]. Caveolin-1 is a structural component of caveolae and is crucial for the stabilization of the specialized membrane domains. Moreover, the caveolae-dependent internalization of TRPV5 was strongly inhibited by PTH-induced phosphorylation of TRPV5 via PKC, indicating that next to its genetic effect on expression, PTH also has a rapid effect on channel abundance [12]. These findings indicate that apical sorting of TRPV5 is likely to be mediated by several mechanisms that could be differentially controlled depending on physiological needs of the body.
Regulation of TRPV5 activity at the membrane
Intracellular Ca2+, Mg2+, and PIP2
Although the Ca2+ concentration in the luminal compartment of DCT2/CNT is in the 1.0–1.5 mM range, the resting [Ca2+]i in these cells is maintained around 100 nM by NCX1 and PMCA1b. TRPV5 has a high Ca2+ selectivity, and at physiological Ca2+ concentrations, its current is mainly carried by Ca2+. In human embryonic kidney (HEK293) cells heterogeneously expressing TRPV5, currents can be activated under conditions of high intracellular Ca2+ buffering by hyperpolarizing voltage steps. Earlier, Nilius et al. suggested that intracellular Ca2+ acts as a negative feedback switch regulating TRPV5 activity. The Ca2+ current through TRPV5 is inhibited by the [Ca2+]i with an IC50 of 82 nM [50]. Considering this high affinity of Ca2+-dependent channel inhibition, the presence of the co-expressed Ca2+ buffer calbindin in DCT2/CNT plays an important role to maintain TRPV5 activity. Conclusively, the [Ca2+]i itself directly regulates channel function in order to maintain optimal Ca2+ reabsorption without excessive influx of Ca2+.
Single TRPV5 channel currents in cell-attached and inside-out patches were only detected in the absence of Ca2+ and had a conductance of ∼75 pS [51]. So far, no reliable single channel measurements have been performed in the presence of extracellular Ca2+. Another interesting feature of TRPV5 is the open pore blockage by intracellular Mg2+. Currents through TRPV5 are small at physiological extracellular Mg2+ and Ca2+ concentrations, but sufficient to increase the [Ca2+]i at hyperpolarized potentials. Therefore, block by Mg2+ and decrease of the current by extracellular Ca2+ might be physiologically important to prevent Ca2+ overload of TRPV5-expressing cells [51, 75]. In addition, Huang and colleagues reported that PIP2 activates TRPV5 and that activation of the channel by PIP2 reduces the sensitivity of TRPV5 to the inhibition by the intracellular [Mg2+] [17, 42]. In this model, hydrolysis of PIP2 by receptor activation of PLC may increase the sensitivity for Mg2+ inhibition.
80K-H
Another protein called 80K-H has been shown to directly interact with TRPV5 in a Ca2+-dependent manner [23]. The 80K-H protein contains two putative EF-hands and an ER-targeting signal. This TRPV5-linked protein was originally cloned as a PKC substrate and was subsequently associated with intracellular signaling [58]. Binding with Ca2+ abolished the inactivation of the two EF-hand motifs of 80K-H, and this, in turn, reduced the TRPV5-mediated Ca2+ current and increased the sensitivity of TRPV5 to the [Ca2+]i, accelerating the feedback inhibition of the channel [23]. Moreover, 80K-H also co-localizes with TRPV5 in the DCT2/CNT. Based on these findings, 80K-H has been hypothesized to act a Ca2+ sensor to regulate the activity of TRPV5 at the plasma membrane [23].
Pro-urinary factors stabilize TRPV5 in the apical membrane
Tissue kallikrein
Tissue kallikrein (TK) is a multifunctional serine protease that is primarily synthesized in the DCT2 and CNT and catalyzes the kininogen kinin conversion [55]. TK is secreted into the pro-urine, where it mediates the formation of bradykinin that binds to the type 2 bradykinin receptor (B2R) [18]. A striking effect of TK has been observed in primary rabbit CNT cells mediating transcellular Ca2+ transport [24]. Apical addition of TK or bradykinin (BK) significantly increased transcellular Ca2+ transport that was prevented by B2R antagonists, whereas basolateral application of either TK or BK had no effect [24]. This stimulatory effect of TK was mediated by the apical B2R signaling through the phospholipase C/diacylglycerol/PKC pathway, resulting in phosphorylation of TRPV5 and subsequent delay in its retrieval from the plasma membrane. Additionally, mice lacking TK (TK−/−) waste a large amount of Ca2+ without any significant alterations in plasma Ca2+, PTH, and vitamin D3 levels or any detectable changes in the expression of Ca2+ transporters in the DCT2/CNT. These observations together highlight the importance of the regulation of the TRPV5 channel abundance in the membrane by the pro-urine TK.
Urinary pH and Mg2+
The acid–base balance has long been known to affect Ca2+ homeostasis. For example, patients with chronic metabolic acidosis waste Ca2+. Pioneering micropuncture studies have shown that chronic metabolic acidosis results in Ca2+ wasting [63]. Recent advances in total reflection fluorescent microscopy analysis allowed the identification of the molecular basis underlying these in vivo observations. Exposure of TRPV5-expressing cells to an alkaline extracellular environment (pH 8.0) caused rapid recruitment of TRPV5-containing vesicles to the cell surface and a consequent increase in TRPV5 activity [40]. In the reciprocal experiment, acidic extracellular milieu (pH 6.5) induced the internalization of TRPV5-containing vesicles from the plasma membrane, resulting in a reduced channel activity [40]. The extracellular acidity clearly affected the current kinetics resulting in diminished single channel conductance as shown by Yeh et al. [77]. Binding of protons to an extracellular glutamate near the pore helix of TRPV5 at position 522 (E522) resulted in decreased channel activity, whereas substitution with a glutamine (E522→Q522) abolished the proton sensitivity. This recognized E522 as the extracellular pH sensor in TRPV5. Based on these experiments, binding of protons to the sensor has been proposed to induce a conformational change of the TRPV5 pore helix, leading to a lowered channel activity. These observations clearly point out that urinary acidification results in decreased channel activity at the apical cell membrane, as well as in a rapid retrieval of TRPV5 from the apical membrane, both of which are likely to account for the renal Ca2+ wasting in metabolic acidosis. Also, urinary Mg2+ is known to modulate urinary Ca2+ excretion, but the mechanism underlying this relationship is unknown. In a recent study by Bonny et al., it was elegantly demonstrated that an alteration in urinary Ca2+ excretion is directly proportional to the change in Mg2+ excretion and inversely proportional to the adjustment in urine pH [5]. Because TRPV5 was inhibited by Mg2+, these data are compatible with the hypothesis that urinary Mg2+ directly inhibits Ca2+ reabsorption in DCT2/CNT, which can be overruled by an alkaline luminal pH.
Klotho
Klotho is a type-I (single-pass) membrane protein predominantly expressed in tissues involved in Ca2+ homeostasis, such as kidney, choroid plexus, and the parathyroid gland [34]. The ablation of klotho causes severe multiple phenotypes in klotho-deficient (klotho−/−) mice, such as short life span associated with infertility and sternly impaired Ca2+ and phosphate metabolism [38, 65]. There is a growing body of evidence that klotho controls active Ca2+ reabsorption in the DCT2/CNT segments through several mechanisms.
In kidney, klotho is exclusively expressed in DCT2/CNT, where following extracellular domain shedding, it is secreted into the circulation and the pro-urine [13]. The secreted form of klotho exerts β-glucuronidase activity [14]. More importantly, klotho was suggested to regulate the apical entry of Ca2+ in the DCT2/CNT region. The presence of extracellular klotho robustly increased the activity of TRPV5 in cultured rabbit primary CNT cells. Moreover, TRPV5-expressing HEK293 cells also showed a significant rise in channel activity after klotho treatment that was accompanied with concomitant increased plasma membrane channel abundance [14]. In addition, removal of the complete N-glycan tree by Endo-F was recently reported to result in a more pronounced increase in TRPV5 activity compared to that of observed upon klotho treatment [44]. Recently, Cha et al. suggested sialidase rather than β-glucuronidase activity for klotho, as they observed a klotho-mediated removal of terminal sialic acids from the N-glycan in TRPV5 [11]. This cleavage exposed the underlying galactose-N-acetylglucoseamine disaccharides in TRPV5, which can directly interact with membrane-bound galectin-1, causing the subsequent plasma membrane retention of TRPV5. Interestingly, treatment with PNGaseF to hydrolyze the entire N-glycan of TRPV5 mimicked the stimulatory effect of klotho, suggesting additional mechanisms besides binding to membrane galectin-1. It should be noted that Cha et al. also reported a comparable increase in TRPV5 activity upon β-glucuronidase treatment; however, this effect could be observed only at much higher (0.1–1 μM) concentrations of the enzyme, suggesting that klotho exhibits primarily sialidase activity at physiologic concentrations (∼20–200 pM) [11]. These observations confirmed the original conclusion that extracellular klotho hydrolyses oligosaccharide chains from the N-glycosylated TRPV5, causing channel retention at the membrane and a subsequent increase in TRPV5-mediated Ca2+ influx [14]. In line with these findings, microperfusion studies have shown that CNTs from klotho−/− mice fail to respond to PTH and, therefore, waste large amount of Ca2+ [66]. Interestingly, klotho was reported to interact and increase Na+/K+-ATPase activity at the plasma membrane and stimulating the Na+/Ca2+ exchange through NCX1 [34]. Taken altogether, there is compelling evidence that klotho is a novel calciotropic factor that, amongst others, can exert its stimulatory effect on TRPV5 cell surface retention from the pro-urine in the DCT2 and the CNT.
Clinical relevance
Several clinical disorders, such as chronic renal failure or diabetes, are associated with the symptoms of dysregulating body Ca2+ homeostasis. Chronic renal failure (CRF) is frequently characterized by hypocalcemia, osteoporosis, growth retardation, and secondary hyperparathyroidism. Remarkably, the phenotype of the klotho−/− mice resembles most of these characteristics [36]. Moreover, CRF patients also have greatly reduced renal klotho levels [36]. Together with the fact that klotho can regulate the activity of TRPV5 and NCX1 via Na+/K+-ATPase, the involvement of klotho in the pathogenesis of Ca2+ abnormalities in CRF may be envisaged.
Hypercalciuria and nephrolithias, disorders with a high prevalence and socio–economic burden in the Western society, are often treated with thiazide diuretics. These drugs are known to affect the Ca2+ balance by inducing hypocalciuria. Over the last decade, it was speculated that thiazide-inhibited NCC activity stimulates active Ca2+ reabsorption in the DCT/CNT. However, recent studies indicated that paracellular Ca2+ transport in the proximal tubule due to extracellular volume contraction explains the hypocalciuria during chronic thiazide treatment [48, 49]. Hypercalciuria is also an early finding in diabetes mellitus patients. Similarly, rats with experimentally induced diabetes display a significant increase in the fractional excretion of Ca2+ [41]. However, these diabetic rats showed increased mRNA and protein levels of both TRPV5 and calbindin-D28K. Additionally, insulin therapy corrected the hyperglycemia-associated hypercalciuria and abolished the upregulation of TRPV5, suggesting that the increased TRPV5 abundance in diabetic rats is due to a compensatory adaptation to an increased load of Ca2+ secondary to hyperglycemia. Although in many pathophysiological conditions the expression level of the Ca2+ transporters is out of balance, mutations in these proteins have not been discovered yet.
Concluding remarks and future perspectives
Ca2+ reabsorption in the kidney and particularly in the distal DCT2/CNT segments of the nephron is critical in the maintenance of the Ca2+ balance. Here, TRPV5 is the gatekeeper of the Ca2+ entry, and therefore, a tight control of its activity enables the organism to adjust Ca2+ reabsorption according to any demands of the body. The available experimental data summarized in this review highlights the most important mechanisms that can actually regulate active Ca2+ reabsorption. Of these, controlling the TRPV5 cell surface expression by extracellular factors in the pro-urine is a newly discovered mechanism. Klotho delays the retrieval of TRPV5 from the cell membrane by modifying the N-glycan composition, whereas the insertion of channels in the cell membrane is promoted by TK-induced phosphorylation or alkaline pH. All mechanisms result in increased TRPV5 abundance and, in turn, regulate the entry of Ca2+ at the gate. However, the canvas is far from complete. The molecular mechanism by which intracellular Ca2+ bound to calbindin-D28K is transported to the basolateral extrusion transporters NCX1 and PMCA1b is largely unknown. It could be envisaged that the local Na+ concentration may promote the release of Ca2+; however, no experimental data are available to support this theory. Therefore, the Ca2+ transfer between calbindin-D28K on one site and NCX1 and PMCA1b on the other site needs special attention. Another interesting question to be solved is the regulation of the basolateral trafficking of NCX1 and PMCA1b in the DCT2 and CNT cells. Certain players, such as the scaffold 14-3-3, phospholemman, ankyrin, or caveolin-3, have been proposed to play a role in the trafficking of the NCX transporters in neurons and cardiac cells. Nevertheless, the basolateral sorting of these transporters in the DCT2/CNT is essentially unknown. Whether there is a crosstalk between the apical Ca2+ entry and the basolateral Ca2+ extrusion regulatory apparatus is not known. The fact that klotho increases both the channel abundance of TRPV5 in the apical membrane and the activity of NCX1 at the basolateral side certainly predicts the existence of such crosstalk [14, 34]. The main question for the coming years is how all of these Ca2+ transport proteins communicate with each other in order to facilitate optimal and regulated transcellular Ca2+ reabsorption in DCT2 and CNT under conditions of disturbed Ca2+ homeostasis.
References
Bianco SD, Peng JB, Takanaga H, Suzuki Y, Crescenzi A, Kos CH, Zhuang L, Freeman MR, Gouveia CH, Wu J, Luo H, Mauro T, Brown EM, Hediger MA (2007) Marked disturbance of calcium homeostasis in mice with targeted disruption of the Trpv6 calcium channel gene. J Bone Miner Res 22:274–285
Bindels RJ, Ramakers PL, Dempster JA, Hartog A, van Os CH (1992) Role of Na+/Ca2+ exchange in transcellular Ca2+ transport across primary cultures of rabbit kidney collecting system. Pflugers Arch 420:566–572
Biner HL, Arpin-Bott MP, Loffing J, Wang X, Knepper M, Hebert SC, Kaissling B (2002) Human cortical distal nephron: distribution of electrolyte and water transport pathways. J Am Soc Nephrol 13:836–847
Blaustein MP, Juhaszova M, Golovina VA, Church PJ, Stanley EF (2002) Na/Ca exchanger and PMCA localization in neurons and astrocytes: functional implications. Ann N Y Acad Sci 976:356–366
Bonny O, Rubin A, Huang CL, Frawley WH, Pak CY, Moe OW (2008) Mechanism of urinary calcium regulation by urinary magnesium and pH. J Am Soc Nephrol 19:1530–1537
Bouhtiauy I, Lajeunesse D, Christakos S, Brunette MG (1994) Two vitamin D3-dependent calcium binding proteins increase calcium reabsorption by different mechanisms. I. Effect of CaBP28K. Kidney Int 45:461–468
Bronner F (1989) Renal calcium transport: mechanisms and regulation—an overview. Am J Physiol 257:F707–711
Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC (1993) Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366:575–580
Cai Q, Chandler JS, Wasserman RH, Kumar R, Penniston JT (1993) Vitamin D and adaptation to dietary calcium and phosphate deficiencies increase intestinal plasma membrane calcium pump gene expression. Proc Natl Acad Sci USA 90:1345–1349
Casanova JE, Wang X, Kumar R, Bhartur SG, Navarre J, Woodrum JE, Altschuler Y, Ray GS, Goldenring JR (1999) Association of Rab25 and Rab11a with the apical recycling system of polarized Madin–Darby canine kidney cells. Mol Biol Cell 10:47–61
Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro-o M, Huang CL (2008) Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci USA 105:9805–9810
Cha SK, Wu T, Huang CL (2008) Protein kinase C inhibits caveolae-mediated endocytosis of TRPV5. Am J Physiol Renal Physiol 294:F1212–F1221
Chang Q, Gyftogianni E, van de Graaf SF, Hoefs S, Weidema FA, Bindels RJ, Hoenderop JG (2004) Molecular determinants in TRPV5 channel assembly. J Biol Chem 279:54304–54311
Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG (2005) The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310:490–493
Christakos S, Gabrielides C, Rhoten WB (1989) Vitamin D-dependent calcium binding proteins: chemistry, distribution, functional considerations, and molecular biology. Endocr Rev 10:3–26
Costanzo LS, Windhager EE, Ellison DH (2000) Calcium and sodium transport by the distal convoluted tubule of the rat. 1978. J Am Soc Nephrol 11:1562–1580
Dodier Y, Banderali U, Klein H, Topalak O, Dafi O, Simoes M, Bernatchez G, Sauve R, Parent L (2004) Outer pore topology of the ECaC-TRPV5 channel by cysteine scan mutagenesis. J Biol Chem 279:6853–6862
Erdos EG, Deddish PA (2002) The kinin system: suggestions to broaden some prevailing concepts. Int Immunopharmacol 2:1741–1746
Erler I, Hirnet D, Wissenbach U, Flockerzi V, Niemeyer BA (2004) Ca2+-selective transient receptor potential V channel architecture and function require a specific ankyrin repeat. J Biol Chem 279:34456–34463
Feher JJ (1983) Facilitated calcium diffusion by intestinal calcium-binding protein. Am J Physiol 244:C303–307
Gerke V, Creutz CE, Moss SE (2005) Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol 6:449–461
Gkika D, Hsu YJ, van der Kemp AW, Christakos S, Bindels RJ, Hoenderop JG (2006) Critical role of the epithelial Ca2+ channel TRPV5 in active Ca2+ reabsorption as revealed by TRPV5/calbindin-D28K knockout mice. J Am Soc Nephrol 17:3020–3027
Gkika D, Mahieu F, Nilius B, Hoenderop JG, Bindels RJ (2004) 80K-H as a new Ca2+ sensor regulating the activity of the epithelial Ca2+ channel transient receptor potential cation channel V5 (TRPV5). J Biol Chem 279:26351–26357
Gkika D, Topala CN, Chang Q, Picard N, Thebault S, Houillier P, Hoenderop JG, Bindels RJ (2006) Tissue kallikrein stimulates Ca2+ reabsorption via PKC-dependent plasma membrane accumulation of TRPV5. Embo J 25:4707–4716
Heller HJ, Sakhaee K, Moe OW, Pak CY (2002) Etiological role of estrogen status in renal stone formation. J Urol 168:1923–1927
Hellwig N, Albrecht N, Harteneck C, Schultz G, Schaefer M (2005) Homo- and heteromeric assembly of TRPV channel subunits. J Cell Science 118:917–928
Hoenderop JG, Dardenne O, Van Abel M, Van Der Kemp AW, Van Os CH, St-Arnaud R, Bindels RJ (2002) Modulation of renal Ca2+ transport protein genes by dietary Ca2+ and 1,25-dihydroxyvitamin D3 in 25-hydroxyvitamin D3-1alpha-hydroxylase knockout mice. Faseb J 16:1398–1406
Hoenderop JG, Hartog A, Stuiver M, Doucet A, Willems PH, Bindels RJ (2000) Localization of the epithelial Ca2+ channel in rabbit kidney and intestine. J Am Soc Nephrol 11:1171–1178
Hoenderop JG, Nilius B, Bindels RJ (2005) Calcium absorption across epithelia. Physiol Rev 85:373–422
Hoenderop JG, van der Kemp AW, Hartog A, van de Graaf SF, van Os CH, Willems PH, Bindels RJ (1999) Molecular identification of the apical Ca2+ channel in 1,25-dihydroxyvitamin D3-responsive epithelia. J Biol Chem 274:8375–8378
Hoenderop JG, van Leeuwen JP, van der Eerden BC, Kersten FF, van der Kemp AW, Merillat AM, Waarsing JH, Rossier BC, Vallon V, Hummler E, Bindels RJ (2003) Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest 112:1906–1914
Hoenderop JG, Vennekens R, Muller D, Prenen J, Droogmans G, Bindels RJ, Nilius B (2001) Function and expression of the epithelial Ca2+ channel family: comparison of mammalian ECaC1 and 2. J Physiol 537:747–761
Hoenderop JG, Voets T, Hoefs S, Weidema F, Prenen J, Nilius B, Bindels RJ (2003) Homo- and heterotetrameric architecture of the epithelial Ca2+ channels TRPV5 and TRPV6. Embo J 22:776–785
Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A, Obuse C, Togashi K, Tominaga M, Kita N, Tomiyama K, Iijima J, Nabeshima Y, Fujioka M, Asato R, Tanaka S, Kojima K, Ito J, Nozaki K, Hashimoto N, Ito T, Nishio T, Uchiyama T, Fujimori T, Nabeshima Y (2007) alpha-Klotho as a regulator of calcium homeostasis. Science 316:1615–1618
Khanal RC, Nemere I (2008) Regulation of intestinal calcium transport. Annu Rev Nutr 28:179–196
Koh N, Fujimori T, Nishiguchi S, Tamori A, Shiomi S, Nakatani T, Sugimura K, Kishimoto T, Kinoshita S, Kuroki T, Nabeshima Y (2001) Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun 280:1015–1020
Koster HP, Hartog A, Van Os CH, Bindels RJ (1995) Calbindin-D28K facilitates cytosolic calcium diffusion without interfering with calcium signaling. Cell Calcium 18:187–196
Kuro-o M (2000) Introduction: aging research comes of age. Cell Mol Life Sci 57:695–697
Lambers TT, Mahieu F, Oancea E, Hoofd L, de Lange F, Mensenkamp AR, Voets T, Nilius B, Clapham DE, Hoenderop JG, Bindels RJ (2006) Calbindin-D28K dynamically controls TRPV5-mediated Ca2+ transport. Embo J 25:2978–2988
Lambers TT, Oancea E, de Groot T, Topala CN, Hoenderop JG, Bindels RJ (2007) Extracellular pH dynamically controls cell surface delivery of functional TRPV5 channels. Mol Cell Biol 27:1486–1494
Lee CT, Lien YH, Lai LW, Chen JB, Lin CR, Chen HC (2006) Increased renal calcium and magnesium transporter abundance in streptozotocin-induced diabetes mellitus. Kidney Int 69:1786–1791
Lee J, Cha SK, Sun TJ, Huang CL (2005) PIP2 activates TRPV5 and releases its inhibition by intracellular Mg2+. J Gen Physiol 126:439–451
Loffing J, Loffing-Cueni D, Valderrabano V, Klausli L, Hebert SC, Rossier BC, Hoenderop JG, Bindels RJ, Kaissling B (2001) Distribution of transcellular calcium and sodium transport pathways along mouse distal nephron. Am J Physiol Renal Physiol 281:F1021–1027
Lu P, Boros S, Chang Q, Bindels RJ, Hoenderop JG (2008) The {beta}-glucuronidase klotho exclusively activates the epithelial Ca2+ channels TRPV5 and TRPV6. Nephrol Dial Transplant 23(11):3397–3402
Lytton J (2007) Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J 406:365–382
Nemere I, Norman AW (1988) 1,25-Dihydroxyvitamin D3-mediated vesicular transport of calcium in intestine: time-course studies. Endocrinology 122:2962–2969
Niemeyer BA, Bergs C, Wissenbach U, Flockerzi V, Trost C (2001) Competitive regulation of CaT-like-mediated Ca2+ entry by protein kinase C and calmodulin. Proc Natl Acad Sci USA 98:3600–3605
Nijenhuis T, Hoenderop JG, Bindels RJ (2005) TRPV5 and TRPV6 in Ca2+ (re)absorption: regulating Ca2+ entry at the gate. Pflugers Arch 451:181–192
Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ (2005) Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 115:1651–1658
Nilius B, Prenen J, Vennekens R, Hoenderop JG, Bindels RJ, Droogmans G (2001) Modulation of the epithelial calcium channel, ECaC, by intracellular Ca2+. Cell Calcium 29:417–428
Nilius B, Vennekens R, Prenen J, Hoenderop JG, Bindels RJ, Droogmans G (2000) Whole-cell and single channel monovalent cation currents through the novel rabbit epithelial Ca2+ channel ECaC. J Physiol 527(Pt 2):239–248
Oz OK, Hajibeigi A, Howard K, Cummins CL, van Abel M, Bindels RJ, Word RA, Kuro-o M, Pak CY, Zerwekh JE (2007) Aromatase deficiency causes altered expression of molecules critical for calcium reabsorption in the kidneys of female mice. J Bone Miner Res 22:1893–1902
Peng JB, Chen XZ, Berger UV, Vassilev PM, Tsukaguchi H, Brown EM, Hediger MA (1999) Molecular cloning and characterization of a channel-like transporter mediating intestinal calcium absorption. J Biol Chem 274:22739–22746
Phelps CB, Huang RJ, Lishko PV, Wang RR, Gaudet R (2008) Structural analyses of the ankyrin repeat domain of TRPV6 and related TRPV ion channels. Biochemistry 47:2476–2484
Proud D, Knepper MA, Pisano JJ (1983) Distribution of immunoreactive kallikrein along the rat nephron. Am J Physiol 244:F510–F515
Reilly RF, Ellison DH (2000) Mammalian distal tubule: physiology, pathophysiology, and molecular anatomy. Physiol Rev 80:277–313
Riccardi D, Lee WS, Lee K, Segre GV, Brown EM, Hebert SC (1996) Localization of the extracellular Ca2+-sensing receptor and PTH/PTHrP receptor in rat kidney. Am J Physiol 271:F951–956
Sakai K, Hirai M, Minoshima S, Kudoh J, Fukuyama R, Shimizu N (1989) Isolation of cDNAs encoding a substrate for protein kinase C: nucleotide sequence and chromosomal mapping of the gene for a human 80K protein. Genomics 5:309–315
Song Y, Peng X, Porta A, Takanaga H, Peng JB, Hediger MA, Fleet JC, Christakos S (2003) Calcium transporter 1 and epithelial calcium channel messenger ribonucleic acid are differentially regulated by 1,25 dihydroxyvitamin D3 in the intestine and kidney of mice. Endocrinology 144:3885–3894
Sooy K, Schermerhorn T, Noda M, Surana M, Rhoten WB, Meyer M, Fleischer N, Sharp GW, Christakos S (1999) Calbindin-D(28k) controls [Ca2+]i and insulin release. Evidence obtained from calbindin-d(28k) knockout mice and beta cell lines. J Biol Chem 274:34343–34349
Suki WN (1979) Calcium transport in the nephron. Am J Physiol 237:F1–F6
Sutton RA, Dirks JH (1975) The renal excretion of calcium: a review of micropuncture data. Can J Physiol Pharmacol 53:979–988
Sutton RA, Wong NL, Dirks JH (1979) Effects of metabolic acidosis and alkalosis on sodium and calcium transport in the dog kidney. Kidney Int 15:520–533
Suzuki Y, Landowski CP, Hediger MA (2008) Mechanisms and regulation of epithelial Ca2+ absorption in health and disease. Ann Rev Physiol 70:257–271
Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y (2003) Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol 17:2393–2403
Tsuruoka S, Nishiki K, Ioka T, Ando H, Saito Y, Kurabayashi M, Nagai R, Fujimura A (2006) Defect in parathyroid-hormone-induced luminal calcium absorption in connecting tubules of Klotho mice. Nephrol Dial Transplant 21:2762–2767
Van Abel M, Hoenderop JG, Dardenne O, St Arnaud R, Van Os CH, Van Leeuwen HJ, Bindels RJ (2002) 1,25-Dihydroxyvitamin D3-independent stimulatory effect of estrogen on the expression of ECaC1 in the kidney. J Am Soc Nephrol 13:2102–2109
van Abel M, Hoenderop JG, van der Kemp AW, Friedlaender MM, van Leeuwen JP, Bindels RJ (2005) Coordinated control of renal Ca2+ transport proteins by parathyroid hormone. Kidney Int 68:1708–1721
Van Baal J, Yu A, Hartog A, Fransen JA, Willems PH, Lytton J, Bindels RJ (1996) Localization and regulation by vitamin D of calcium transport proteins in rabbit cortical collecting system. Am J Physiol 271:F985–F993
van de Graaf SF, Bindels RJ, Hoenderop JG (2007) Physiology of epithelial Ca2+ and Mg2+ transport. Rev Physiol, Biochem Pharmacol 158:77–160
van de Graaf SF, Chang Q, Mensenkamp AR, Hoenderop JG, Bindels RJ (2006) Direct interaction with Rab11a targets the epithelial Ca2+ channels TRPV5 and TRPV6 to the plasma membrane. Mol Cell Biol 26:303–312
van de Graaf SF, Hoenderop JG, Gkika D, Lamers D, Prenen J, Rescher U, Gerke V, Staub O, Nilius B, Bindels RJ (2003) Functional expression of the epithelial Ca2+ channels (TRPV5 and TRPV6) requires association of the S100A10–annexin 2 complex. Embo J 22:1478–1487
van de Graaf SF, Rescher U, Hoenderop JG, Verkaart S, Bindels RJ, Gerke V (2008) TRPV5 is internalized via clathrin-dependent endocytosis to enter a Ca2+-controlled recycling pathway. J Biol Chem 283:4077–4086
Vassilev PM, Peng JB, Hediger MA, Brown EM (2001) Single-channel activities of the human epithelial Ca2+ transport proteins CaT1 and CaT2. J Membr Biol 184:113–120
Vennekens R, Hoenderop JG, Prenen J, Stuiver M, Willems PH, Droogmans G, Nilius B, Bindels RJ (2000) Permeation and gating properties of the novel epithelial Ca2+ channel. J Biol Chem 275:3963–3969
Wang X, Kumar R, Navarre J, Casanova JE, Goldenring JR (2000) Regulation of vesicle trafficking in madin–darby canine kidney cells by Rab11a and Rab25. J Biol Chem 275:29138–29146
Yeh BI, Sun TJ, Lee JZ, Chen HH, Huang CL (2003) Mechanism and molecular determinant for regulation of rabbit transient receptor potential type 5 (TRPV5) channel by extracellular pH. J Biol Chem 278:51044–51052
Yue L, Peng JB, Hediger MA, Clapham DE (2001) CaT1 manifests the pore properties of the calcium-release-activated calcium channel. Nature 410:705–709
Acknowledgments
This work was financially supported in part by grants from the Dutch Kidney Foundation (C03.6017, C06.2170), the Netherlands Organization for Scientific Research (NWO-ALW 814.02.001, NWO-CW 700.55.302, ZonMw 9120.6110). J. Hoenderop is supported by an EURYI award.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Boros, S., Bindels, R.J.M. & Hoenderop, J.G.J. Active Ca2+ reabsorption in the connecting tubule. Pflugers Arch - Eur J Physiol 458, 99–109 (2009). https://doi.org/10.1007/s00424-008-0602-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-008-0602-6