
Abstract
Medulloblastoma, a small blue cell malignancy of the cerebellum, is a major cause of morbidity and mortality in pediatric oncology. Current mechanisms for clinical prognostication and stratification include clinical factors (age, presence of metastases, and extent of resection) as well as histological subgrouping (classic, desmoplastic, and large cell/anaplastic histology). Transcriptional profiling studies of medulloblastoma cohorts from several research groups around the globe have suggested the existence of multiple distinct molecular subgroups that differ in their demographics, transcriptomes, somatic genetic events, and clinical outcomes. Variations in the number, composition, and nature of the subgroups between studies brought about a consensus conference in Boston in the fall of 2010. Discussants at the conference came to a consensus that the evidence supported the existence of four main subgroups of medulloblastoma (Wnt, Shh, Group 3, and Group 4). Participants outlined the demographic, transcriptional, genetic, and clinical differences between the four subgroups. While it is anticipated that the molecular classification of medulloblastoma will continue to evolve and diversify in the future as larger cohorts are studied at greater depth, herein we outline the current consensus nomenclature, and the differences between the medulloblastoma subgroups.
Similar content being viewed by others

Introduction
Current classification schemes for medulloblastoma are based primarily on morphology (histopathology), and include variants such as desmoplastic/nodular, MBEN (medulloblastoma with extensive nodularity), classic medulloblastoma, large cell, and anaplastic medulloblastoma. With recent developments in the ability to monitor transcription across the genome in the setting of a single experiment, various groups have started to sub-classify medulloblastoma on the basis of differences in the transcriptome [36]. Several laboratories across the globe have published on this topic in the recent past, with largely convergent conclusions [2, 7, 14, 22, 28, 30, 48]. This transcriptional approach to tumor sub-classification has also been applied to a number of different non-medulloblastoma histologies [4, 16, 18, 31, 35, 43], with the underlying assumption that tumors with similar transcriptomes will behave in a biologically similar manner, allowing a transcriptionally driven classification to serve as a guide for successful anti-neoplastic therapy.
The number of ‘subgroups’ of medulloblastoma identified among cohorts of medulloblastoma is largely dependant on the number of individual tumors within a given cohort, with larger cohorts identifying additional levels of hierarchical complexity [7, 22, 28, 36]. On the basis of the published literature, and some unpublished data presented at a recent consensus conference in Boston, Massachusetts, members from many of the laboratories publishing on this topic agreed that there were four principal transcriptional subgroups of medulloblastoma, with many of these subgroups showing a subsequent level of hierarchical structure that will be designated the subtypes of the subgroups (Fig. 1). The true number of subtypes for each of the subgroups is currently unknown, but it is likely more than one subtype for each subgroup. The four principal subgroups of medulloblastoma were named as follows: Wnt, Shh, Group 3, and Group 4 (Fig. 2). The Wnt and Shh (Sonic Hedgehog) were named for the signaling pathways thought to play prominent roles in the pathogenesis of that subgroup. Since less is known about the biology of the remaining two subgroups, the consensus was to retain generic names for the present until the underlying biology driving these subgroups was better delineated. There is evidence for the existence of subtypes within the subgroups, particularly for Group 3, which is also reflected by the fact that among Non-WNT, Non-SHH tumors, there may be a small fraction of “intermediate” tumors that would be assigned to Group 3 or Group 4 based on the clustering algorithm applied [7]. While the subsets of the subgroups seem readily apparent, they are not well characterized at this point and as such the consensus was to name them using Greek letters (α, β, γ, etc.) until additional characterization was available from multiple centers, on much larger cohorts (Fig. 1). Since the four groups as proposed are clearly distinct in terms of demographics, histology, DNA copy-number aberrations, and clinical outcome as also nicely summarized in a meta-analysis paper in the same issue of this journal [21], the authors consider it highly useful to start using these subgroups in the clinic. More specifically, molecular subgrouping will not only contribute to identify target cohorts for certain drugs (e.g., SHH inhibitors), but also add significantly to outcome prediction by the time of diagnosis [21], much more than any of the established clinical markers such as patient age, metastatic stage at diagnosis, level of resection, and histological subtype according to the WHO classification [25]. To this end, Table 1 summarizes markers that have been proposed to perform subgrouping by immunostaining from various groups. Although it is evident that few tumors will either be positive for more than one marker or negative for all markers [28], it is encouraging that using two markers that are supposed to select the same subgroup (such as SFRP1 and GAB1 for SHH tumors) resulted in a very high overlap in a study published in the same issue of this journal [23]. In the following, the distinctive characteristics of these four core subgroups are described in more detail.

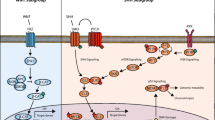
Dendrogram depicting the classification of embryonal tumors of the cerebellum. Medulloblastomas should be differentiated from the less common ATRTs and ETANTRs of the cerebellum. Under the current consensus classification of medulloblastoma four principle subgroups are identified: Wnt, Shh, Group 3, and Group 4. The evidence suggests that each of the four principle subgroups will likely have distinct ‘subsets’ that are biologically and clinically homogeneous as compared to other subsets from within the same subgroup. As the nature and number of subsets for each subgroup are currently unknown, the consensus classification suggests that each subset be named using a Greek letter (α, β, γ, etc.) until such time as they are sufficiently characterized to be named based on their molecular etiology

Comparison of the various subgroups of medulloblastoma including their affiliations with previously published papers on medulloblastoma molecular subgrouping
Wnt subgroup
The best known of the medulloblastoma subgroups is the Wnt subgroup due to its very good long-term prognosis in comparison to other subgroups [8, 11, 13, 15, 22, 28, 39, 48]. Indeed, long-term survival rates for patients with Wnt medulloblastoma likely exceed 90%, with those patients who die succumbing more often to complications of therapy or secondary neoplasms rather than to recurrent Wnt medulloblastoma [12]. Germline mutations of the Wnt pathway inhibitor APC predispose to Turcot syndrome, which includes a proclivity to medulloblastoma, in addition, somatic mutations of CTNNB1 encoding β-catenin have been found in sporadic medulloblastomas [19, 50]. These strong germline and somatic genetic data strongly support an etiological role for canonical Wnt signaling in the pathogenesis of this group of tumors, and lead to the nomenclature of ‘Wnt subgroup medulloblastomas’.
Nearly all of the Wnt medulloblastomas studied to date have classic histology. Wnt medulloblastomas are frequently described as having CTNNB1 mutations, nuclear immunohistochemical staining for β-catenin, and monosomy six (deletion of one copy of chromosome 6 in the tumor). Which, if any of these markers is a gold standard for the diagnosis of Wnt medulloblastoma is unclear, as medulloblastomas with a clear Wnt transcriptional signature that do not have monosomy six have been reported [28]. The recent report of a single medulloblastoma with mutations in both CTNNB1 and PTCH also complicates the story [32]. Indeed, over-representation of genes in the Wnt pathway has also been reported in Shh and Group 3 medulloblastomas, although this finding is of unknown significance [28]. Medulloblastomas with large cell/anaplastic histology have also been reported in the Wnt subgroup, although they appear to maintain the excellent prognosis associated with the Wnt subgroup [12]. Which of monosomy 6, nuclear staining for β-catenin, mutation of CTNNB1, immunohistochemical staining for DKK1 [28, 48], or a transcriptional signature that clusters with other ‘Wnt’ tumors should be used as a gold standard for the diagnosis of Wnt medulloblastoma awaits further validation on larger cohorts of well-characterized medulloblastomas.
Overall medulloblastoma is more common in males, however, the gender ratio for Wnt medulloblastomas is about 1:1 male:female. Wnt medulloblastomas can occur at all ages, but are uncommon in infants. Other than monosomy six, there are few other regions of genetic amplification or deletion in the genome of Wnt medulloblastomas [28]. Recently a mouse model of Wnt medulloblastoma was published, which suggested that Wnt medulloblastomas arise from the lower rhombic lip of the cerebellum [17]. This mouse will undoubtedly serve as a valuable tool in the preclinical assessment of novel therapeutics. As most patients with Wnt medulloblastoma survive, it is possible that they are being over treated with current therapies which are quite morbid, and there is an active discussion of a clinical trial of therapy de-escalation in this patient population.
Sonic hedgehog subgroup
The Shh group of medulloblastomas are named after the Sonic Hedgehog signaling pathway, which is thought to drive tumor initiation in many, if not all such cases. Individuals with germline mutations in the Shh receptor PTCH have Gorlin syndrome, which includes a predisposition to medulloblastoma [3, 47]. Similarly, individuals with germline mutations of the Shh inhibitor SUFU are predisposed to medulloblastoma, particularly infantile medulloblastoma [5, 33, 42, 46]. Similarly, somatic mutations of PTCH, SMO, and SUFU, as well as amplifications of GLI1 and GLI2 have been found in sporadic medulloblastoma [27, 30, 46]. These genetic data implicating Shh signaling in the etiology of this group of tumors has lead to the current proposal to formally name them ‘Sonic Hedgehog’ subgroup medulloblastomas.
Sonic hedgehog subgroup medulloblastomas have largely been identified on the basis of transcriptional profiling [7, 22, 28, 29, 41, 48]. Others approaches to identify Shh medulloblastomas have included immunohistochemical staining for SFRP1 [2, 28, 48], or GAB1 [11]. Deletion of chromosome 9q appears to be limited to Shh medulloblastomas, which is appropriate as the PTCH gene is located at chromosome 9q22 [28]. The vast majority of published mouse models of medulloblastoma belong to the Shh subgroup of tumors [20]. The temporal incidence of human Shh medulloblastoma is curiously dichotomous, in that it is very frequent in both infants (0–3 years) and adults (>16 years), but much less frequent in children (3–16 years). The extent to which adult Shh medulloblastomas resemble infant Shh medulloblastoma remains to be determined by the analyses of larger cohorts of tumors. Some recent series [28], but not others [7] have found a preponderance of females among Shh medulloblastoma. Taken into account all published studies, the gender ratio is approximately 1:1. Most, if not all nodular/desmoplastic medulloblastomas belong to the Shh subgroup. However, this is not an effective marker for the subgroup as up to 50% of Shh subgroup medulloblastomas are not nodular/desmoplastic. The prognosis of Shh medulloblastoma appears to be similar to Group 4 medulloblastomas, and intermediate between that Wnt medulloblastomas (good) and Group 3 medulloblastomas (poor). The recent demonstration that small molecules targeting smoothened (SMO) are highly effective, albeit temporarily, against Shh medulloblastoma underlines the urgent need to develop effective and practical markers for Shh medulloblastoma [6, 40, 49].
Group 3
Group 3 tumors are mostly ‘classic’ medulloblastomas, although they do encompass the majority of the LCA tumors. The current gold standard for diagnosis of a Group 3 tumor is a transcriptional profile that clusters with other Group 3 tumors [7, 22, 28]. Immunohistochemical positivity for NPR3 has been suggested as a Group 3 marker [28]. While Shh subgroup tumors have high levels of expression of MYCN, and Wnt subgroup and Group 3 tumors have high levels of expression of MYC, whereas Group 4 tumors have relatively low expression of both MYC and MYCN, apart from the few cases that have MYCN amplification [20, 22, 28]. As such, it has been suggested by one group that Group 3 tumors could be called the MYC group [20]. Similarly, MYC amplification (but not MYCN amplification) appears to be almost always limited to Group 3 [7, 22, 28]. Amplification and over-expression of the medulloblastoma oncogene OTX2 appears to be restricted to Group 3 and Group 4 tumors [1, 9, 10, 28]. Group 3 tumors over-express a number of genes that were initially identified through their role in retinal development, although the role of these genes in the pathogenesis in Group 3 tumors is currently opaque [7, 22, 28]. Group 3 tumors are much more likely than Group 4 tumors to show gain of chromosome 1q, and/or loss of chromosome 5q and chromosome 10q.
Group 3 tumors occur more commonly in males than females, and are found in infants and children, but are almost never observed in adults. Group 3 tumors have a high incidence of LCA histology, and are very frequently metastatic [28]. Indeed, it has been suggested that prior identification of metastatic status as risk factor for poor prognosis in medulloblastoma was in fact identifying a group of patients enriched for Group 3 patients [28]. The best evidence for a clear ‘subset of a subgroup’ in medulloblastoma to date is found in Group 3, in which one subset (Group 3α) includes all of the patients with MYC amplifications, and which assumes that most of the high risk of recurrence and death associated with a Group 3 diagnosis. Conversely, Group 3β patients were not found to harbor MYC amplifications, and had a clinical outcome similar to Group 4 patients [7]. This exciting finding awaits validation in additional cohorts, and the development of clinically expedient markers for the identification of Group 3α and Group 3β patients. Although Group 3 tumors are likely more similar to Group 4 tumors than to Shh or Wnt tumors, the demographic, clinical, transcriptional, and genetic differences between the Group 3 and Group 4 suggest that they are indeed distinct entities [22, 28]. Clearly, the terrible prognosis of Group 3 patients indicates that the medulloblastoma community needs to focus further on this subgroup to develop practical biomarkers, understand its underlying pathogenesis, and develop accurate mouse models.
Group 4
Group 4 medulloblastomas are the prototypical medulloblastoma: a 7-year-old boy with a classic histology medulloblastoma that has an isochromosome 17q. As the molecular pathogenesis of Group 4 tumors is not currently clear, the generic name ‘Group 4’ has been chosen for the current consensus nomenclature pending further insights. Currently, Group 4 medulloblastomas are identified through a transcriptional profile that clusters with other Group 4 medulloblastomas. KCNA1 has been suggested as an immunohistochemical marker for Group 4 tumors but requires validation in additional cohorts [7, 28]. Although isochromosome 17q is also seen in Group 3 tumors (26%), it is much more common in Group 4 tumors where it is the most common cytogenetic change observed (66%) as also seen in the meta-analysis paper by Kool et al. on all published data sets in this issue of the journal [21, 28]. Similarly, isolated 17p deletion is seen in both Group 3 and Group 4, but almost never in Wnt or Shh subgroup medulloblastomas. The only other notable cytogenetic change among Group 4 tumors is loss of the X chromosome, which is seen in 80% of females with Group 4 medulloblastoma. The high incidence of X chromosome loss in females with Group 4 medulloblastomas is particularly poignant in light of the high male:female ratio in Group 4 patients (2:1). Multiple publications have identified over-representation of genes involved in neuronal differentiation and neuronal development in Group 4 tumors, although neither the genetic basis, nor the clinical relevance of this is yet apparent [7, 22, 28]. Group 4 patients have an intermediate prognosis, similar to individuals with Shh tumors. Although Group 4 tumors make up >30% of all medulloblastomas, we probably understand the least about the molecular pathogenesis of this subgroup, and no mouse models of Group 4 have been reported.
Histology: special considerations
While most investigators in the field feel strongly that molecular markers will assume increasing importance in the diagnosis and classification of medulloblastoma in the near future, still there will clearly be a role for histopathology. True nodular/desmoplastic tumors, and likely all of the MBENs belong to the Shh subgroup of tumors [11, 26]. Classic histology, however, is spread over all four of the principal subgroups, and can even be difficult to distinguish from atypical teratoid/rhaboid tumors on the basis of morphology alone. Large cell/anaplastic tumors also appear to be found in all four principal subgroups, although the majority are Group 3. However, LCA histology maintains its prognostic significance after accounting for subgroup, and the diagnosis of LCA histology may therefore retain significance in a ‘post-subgroup’ world [28]. We would suggest, however, that the prognostic significance of LCA histology needs to be assessed separately in large cohorts of each subgroup as the field moves forward.
Adult medulloblastoma
The degree of molecular and clinical similarity between adult and pediatric medulloblastoma is not entirely clear, yet. Some authors have certainly reported distinct molecular profiles [24] and clinical behavior [44, 45] when comparing adults and children/infants. However, the distribution of cases across the subgroups is very different in adults as compared to children [2, 28, 37, 38]. The most comprehensive study on adult MB published to date suggests that group 3 tumors are exceptionally rare in adults, whereas SHH tumors comprise about two-thirds of cases in this age group [38], which makes adult MB patients a particularly attractive cohort for molecular-targeted therapies. However, the exact proportions of subgroups and genetic as well as transcriptomic differences between adult and pediatric patients attributed to the same subgroups have yet to be studied in more detail in larger cohorts.
Conclusions
While histology and the WHO classification have not been supplanted, it has become clear that histology alone is inadequate for the diagnosis and classification of ‘medulloblastoma’. The current generation of studies supports a model in which transcriptionally similar tumors share clinical and molecular features that will be useful in the clinic. The identification of molecular subgroup will likely assume great importance in the design and implementation of targeted therapies. While transcriptionally driven classifications of medulloblastoma will increase in importance over time, we predict that classification based on whole genome or exome sequencing of tumor DNA will likely assume additional importance as sequencing technologies become less expensive, more widely available, and more reliable. While it is clear that distinct molecular subgroups of medulloblastoma exist and are clinically important, we should not lose sight of the fact that there are also commonalities seen across the subgroups, such as mutations in TP53 and MLL2 which are seen in several subgroups [32, 34]. The challenges facing the medulloblastoma community in moving forward are (1) to achieve sufficient inter-center co-operation to assemble large cohorts of tumors for both discovery and validation, (2) determine the true extent and nature of inter-tumoral heterogeneity as defined by transcriptional profiling, and (3) development of clinically practical tests for medulloblastoma subgroup assignment that will be feasible for both clinical trials, and inclusion in the day to day clinical care of medulloblastoma patients across the globe.
References
Adamson DC, Shi Q, Wortham M, Northcott PA, Di C, Duncan CG, Li J, McLendon RE, Bigner DD, Taylor MD, Yan H (2010) OTX2 is critical for the maintenance and progression of Shh-independent medulloblastomas. Cancer Res 70:181–191
Al-Halabi H, Nantel A, Klekner A, Guiot MC, Albrecht S, Hauser P, Garami M, Bognar L, Kavan P, Gerges N, Shirinian M, Roberge D, Muanza T, Jabado N (2011) Preponderance of sonic hedgehog pathway activation characterizes adult medulloblastoma. Acta Neuropathol 121:229–239
Bale SJ, Falk RT, Rogers GR (1998) Patching together the genetics of Gorlin syndrome. J Cutan Med Surg 3:31–34
Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, Lander ES, Wong W, Johnson BE, Golub TR, Sugarbaker DJ, Meyerson M (2001) Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci USA 98:13790–13795
Brugieres L, Pierron G, Chompret A, Paillerets BB, Di Rocco F, Varlet P, Pierre-Kahn A, Caron O, Grill J, Delattre O (2010) Incomplete penetrance of the predisposition to medulloblastoma associated with germ-line SUFU mutations. J Med Genet 47:142–144
Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, Hsiao K, Yuan J, Green J, Ospina B, Yu Q, Ostrom L, Fordjour P, Anderson DL, Monahan JE, Kelleher JF, Peukert S, Pan S, Wu X, Maira SM, Garcia-Echeverria C, Briggs KJ, Watkins DN, Yao YM, Lengauer C, Warmuth M, Sellers WR, Dorsch M (2010) Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med 2:51ra70
Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, Berhoukim R, Amani V, Goumnerova L, Eberhart CG, Lau CC, Olson JM, Gilbertson RJ, Gajjar A, Delattre O, Kool M, Ligon K, Meyerson M, Mesirov JP, Pomeroy SL (2011) Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29:1424–1430
Clifford S, Lusher M, Lindsey J, Langdon J, Gilbertson R, Straughton D, Ellison D (2006) Wnt/wingless pathway activation and chromosome 6 loss characterise a distinct molecular sub-group of medulloblastomas associated with a favourable prognosis. Cell Cycle 5:2666–2670
de Haas T, Oussoren E, Grajkowska W, Perek-Polnik M, Popovic M, Zadravec-Zalatel L, Perera M, Corte G, Wirths O, van Sluis P, Pietsch T, Troost D, Baas F, Versteeg R, Kool M (2006) OTX1 and OTX2 expression correlates with the clinicopathologic classification of medulloblastomas. J Neuropathol Exp Neurol 65:1–11
Di C, Liao S, Adamson DC, Parrett TJ, Broderick DK, Shi Q, Lengauer C, Cummins JM, Velculescu VE, Fults DW, McLendon RE, Bigner DD, Yan H (2005) Identification of OTX2 as a medulloblastoma oncogene whose product can be targeted by all-trans retinoic acid. Cancer Res 65:919–924
Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G, Kenney AM, Brat DJ, Perry A, Yong WH, Taylor RE, Bailey S, Clifford SC, Gilbertson RJ (2011) Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol 121:381–396
Ellison DW, Kocak M, Dalton J, Megahed H, Lusher ME, Ryan SL, Zhao W, Nicholson SL, Taylor RE, Bailey S, Clifford SC (2011) Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol 29:1400–1407
Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE, Pearson AD, Clifford SC (2005) ß-catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol 23:7951–7957
Fattet S, Haberler C, Legoix P, Varlet P, Lellouch-Tubiana A, Lair S, Manie E, Raquin MA, Bours D, Carpentier S, Barillot E, Grill J, Doz F, Puget S, Janoueix-Lerosey I, Delattre O (2009) Beta-catenin status in paediatric medulloblastomas: correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J Pathol 218:86–94
Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, Woo S, Wheeler G, Ahern V, Krasin MJ, Fouladi M, Broniscer A, Krance R, Hale GA, Stewart CF, Dauser R, Sanford RA, Fuller C, Lau C, Boyett JM, Wallace D, Gilbertson RJ (2006) Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol 7:813–820
Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, Datto MB, Kelley M, Mathey-Prevot B, Potti A, Nevins JR (2010) A pathway-based classification of human breast cancer. Proc Natl Acad Sci USA 107:6994–6999
Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, Finkelstein D, Pounds S, Weiss A, Patay Z, Scoggins M, Ogg R, Pei Y, Yang ZJ, Brun S, Lee Y, Zindy F, Lindsey JC, Taketo MM, Boop FA, Sanford RA, Gajjar A, Clifford SC, Roussel MF, McKinnon PJ, Gutmann DH, Ellison DW, Wechsler-Reya R, Gilbertson RJ (2010) Subtypes of medulloblastoma have distinct developmental origins. Nature 468:1095–1099
Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286:531–537
Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B et al (1995) The molecular basis of Turcot’s syndrome. N Engl J Med 332:839–847
Hatten ME, Roussel MF (2011) Development and cancer of the cerebellum. Trends Neurosci 34(3):134–142
Kool M, Korshunov A, Remke M, Jones D, Schlanstein M, Northcott P, Cho Y, Schouten - van Meeteren N, van Vuurden D, Clifford S, Pietsch T, von Bueren A, Rutkowski S, McCabe M, Collins P, Bäcklund M, Haberler C, Bourdeaut F, Delattre O, Doz F, Ellison D, Gilbertson R, Pomeroy S, Taylor M, Lichter P, Pfister S (2011) Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of wnt, shh, group 3, and group 4 medulloblastomas. Acta Neuropathol (under review)
Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, Troost D, Meeteren NS, Caron HN, Cloos J, Mrsic A, Ylstra B, Grajkowska W, Hartmann W, Pietsch T, Ellison D, Clifford SC, Versteeg R (2008) Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE 3:e3088
Korshunov A, Remke M, Kool M, Hielscher T, Northcott P, Williamson D, Pfaff E, Witt H, Jones D, Ryzhova M, Cho Y, Wittmann A, Benner A, Weiss W, von Deimling A, Scheurlen W, Kulozik A, Clifford S, Collins V, Westermann F, Taylor M, Lichter P, Pfister S (2011) Biological and clinical heterogeneity of MYCN-amplified medulloblastoma. Acta Neuropathol. doi:10.1007/s00401-011-0918-8
Korshunov A, Remke M, Werft W, Benner A, Ryzhova M, Witt H, Sturm D, Wittmann A, Schöttler A, Felsberg J, Reifenberger G, Rutkowski S, Scheurlen W, Kulozik A, von Deimling A, Lichter P, Pfister S (2010) Adult and pediatric medulloblastomas are genetically distinct and require different algorithms for molecular risk stratification. J Clin Oncol 28:3054–3060
Louis D, Ohgaki H, Wiestler O, Cavenee W, Burger P, Jouvet A, Scheithauer B, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
McManamy CS, Pears J, Weston CL, Hanzely Z, Ironside JW, Taylor RE, Grundy RG, Clifford SC, Ellison DW (2007) Nodule formation and desmoplasia in medulloblastomas—defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol 17:151–164
Northcott P, Hielscher T, Dubuc A, Mack S, Shih D, Remke M, Al-Halabi H, Albrecht S, Jabado N, Eberhart C, Grajkowska W, Weiss W, Clifford S, Bouffet E, Rutka J, Korshunov A, Pfister S, Taylor M (2011) Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol 122:231–240
Northcott P, Korshunov A, Witt H, Hielscher T, Eberhart C, Mack S, Bouffet E, Clifford S, Hawkins C, French P, Rutka J, Pfister S, Taylor M (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29:1408–1414
Northcott PA, Fernandez LA, Hagan JP, Ellison DW, Grajkowska W, Gillespie Y, Grundy R, Van Meter T, Rutka JT, Croce CM, Kenney AM, Taylor MD (2009) The miR-17/92 polycistron is up-regulated in sonic hedgehog-driven medulloblastomas and induced by N-myc in sonic hedgehog-treated cerebellar neural precursors. Cancer Res 69:3249–3255
Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, Ra YS, Zilberberg K, McLeod J, Scherer SW, Sunil Rao J, Eberhart CG, Grajkowska W, Gillespie Y, Lach B, Grundy R, Pollack IF, Hamilton RL, Van Meter T, Carlotti CG, Boop F, Bigner D, Gilbertson RJ, Rutka JT, Taylor MD (2009) Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet 41:465–472
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522
Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, Gallia GL, Jallo GI, Binder ZA, Nikolsky Y, Hartigan J, Smith DR, Gerhard DS, Fults DW, VandenBerg S, Berger MS, Marie SK, Shinjo SM, Clara C, Phillips PC, Minturn JE, Biegel JA, Judkins AR, Resnick AC, Storm PB, Curran T, He Y, Rasheed BA, Friedman HS, Keir ST, McLendon R, Northcott PA, Taylor MD, Burger PC, Riggins GJ, Karchin R, Parmigiani G, Bigner DD, Yan H, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE (2011) The genetic landscape of the childhood cancer medulloblastoma. Science 331:435–439
Pastorino L, Ghiorzo P, Nasti S, Battistuzzi L, Cusano R, Marzocchi C, Garre ML, Clementi M, Scarra GB (2009) Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A 149A:1539–1543
Pfaff E, Remke M, Sturm D, Benner A, Witt H, Milde T, von Bueren AO, Wittmann A, Schottler A, Jorch N, Graf N, Kulozik AE, Witt O, Scheurlen W, von Deimling A, Rutkowski S, Taylor MD, Tabori U, Lichter P, Korshunov A, Pfister SM (2010) Tp53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J Clin Oncol 28:5188–5196
Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K (2006) Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9:157–173
Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, Allen JC, Zagzag D, Olson JM, Curran T, Wetmore C, Biegel JA, Poggio T, Mukherjee S, Rifkin R, Califano A, Stolovitzky G, Louis DN, Mesirov JP, Lander ES, Golub TR (2002) Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415:436–442
Remke M, Hielscher T, Korshunov A, Northcott P, Bender S, Kool M, Westermann F, Benner A, Cin H, Ryzhova M, Sturm D, Witt H, Haag D, Toedt G, Wittmann A, Schöttler A, von Bueren A, Rutkowski S, Scheurlen W, Kulozik A, Taylor M, Lichter P, Pfister S (2011) FSTL5 is a marker of poor prognosis in non-WNT/non-SHH medulloblastoma. J Clin Oncol 29:3852–3861
Remke M, Hielscher T, Northcott P, Witt H, Ryzhova M, Wittmann A, Benner A, von Deimling A, Scheurlen W, Perry A, Croul S, Kulozik A, Lichter P, Taylor M, Pfister S, Korshunov A (2011) Adult medulloblastoma is comprised of three major molecular variants. J Clin Oncol 29:2717–2723
Rogers HA, Miller S, Lowe J, Brundler MA, Coyle B, Grundy RG (2009) An investigation of WNT pathway activation and association with survival in central nervous system primitive neuroectodermal tumours (CNS PNET). Br J Cancer 100:1292–1302
Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, Low JA (2009) Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 361:1173–1178
Schwalbe EC, Lindsey JC, Straughton D, Hogg TL, Cole M, Megahed H, Ryan SL, Lusher ME, Taylor MD, Gilbertson RJ, Ellison DW, Bailey S, Clifford SC (2011) Rapid diagnosis of medulloblastoma molecular subgroups. Clin Cancer Res 17:1883–1894
Slade I, Murray A, Hanks S, Kumar A, Walker L, Hargrave D, Douglas J, Stiller C, Izatt L, Rahman N (2010) Heterogeneity of familial medulloblastoma and contribution of germline PTCH1 and SUFU mutations to sporadic medulloblastoma. Fam Cancer 10(2):337–342
Stegmaier K, Ross KN, Colavito SA, O’Malley S, Stockwell BR, Golub TR (2004) Gene expression-based high-throughput screening(GE-HTS) and application to leukemia differentiation. Nat Genet 36:257–263
Tabori U, Sung L, Hukin J, Laperriere N, Crooks B, Carret AS, Silva M, Odame I, Mpofu C, Strother D, Wilson B, Samson Y, Bouffet E (2005) Medulloblastoma in the second decade of life: a specific group with respect to toxicity and management: a Canadian Pediatric Brain Tumor Consortium Study. Cancer 103:1874–1880
Tabori U, Sung L, Hukin J, Laperriere N, Crooks B, Carret AS, Silva M, Odame I, Mpofu C, Strother D, Wilson B, Samson Y, Bouffet E (2006) Distinctive clinical course and pattern of relapse in adolescents with medulloblastoma. Int J Radiat Oncol Biol Phys 64:402–407
Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, Hao A, Goldstein AM, Stavrou T, Scherer SW, Dura WT, Wainwright B, Squire JA, Rutka JT, Hogg D (2002) Mutations in SUFU predispose to medulloblastoma. Nat Genet 31:306–310
Taylor MD, Mainprize TG, Rutka JT (2000) Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery 47:888–901
Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, Chintagumpala M, Adesina A, Ashley DM, Kellie SJ, Taylor MD, Curran T, Gajjar A, Gilbertson RJ (2006) Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24:1924–1931
Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, Bazan JF, Kan Z, Seshagiri S, Hann CL, Gould SE, Low JA, Rudin CM, de Sauvage FJ (2009) Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 326:572–574
Zurawel RH, Chiappa SA, Allen C, Raffel C (1998) Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res 58:896–899
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Taylor, M.D., Northcott, P.A., Korshunov, A. et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123, 465–472 (2012). https://doi.org/10.1007/s00401-011-0922-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-011-0922-z