
Abstract
Type 1 diabetes (T1D) is considered a disorder whose pathogenesis is autoimmune in origin, a notion drawn in large part from studies of human pancreata performed as far back as the 1960s. While studies of the genetics, epidemiology, and peripheral immunity in T1D have been subject to widespread analysis over the ensuing decades, efforts to understand the disorder through analysis of human pancreata have been far more limited. We have reviewed the published literature pertaining to the pathology of the human pancreas throughout all stages in the natural history of T1D. This effort uncovered a series of findings that challenge many dogmas ascribed to T1D and revealed data suggesting the marked heterogeneity in terms of its pathology. An improved understanding and appreciation for pancreatic pathology in T1D could lead to improved disease classification, an understanding of why the disorder occurs, and better therapies for disease prevention and management.
Similar content being viewed by others

Type 1 diabetes
The insulin deficiency associated with type 1 diabetes (T1D) is thought to be due to an autoimmune-mediated destruction of pancreatic β-cells; this is the result of an (still) ill-defined combination of genetic susceptibilities alongside environmental factors [59, 65]. Despite the availability of exogenous insulin therapy, T1D patients remain at risk for a number of chronic complications that result from poorly controlled blood glucose concentrations [1, 26, 63]. For this reason, as well as other issues of increased morbidity and mortality, an immense need exists for studies capable of shedding light onto the pathogenesis of T1D including an elucidation to the question of how and why the pancreatic β-cell becomes a target.
Beyond genetic (HLA) susceptibility, the autoimmune etiology of T1D has primarily been based on the identification of islet cell reactive autoantibodies in the serum of patients with or at genetic risk for T1D [6, 48, 50, 52, 61, 83, 87, 100], as well as lymphocytic infiltrates in the pancreatic islets of patients who died shortly after disease onset [12, 17, 21, 22, 28, 29, 46]. Little is known about the etiological underpinnings for pathogenesis of T1D in humans, but a number of theories, based on data from humans and animal models, have been proposed to explain how β-cell autoimmunity is initiated and eventually, progresses to a degree of insulin deficiency, which expresses itself as hyperglycemia. Some of the more popular theories include molecular mimicry [7] resulting from cross-reactivity of viral [77, 92] or dietary antigens [3, 55, 57] with β-cell antigens, gut microflora that drive deleterious immune responsiveness [15, 25, 80], dysfunctional β-cell antigen expression in the thymus [19] and pancreatic lymph nodes [99], among others. With so many etiologies that could potentially lead to loss of immunological tolerance to β-cell antigens and subsequent cellular destruction, attention has recently been directed towards investigations of the human pancreas in T1D. To be clear, this notion is not a new one since as mentioned previously, studies of human pancreata from individuals with diabetes date back many decades.
With this, it is fair to ask the question of what can re-examination of the literature pertaining to human pancreas pathology reveal regarding the pathogenesis of T1D that may not have been evident in the past? Insights gained from recent epidemiological studies, when combined with histopathology, may provide valuable information regarding the factors involved in the incidence of this disease, with the pathologic features either supporting or refuting hypothetical relationships. As but one example, the steady increase in T1D incidence in most developed countries [68, 74, 82, 86, 89, 90] suggests that new, or more widespread exposure to environmental agents (dietary, virus), may initiate or accelerate β-cell autoimmunity. Indeed, worldwide epidemiological data indicate that the proportion of newly diagnosed children with genotypes considered at high risk for T1D are decreasing (i.e., the disease is more often diagnosed in those who, in previous generations, may not have been considered as having an exceedingly high genetic risk for T1D), which as Borchers et al. [11] and others [24] have pointed out, suggests that environmental factors are now of increasing importance in the development of β-cell autoimmunity. In terms of actual environmental factors that may be driving such processes, there are no shortage of candidates as increased consumption of milk, cereals, and meat (among other dietary factors) have been associated with the increased T1D incidence [67]. Other studies have noted that children who developed T1D had higher energy intake, disaccharides and sucrose in particular, and larger body size in the years preceding disease onset [73]. Taken collectively, these studies support the so-called “overload hypothesis” proposed by Dahlquist [18], which suggests that environmental factors which increase insulin demand will accelerate the β-cell destruction following the initial triggering of autoimmunity. In an alternate scenario for this model, heightened insulin secretion “injures” β-cells, providing the initial trigger (i.e., expression of autoantigens [16, 33], class I HLA [70], cytokines [23]) that provokes a self-destructive immune response.
As this review discusses the pancreatic pathology of T1D, it is important to begin by briefly describing previous attempts to categorize this heterogeneous disease, both with and without the aid of pancreatic histology. The clinical diagnosis of T1D is based primarily on a combination of hyperglycemia, an absolute or severe loss of C-peptide secretion and dependence on exogenous insulin for survival [42]. The etiology of this loss of C-peptide secretion and, presumably, β-cell destruction has been divided into autoimmune-mediated (i.e., type 1A; based on the presence of islet cell autoantibodies), as well as idiopathic (type 1B) diabetes [42]. Currently, islet autoantibodies (e.g., glutamic acid decarboxylase, insulin, IA-2, zinc transporter) are not routinely performed in most clinical settings, making it difficult to differentiate between type 1A and type 1B diabetes. This fact complicates the interpretation of (already confusing) past pathologic studies of what has, over the years, been referred to as juvenile diabetes, or insulin-dependent diabetes. Hence, it is not possible to definitively differentiate between cases that are true type 1A in origin without histologic evidence of autoimmunity from those with type 1B.
The conundrum of evaluating human pancreas pathology
While the study of pancreata from individuals with T1D would be expected to provide important clues as to T1D pathophysiology, limitations in access to research quality samples are hampered by the very nature of the organ; namely, to function as a potent digestive tissue. Another major limitation, however, is access to the earliest stages of T1D when β-cell loss begins. Since (and fortunately) T1D is generally not an acutely fatal disease at its diagnosis, pancreatic tissue samples at clinical onset are not generally available nor can biopsy be safely performed without a great risk for pancreatitis. In addition to the inaccessibility of the pancreas, T1D is a difficult disease to investigate due to a long, clinically silent period that precedes disease onset (i.e., symptomatic hyperglycemia), during which time, lymphocytes traffic to the islets and mediate the progressive destruction of β-cells. Elucidating the early events that occur prior to, and presumably stimulate this self-destructive process, is therefore an important goal for evaluating human pancreatic specimens to identify disease initiation and progression. With longer disease duration, evidence of active β-cell autoimmunity (i.e., insulitis) appears to become increasingly rare. In addition to these T1D-specific caveats, the very nature of cadaveric histopathology studies in which pathologic information is only available for a single time point makes it difficult to understand the pattern and rate of β-cell loss or, alternately, β-cell regeneration from one individual to another. Finally, unknown chronic effects of fluctuating blood glucose and insulin concentrations on β-cell survival and autoimmunity complicate interpretation of pancreatic histopathology. These multiple factors obscure the interpretation from human pancreatic specimens but nevertheless, such are the primary means for elucidating T1D pathogenesis in humans.
Study descriptions (case reports and beyond)
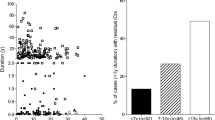
To initiate our presentation of the pancreatic pathology of human T1D, patient information was compiled from six T1D histopathology studies (Fig. 1) and categorized by the presence or absence of β-cells or insulitis in patients who underwent either a short (Tables 1 and 2) or long disease duration (Tables 3 and 4), all as defined in the respective studies [17, 21, 22, 28, 29, 46]. These studies were chosen because data on age, disease duration, and islet pathology (i.e., β-cells, insulitis) were available for each individual case. Pancreatic pathology prior to disease onset has also been described in a small number (n = 6) of autoantibody-positive nondiabetic individuals drawn from three additional studies (Fig. 1) [31, 40, 93].

Comparison of histopathological studies of type 1 diabetes by stage of disease course. The region in blue corresponds to the narrow range of disease duration (<2 months) that is represented across all six studies indicated at right. The region in gray corresponds to the period prior to disease onset (pre-clinical phase) and lists the three studies that examined pancreata from nondiabetic individuals with islet autoantibodies
From a historical perspective, the original work of Gepts [28] is considered by most to represent the seminal work with examination of autopsy pancreatic specimens from 54 diabetic patients who developed the disease prior to age of 31 years. These cases were obtained from hospitals in Brussels, Belgium and the USA (Philadelphia, Boston, Winston-Salem, Keene). Twenty-two cases were considered “acute”, since duration from first appearance of diabetic symptoms (i.e., polydipsia, weight loss, polyuria) to death was less than 6 months. The remaining 32 cases were considered “chronic” as disease duration ranged from 2 to 37 years. In addition to these studies of Gepts, Junker et al. [46] examined autopsy material from 11 patients with reported diabetes of ages 30 years or less who also resided in Denmark. The first appearance of diabetic symptoms in each of these Danish patients occurred within 2 months of death, and none of the patients received insulin therapy prior to admission; however, all received insulin at hospital admission. The cause of death for each of these 11 patients was related to coma most likely secondary to diabetic ketoacidosis (i.e., hypopotassemia, cerebral edema, ketoacidosis). Later, Gepts and De Mey [29] studied pancreatic specimens, obtained from hospitals in Brussels, Belgium and the USA, from 58 patients with diabetes under 40 years of age who were divided into three groups based on disease duration: 1–90 days (n = 16), 0.5–10 years (n = 14), and greater than 10 years (n = 28). Foulis and Stewart [21] examined pancreata from 11 patients with diabetes, obtained from the Royal Hospital for Sick Children in Glasgow, UK. Of these 11 patients, 9 had diabetes symptoms for 1–12 weeks and died within 24 h of clinical diagnosis, while the other two patients had diabetes for 2 and 6 years and received injections of insulin to control symptoms. Death in all patients, with the exception of one patient with a 2-year diabetes duration who died of lobar pneumonia, was caused by complications associated with ketoacidotic coma. Later, Foulis et al. [22] studied pancreatic autopsy specimens from 119 patients with diabetes under 20 years of age from Scotland, Wales, and England. Among the 95 patients for which duration of diabetes was known, disease duration was less than a year for 60 patients and 1–14 years in the remaining 35. Complications of ketoacidosis was the cause of death among all recent-onset cases in this series except one, and the most common cause of death among the long duration cases. Finally and far more recently, Butler et al. [17] examined pancreatic autopsy specimens from patients diagnosed with diabetes and with a disease duration of less than 3 years, who died of ketoacidosis.
Pathology
Islet morphology
Gepts described the appearance of islets from individuals with juvenile diabetes mellitus, classifying them into one of three categories [28]. So-called “type I islets” were distinguished primarily by their irregular outline and continuity between β-cells and the surrounding acinar tissue, based on staining by Gomori's chromium hematoxylin phloxine. Most islets of this type are small in size, though variable, with thin cords of endocrine cells within a fibrous stroma. The cells that comprise these type I islets were also small, having small nuclei containing dense chromatin and cytoplasm that was not abundant and did not show evidence of granulation. These were considered by Gepts to be the most prevalent type of islets in juvenile diabetics in general, and accounted for the vast majority of islets found in chronic cases [28]. Type II islets were more clearly defined by Gomori's stain in the acinar tissue than type I islets [28]. While type II islets were also variable in size like type I islets, type II islets tended to be large. Likewise, cells within these islets were large in size, with large nuclei and unlike type I islets; their cytoplasm was abundant, indicating hypertrophy. While the cytoplasm was often free of granules like type I islets, some β-cells could be found with granulations, some irregularly shaped. Variable numbers of type II islets were found in acute cases only [28]. Finally, type III islets were considered to resemble normal islets and have variable proportions of β-cells that were marginally hypertrophic and appeared degranulated to differing degrees. These types of islets were infrequent in both acute and chronic cases [28].
Pathologic features of short disease duration
β-cell survival
The great majority (i.e., 87–100%) of recent-onset cases with diabetes were noted to contain insulin-positive β-cells, as reported in five [17, 21, 22, 28, 29] of the six studies (Table 1). Junker et al. [46] reported finding only one case among 11 studied with β-cells containing insulin granules. However, as Gepts and De Mey [29] pointed out, this inability to identify β-cell granules, especially in cases of such short duration (i.e., 1–8 weeks), may have been due to the relative insensitivity of the Gomori staining method in comparison to more modern immunohistochemical techniques. In contrast, Gepts reported the observation of β-cells in 16 out of 17 acute cases. Onset age of the single case without any detectable β-cells was much younger (2.9 years) in comparison to those that had β-cells (12.5 years). Similarly, Gepts and De Mey reported that β-cells were observed in 14 out of 16 short duration cases, with the onset age in the two cases devoid of detectable β-cells occurring somewhat earlier (7.9 years) than those cases with β-cells (12.8 years).
Lobular distribution of insulin-containing islets
In those patients with insulin-containing islets remaining, although greatly reduced in proportion, a distinct lobular distribution of these islets has consistently been reported by several studies [17, 21, 22, 28, 29]. In other words, multiple insulin-positive islets tend to be found within one or a few lobules, as opposed to scattered throughout all acinar regions. This can result in an adjacent lobule being entirely deficient of insulin-positive islets. The biological significance of this lobular distribution is unclear, but may have implications for T1D pathogenesis such as lobular differences in islet autoreactivity, blood supply to and/or within lobules, trafficking of autoreactive T cells, and other factors may be of significance. However, it must also be clearly defined whether the pancreatic uncinate lobe was examined, since that lobe contains islets with a much lower proportion of β-cells within islets [30].
Insulitis
The term insulitis was coined by Von Meyenburg in 1940 to describe the mononuclear inflammatory infiltrate surrounding and within the islets of individuals with juvenile diabetes [91]. The insulitis lesion represents the predominant histopathological feature of T1D as it is frequently observed in pancreata from T1D patients after a short disease course (Fig. 2) [12, 17, 21, 22, 28, 29, 46]. The presence of insulitis is, with near uniformity, associated with a marked reduction in the number of insulin-containing islets and a dependence on exogenous insulin. Therefore, it is thought to represent a process whereby inflammatory cells directly cause the destruction and/or dysfunction of β-cells. For these reasons, much effort has been focused on identifying the cell types involved, as well as the various stimuli that provoke progression of insulitis to understand pathogenesis as well as to identify potential therapies for this disease.

Insulin-containing islet affected by insulitis in recent onset patient with type 1 diabetes. Pancreatic section, from nPOD, of a 12-year old recent-onset (1 year duration) T1D patient stained with antiserum against glucagon (red) and CD3 (brown). Inset image, serial section of the same islet stained with antiserum against insulin (red) and Ki-67 (brown). Insulitis with CD3+ T cells is observed around periphery and within the islet
Insulitis was found in the majority of recent-onset cases (54–89%) in the six studies [17, 21, 22, 28, 29, 46] shown in Table 2. Based on a comparison of these studies, the presence of insulitis appeared to be more dependent on earlier onset age than the variable of disease duration. In the Gepts study, for example, onset age among those cases without insulitis was 17.1 years (n = 7), while among those with insulitis, it was 7.9 years of age (n = 15). Similarly, in the study by Junker et al. [46] there was no insulitis observed in cases with an onset of 23.6 years of age (n = 5) in comparison to cases with insulitis, in which the onset age was 8.5 years of age (n = 6). This relationship was also shared in the study by Gepts and De Mey [29]. The one exception to this relationship between insulitis and earlier onset age was seen in the Butler study, where in which onset age was very similar between those cases with and without insulitis [17]. This may have been due to the older patient population or the longer disease duration (<3 years), which was longer in cases without insulitis, compared with the studies of patients of shorter duration. There is the possibility that some of the cases in these studies, especially those of late onset age, were of the idiopathic, non-autoimmune, subtype of T1D or even type 2 diabetes which would certainly alter the proportion of insulin + insulitis cases.
Immune cells of the insulitis lesion
Bottazzo et al. reported that lymphocytes were the primary cell type occupying the insulitis lesions of a 12-year-old girl who died within 24 h of diagnosis [12]. More specifically, the lymphocytes were considered predominately cytotoxic/suppressor T cells, with fewer numbers of helper T cells and natural killer (NK) cells. The majority of T cells within and around the islets were immunoreactive for HLA-DR. Class I HLA expression was clearly increased compared with normal tissues, in islets, and interestingly, this occurred whether or not an islet was affected by insulitis. β-cells, identified by C-peptide positivity, were immunoreactive for HLA-DR, but glucagon+ and somatostatin+ cells were not. Capillaries within and surrounding islets were stained for HLA-DR as well. Foulis et al. described a peripheral infiltrate of cells immunoreactive for T200 leukocyte common antigen, which labels all cells of hematopoietic origin, in early insulitis and a more extensive T200+ cell infiltrate throughout the islets with advanced insulitis [22].
Itoh et al. examined pancreata from 18 newly diagnosed patients with T1D for expression of the intercellular adhesion molecule-1 (ICAM-1) and MHC classes I and II, as well as immune cell types present in insulitis lesions [41]. MHC class I hyperexpression was observed in islets and endothelial cells from nine patients, and MHC class II hyperexpression was observed in endothelial cells of those same patients. In the eight patients with insulitis, CD8+ cells predominated along with macrophages and lesser numbers of CD4+ cells and B cells. Seven of the eight patients with insulitis showed hyperexpression of MHC class I. Expression of the adhesion molecule ICAM-1 was increased in endothelial cells from two of nine patients with MHC class I hyperexpression. Their data indicated that adhesion molecule expression was not associated with insulitis but confirmed that CD8+ cells were associated with MHC class I hyperexpression in insulitis lesions from new-onset T1D patients [41].
Insulitis preferentially affects insulin-containing islets
In most studies where insulitis was found, islets with insulin-containing β-cells were much more frequently affected than insulin-deficient islets. Specifically, Foulis and Stewart reported that 18% of insulin-containing versus 1% of insulin-deficient islets were affected by insulitis in eight recent-onset children who died within 24 h of initial presentation [21]. Foulis et al. observed a similar frequency with 23% of insulin-containing islets and 1% of insulin-deficient islets affected among recent-onset patients [22]. In the aforementioned case report by Bottazzo et al., insulitis affected 24% of the islets examined [12]. Additionally, the number of insulin-containing islets found and the percentage that was affected by insulitis were 2 (100%) in the pancreas head, 30 (43%) in the body, 53 (28%) in the tail, and 85 (35%) in the pancreas as a whole. The frequency and distribution of insulitis were described as “patchy” (i.e., heterogeneous), in that all insulin-containing islets were not uniformly affected. The apparent preference towards insulin-containing islets could indicate that the inflammatory lesion is targeted against the β-cells and not islets in general.
This relationship between insulin-containing islets and insulitis was further investigated by Willcox et al. [96], who described the association between the degree of islet β-cell loss and the type and number of immune cells in infiltrates from 29 recent-onset T1D patients, which was part of the larger cohort examined by Foulis et al. [22]. The mean age of the cases was 11.7 years with a duration of less than 18 months for 24 cases and unknown in the remaining three. At all stages of β-cell decline, CD8+ T cells were the predominant cell type. In early insulitis, i.e., when a large percentage of islet insulin area still remained, CD68+ macrophages were present in relatively larger numbers. CD20+ cells, in contrast, were present in low numbers in early insulitis but increased as β-cell loss progressed. At all stages, CD138+ plasma cells were rare. CD4+ T cells were present at all stages of β-cell decline in similar numbers, though far less than CD8+ and CD68+ cells, regardless of the degree of insulin area. Interestingly, FoxP3+ regulatory T cells were only found in islets from a single patient. These data indicate that CD8+ T cells and CD68+ macrophages are recruited early to the insulitis lesion and could contribute to β-cell degeneration. This immune-mediated process could progressively bring greater numbers of cytotoxic CD8+ cells to the islet until all β-cells, and the auto-antigen(s) driving this process, have been eliminated. After this stage, insulitis is no longer observed.
Insulitis and insulin secretion
A number of studies in animals [5, 13, 75, 85] and humans [10, 69] have suggested that β-cell autoimmunity is dependent on, or amplified by, insulin secretion. Do any of the histopathological data support this hypothesis? The data described above indicate that insulin-containing islets are preferentially targeted by lymphocytes but do not reveal whether or not insulin-secreting islets are the true targets. Foulis and Stewart noted that insulin-containing islets, even those affected by insulitis, were surrounded by normal, as opposed to atrophic, acinar tissue [21]. This indirectly suggests that insulitis-affected islets are actively secreting insulin and that insulin secretion, or metabolic functions associated with insulin secretion, may play a role in the attraction of antigen-presenting cells (APCs) and/or lymphocytes based on data from mouse studies (discussed below). The fact that diabetic ketoacidosis contributed to death in a large number of these cases presents a significant obstacle to linking heightened insulin secretion, or some function associated with it such as expression of HLA class I [12] or IAPP [70], to β-cell autoimmunity since ketoacidosis implies systemic insulin levels are sufficiently reduced to promote insulin secretion and, consequently, insulitis in these cases. These questions may be better addressed in animal models where insulin therapy and glycemia can be more tightly regulated.
Insulitis and β-cell hyperplasia/hypertrophy
Several studies [21, 22, 28, 29] reported β-cell hypertrophy in pancreata from T1D patients of short duration. As mentioned above, a general feature of type II islets that Gepts described is the presence of hypertrophic β-cells with nuclear hypertrophy [28]. Additionally, some cases had type II islets containing β-cells with irregularly shaped nuclei that had greater affinity for hematoxylin. The cytoplasmic granulations that Gepts observed in some of these large β-cells [28] were previously described by Weichselbaum, and are referred to as “Körnchen” [94]. These structures appeared to be rich in ribonucleic acids based on their loss of affinity for toluidine blue after sections were subjected to ribonuclease digestion. Gepts and De Mey reported routinely finding enlarged β-cells with large nuclei and cytoplasm containing many Körnchen [29]. Foulis and Stewart found that many of the large islets observed in recent-onset cases contained hypertrophic β-cells and some had evidence of polyploidy [21]. Foulis et al. described islet hyperplasia with polyploid β-cells in some of the recent-onset cases they examined [22]. Taken together, these findings suggest that β-cells are in a hyperactive state during the period immediately following disease onset.
Several studies have reported that islets are larger in size in pancreata from T1D patients of short duration [12, 21, 28, 46]. Gepts reported a proportionally greater number of medium and large size islets in pancreata from young diabetics after a short disease duration compared with age-matched controls [28]. Of these acute cases described by Gepts, 15 out of 22 (68%) had insulitis. Interestingly, it was also reported that type II islets, which tended to be large in size, were frequently affected by insulitis and observed in 77% of the short duration cases but none of the long duration cases [28]. Similarly, Foulis and Stewart reported that eight out of nine recent-onset subjects (2–12 years of age) had islets that were larger than normal, and islets affected by insulitis were also found in eight out of nine subjects [21]. The frequency of normal versus large islets that were affected by insulitis was however not quantified in either of these studies. Islet hypertrophy was also reported by Junker et al. in a few of the 11 recent-onset cases examined, however it cannot be ruled out that some of these cases, especially those that were older (25–30 years of age) and showed no signs of insulitis, may have had type 2 diabetes. Foulis et al. reported finding hyperplastic islets unaffected by insulitis that contained polyploid β-cells [22]. Bottazzo et al. also reported finding a few hypertrophied islets in the pancreas from a single recent-onset case [12].
The most straightforward explanation for these findings is that declining β-cell numbers occurring around the time of onset induces a compensatory increase in β-cell function (secretory capacity) and mass [95], although as far back as 1965 Gepts suspected that it was unlikely that the apparent β-cell hyperactivity represents a mechanism to compensate for decreased β-cell mass [28]. An alternate explanation is that dysfunctional glucose-stimulated insulin secretion in individuals susceptible to develop T1D promotes β-cell hypertrophy or hyperplasia which then, through an unknown mechanism, leads to autoimmunity. This can be indirectly drawn from two studies, one in which it was demonstrated that glucose sensitivity was impaired in relatives of T1D patients that progressed to disease [20], and another that reported a decrease in ICA titers, despite no change in progression to disease, in a small group of nondiabetic relatives of T1D patients that were treated with prophylactic insulin [76].
Insulitis in autoimmune vs. fulminant T1D
Fulminant T1D is a subtype of idiopathic (type 1B) diabetes characterized by rapid onset of symptoms of hyperglycemia, absence of autoantibodies, and flu-like symptoms that often occur before disease onset [38, 39]. Hanafusa and Imagawa examined pancreatic samples from patients that presented clinically as autoimmune (biopsy samples obtained for gastric cancer) or fulminant (biopsy and autopsy) T1D [34]. β-cells were reported to be markedly decreased in patients with autoimmune T1D without a significant change in α-cell numbers. As found by others, islet lymphocytic infiltrates consisted primarily of CD8+ T cells with fewer numbers of macrophages and dendritic cells. Expression of MHC class I was increased in all islet cells, Fas expression was evident mostly on β-cells, and infiltrating lymphocytes expressed Fas ligand. In patients with fulminant T1D, in contrast, β-cells were almost entirely gone, and α-cell numbers were significantly decreased. Insulitis, consisting primarily of macrophages and lesser numbers of T cells, was only observed in patients in whom disease duration was less than a month. Lymphocytic infiltrates in the remaining patients with longer disease duration were restricted to the exocrine pancreas. MHC class I hyperexpression was not observed in islet cells and Fas and Fas ligand expression were not observed in islet cells and lymphocytes, respectively. The authors conclude that β-cell death in autoimmune T1D is attributed to a long duration autoimmune process compared to fulminant T1D in which β-cell death proceeds quickly, possibly caused by viral infection [34].
β-cell replication and apoptosis
Characterization of β-cell replication and apoptosis rates in T1D is of great importance in understanding mechanisms of disease and potential therapies. These rates identify whether or not the response to the inflammatory microenvironment and/or metabolic demand is one of regeneration (replication) or injury (apoptosis), as well as the window of time in which these responses are occurring. In order to assess β-cell replication rates, Butler et al. stained sections of pancreatic autopsy samples from nine new-onset T1D cases for insulin and Ki67, a cell cycle protein expressed in all stages except resting (G0), and found no differences in the number of Ki67-positive β-cells compared to those in nondiabetic control cases [17]. Apoptosis was also measured in these samples by staining for insulin and TUNEL (terminal deoxynucleotidyl transferase dUTP neck end labeling). There was a significant increase in the frequency of TUNEL-positive β-cells in T1D cases compared to controls. As the authors noted, all the T1D cases in this study died of diabetic ketoacidosis and five of the nine contained islets affected by insulitis, making it difficult to identify whether these findings are representative of β-cells exposed to the inflammatory milieu found in recent-onset T1D pancreata or the systemic toxicity imposed by ketoacidosis [17]. Regardless, their data confirm that β-cell proliferative capacity was not able to balance destructive processes and indeed was not changed from normal levels.
Pathologic features of long disease duration
β-cell survival
The four studies [21, 22, 28, 29] that included pancreatic specimens from long duration T1D patients reported a surprisingly high percentage (18–100%) of cases with β-cells remaining compared to patients with acute onset (Table 3). This would indicate that the loss seen in patients with young onset or with acute onset is able to rebound or perhaps some of these patients had a clinical course more likely due to type 2 diabetes. This would certainly be a higher possibility for those cases diagnosed several decades ago. The presence of β-cells in long duration cases appears more associated with a later onset age than disease duration since, within two of these studies [28, 29], durations were comparable, but onset age tended to be later among those with β-cells. One possible explanation for this finding, if truly reflective of T1D, is that a greater percentage of β-cell mass remains at clinical onset when that occurs in late adolescence/early adulthood versus childhood because of the age-dependent increase in the ratio of insulin demand (i.e., body weight): insulin production capacity (i.e., β-cell mass) [49]. In other words, β-cell mass increases, albeit slowly, along with the increase in body weight during childhood and adolescence; whereas in early adulthood, β-cell mass has already plateaued despite the continued increase in body weight [66]. This translates into a very different tipping point, in terms of the β-cell mass loss, required to precipitate hyperglycemia in late versus young onset.
Insulitis
Insulitis appears rare among the long duration cases, an example being the study by Foulis et al. [22], in which only three out of 32 cases (9%) were affected (Table 4). The reason that these three cases had insulitis is not clear since the age, duration, and age of onset were essentially identical to the cases in this study that had no insulitis. The relative absence of islets affected by insulitis after a long disease course (Table 4), despite the presence of β-cells (Table 2) in many of these patients, may be due to the suppression of endogenous insulin secretion (β-cell rest [14]) by long-term insulin therapy [4]. Alternatively, some of these cases with remaining β-cells after a long disease course may have been insulin-requiring type 2 diabetics as it is difficult, still today and more so in the past, to retrospectively assign a T1D or type 2 diabetes diagnosis based on limited clinical information such as autoantibody status, HLA genotype, and C-peptide levels.
Pattern A and pattern B histopathology
T1D is, without question, an incredibly heterogeneous clinical disease with variations in autoantibody status, C-peptide level, HLA, onset age, ethnicity, and rate of progression. Gianani et al. evaluated pancreatic specimens from 20 cadaveric organ donors with childhood-onset long duration (mean duration 14 years) T1D in order to identify if there is any relationship between pathologic findings and the C-peptide and autoantibody status of the donors [32]. Among the 20 cases, only six had insulin-containing islets. Three were designated as having the typical pattern of autoimmune T1D (pattern A) with the majority of islets insulin-deficient. The other three patients displayed a very different pattern with all islets having β-cells though reduced islet insulin area (so-called pattern B). Additionally, class I HLA expression was detected in β-cells of donors displaying histopathology pattern A as well as survivin expression, an apoptosis inhibitor that is expressed in human islets during development fetal [56] and in patients with pancreatitis [35]. Neither class I HLA nor survivin expression was detected in β-cells from pattern B donors [32]. Two of the three donors with pattern A maintained minimal C-peptide secretion (both 0.06 nmol/l), one donor after 20-year disease duration, and two of the three were double positive for autoantibodies (i.e., autoantibodies against ICA512, IA-2ic; ICA512, ZnT8). Among the three donors exhibiting pattern B, two maintained C-peptide secretion (7.32, 0.67 nmol/l), and all were autoantibody-negative [32]. Interestingly, insulitis was found in only two of the 20 donors, both pattern A and double positive for autoantibodies. These data suggested that insulin deficiency can result from two distinct etiologies, one autoimmune in origin and one presumably due to β-cell dysfunction. Another explanation for these findings is that both type A and type B have the same etiology, β-cell dysfunction, and the severity of dysfunction combined with an individual’s HLA susceptibility determine if there is an immune response directed against the β-cell.
Pre-clinical pathology
A wealth of information about the early stages of T1D pathogenesis, particularly the antigen specificity of lymphocyte populations, could be gained by studying pancreata from individuals at risk for developing T1D yet before clinical onset. Wagner et al. examined pancreata from three nondiabetic patients with autoimmune polyendocrine syndrome that were positive for islet cell autoantibodies (ICA) and glutamic acid decarboxylase autoantibodies (GADA) [93]. In two of the patients, islet morphology and insulin content appeared normal, and the third had slightly reduced insulin content possibly related to the longer postmortem duration. There was no evidence of insulitis, i.e., T cell, B cell, or macrophage infiltration, no HLA class I or class II expression, and no immunoglobulin or complement deposition in the three cases [93].
Gianani et al. screened a population of 112 organ donors for autoantibodies to IA-2 (IA2A), GAD, and insulin (IAA) [31]. Three subjects were identified as positive, one for IA2A, one for GADA, and one for both IA2A and GADA. The pancreas of the IA2A-positive 47-year-old male donor was available for further study and exhibited fibrosis as was also found in autoantibody-negative donors. This patient also had large numbers of small- and medium-sized islets that were associated with ducts. Glucagon- and insulin-positive cells were seen closely associated with ductal epithelium, and some ducts exhibited cells positive for both CK-19 (i.e., a ductal marker) and chromogranin (i.e., an endocrine marker). The number of CD45+ leukocytes in the IA2A-positive individual was not statistically greater than in controls [31]. Despite the lack of any evidence for insulitis in the IA2A-positive donor, the apparent neogenesis of endocrine cells and islets from precursors within the ductal epithelium is a striking finding that may suggest the possibility that regeneration of endocrine mass [88], prompted by an unknown environmental stimulus or genetic defect, precedes the development of β-cell autoimmunity.
In't Veld et al. screened a large population (n = 1,507) of adult organ donors for ICA, GADA, IA2A, and IAA [40]. Pancreatic biopsies (∼0.5 cm3) from 62 autoantibody-positive donors were examined, and insulitis was found in two donors, who were both positive for ICA, GADA, and IA2A and the HLA-DQ susceptibility genotype. In one case, 9% of islets (5 of 58 islets) were affected by either peri- or central insulitis including one insulin-negative islet. In the second case, 3% of islets (27 of 917 islets) were affected by insulitis, which were all insulin-containing, but a distinct population of insulin-negative islets (3%) were unaffected by insulitis. The infiltrate in both cases consisted largely of CD3+/CD8+ T cells and CD68+ macrophages with few CD20+ B cells and CD3+/CD4+ T cells. The β-cell area was unchanged in the cases with insulitis compared with control cases. Interestingly, one quadruple autoantibody-positive donor with insulitis exhibited Ki67+ β-cells in many of the infiltrated islets, which could either represent a proliferative response to the stimuli imposed by the infiltrate or, alternatively, the stimulus for the infiltration itself.
Comparison of human and NOD insulitis
The non-obese diabetic (NOD) mouse represents the most valuable animal model for human T1D research [37, 47, 51, 71, 84, 97, 98]. The NOD model replicates the age range in which human diabetes incidence occurs, corresponding to adolescence in humans [54] and most importantly, insulitis is the hallmark feature in the destruction of beta cells [62]. It is important to compare and contrast the histopathology of the insulitis lesion in humans and NOD mice since the mouse is so highly utilized in studies aimed at understanding human T1D pathogenesis as well as translation of potential therapies to humans. This comparison is obviously complicated by the scarcity of human data pertaining to the pancreatic pathology prior to disease onset and the inability to accurately predict time-to-onset in individuals at risk (HLA, autoantibodies) of developing T1D. This shortcoming makes the NOD mouse a particularly useful animal model for investigating processes leading up to disease onset, possibly reflective of events occurring in humans.
The earliest evidence of islet-associated inflammation in the NOD mouse, stage I, is identified by swelling of blood vessels adjacent to islets that occurs at about 3 weeks of age in female mice [43] (Table 5). Stage II occurs at approximately 7 weeks, and is characterized by increased numbers of ER-MP23+ dendritic-like cells and MOMA-1+ or BM8+ macrophages that begin to appear at the islet periphery near blood vessels. At this stage, some islets already display infiltrating BM8+ macrophages. At stage III, CD4+ and CD8+ T cells can be seen moving out of blood vessels near islets. Most lymphocyte aggregates also contain some number of B cells and both ER-MP23+ and BM8+ cells with MOMA-1+ cells situated at the outer border. Stage IV lesions are those that completely surround the islet and consist predominately of the same cell types as in stage III, as well as infiltrating BM8+ cells in islets. Stage IV lesions are characterized by a massive infiltration of BM8+ cells, T cells, and B cells. In addition, insulin-positive β-cell numbers are decreased. In stage VI lesions, no β-cells are detectable, and few glucagon-positive α-cells are seen, similar to the insulitis observed in fulminant T1D [34]. Immune cells are still present in varying numbers in some islets and other large islets can be found without any β-cells or immune cells [43].
The earliest pathologic evidence of insulitis in humans comes from two nondiabetic individuals (ICA+ GADA+ IA2A+) in whom a small number of infiltrates were found with predominately CD8+ and CD68+ cells, as well as fewer numbers of CD20+ cells [40] (Table 5). This composition of immune cells corresponds to a stage III lesion in NOD mice [43], a much later “early” phase of insulitis, due to the presence of both CD8+ T cells and CD68+ macrophages as well as the peri- and central-architecture of the infiltrates. Following disease onset, the variety of islets, in terms of insulin area, examined in the 29 recent-onset cases [96] likely represent stage III, IV, V, and VI lesions in NOD mice where stage III corresponds to an islet with 50–69 or 30–49% insulin area, stage IV to an islet with 10–29% insulin area, stage V to an islet with >0–9% islet area, and stage VI to an islet with no detectable insulin area. One obvious difference between insulitis in the NOD mouse and humans is the greater size of insulitis area relative to islet area [8].
β-cell hyperplasia/hypertrophy and attraction of antigen presenting cells
Could the reported larger islets observed in recent-onset T1D patients, due either to β-cell hyperplasia or hypertrophy, be evidence of a process that starts long before disease onset and culminates with autoimmunity and β-cell destruction? Although this chapter is focused on characterization of the human pathology of T1D, a body of literature in the NOD mouse describes a number of observations prior to onset that may parallel some of the human findings seen after onset. As it's possible to longitudinally follow islet changes in NOD mice that occur prior to disease onset, these studies draw attention to a relationship between the progressive change in islet morphology and the initiation of insulitis. From birth there are twice as many very small islets in female NOD and NOD-severe combined immunodeficient (scid) mice, compared with C57BL/6 mice [72]. Glucagon area in these very small islets is significantly greater in NOD mice, with a trend in NODscids, compared with C57BL/6 mice, a feature that could be indicative of immature islets [64]. This resembles the large numbers of small and medium-sized islets observed in an autoantibody-positive nondiabetic donor [31]. Gepts also described what appeared to be new islet formation in many recent-onset cases but not in pancreata from long duration or control cases [28]. This is suggestive of an adaptive response to meet metabolic demand that may be caused by β-cell dysfunction of genetic and/or environmental origin (in utero [27], early in life [55, 67]). By 4 weeks of age, around the time of weaning, there are greater numbers of ER-MP23+ dendritic-like cells and MOMA-1+ macrophages within and near to vessels bordering islets in NODs compared to C57BL/6 mice [43]. Strangely, at 6 weeks of age, “mega-islets” are seen in NOD and, to a lesser extent, NODscid mice with APCs primarily found around these large islets [78]. Since both NOD and NODscid mice had larger islets, this may be suggestive of an adaptive response to β-cell dysfunction, rather than insulitis, as β-cell and islet hypertrophy has been reported in transgenic mice with impaired glucose-stimulated insulin secretion [58]. Following prophylactic insulin treatment, however, the number of large islets at 6 weeks of age was significantly decreased in both NOD and NODscid mice [78]. By 9 weeks of age, 51% of the islets in untreated female NOD mice are greater than 10 μm2 in size, compared to 9% in BALB/c mice [44]. Following prophylactic insulin treatment, the percentage of large islets (>10 μm2) in NOD mice was reduced to the prevalence found in BALB/c mice at 9 weeks of age [44]. Additionally, while the degree of invasive insulitis was similar at 9 weeks between mice that did and did not receive insulin, the numbers of peri-islet ER-MP23+ dendritic-like cells were significantly reduced in insulin-treated mice. This effect translated into significantly less infiltrative insulitis (42%) in the insulin-treated mice compared to the placebo-treated mice (75%) at 13 weeks [44]. These studies in NOD mice may be describing a similar phenomenon to that which gives rise to large islets in T1D patients during early stages, when insulitis is most aggressive, but not after years of insulin therapy when insulitis is rarely seen.
Conclusions
Despite the lack of human data related to alterations in islet morphology prior to T1D onset, In't Veld et al. reported findings wherein islets with high numbers of Ki-67+ β-cells, some of which were affected by insulitis, in a single autoantibody-positive nondiabetic case [40]. In addition, unpublished data from the Network for Pancreatic Organ Donors with Diabetes (nPOD) effort found high numbers of Ki-67+ islet cells including β-cells in islets unaffected by insulitis from a single autoantibody-positive nondiabetic case (Campbell-Thompson et al., personal communication). These very limited studies in humans suggest the possibility that β-cell proliferation may occur prior to insulitis in individuals genetically susceptible to T1D. It is clearly difficult to identify factors, genetic and/or environmental, that may promote β-cell proliferation and recognize when they occur (before or after β-cell autoimmunity is initiated) in human T1D cases. Studies in NOD mice, however, may offer the best alternative to uncover the potential relationship between islet/β-cell function, β-cell adaptive responses, and environmental factors that further burden this interaction and initiate autoimmune responses.
Insulitis is commonly observed in new-onset patients, but it does not uniformly affect all insulin-containing islets (18–34% [21, 22, 96]). If early stages in the development of T1D alter islet size/function, prompted by unknown factors, this non-uniform distribution maybe best explained by differences in islet function (i.e., glucose sensitivity, Ca2+ elevation, oxygen consumption, insulin release) if the in vitro heterogeneity [2, 36] is any reflection of the in vivo environment. Small islets, for example, isolated from humans [53] and rats [60] are superior to large islets in terms of their resistance to cell death in vitro, as well as insulin secretion when transplanted. This suggests that under increased insulin demand, such as that which occurs during puberty/adolescence [81], or high intake of sugars [73], a population of islets may be more prone to dysfunction or death, thereby attracting APCs [40, 78, 96] and promoting insulitis in genetically susceptible individuals. Although difficult to translate to the in vivo T1D environment, functional differences may explain the apparent preference of autoreactive lymphocytes for certain islets.
The best means to identify the factor(s) responsible for stimulating lymphocyte trafficking to and the destruction of β-cells, as well as the specific antigens that drive this process, is still very much a matter of debate. Clearly, earlier studies have paved the way for such new endeavors, yet there remains an overwhelming need for novel strategies that will reveal the nature of early events in T1D progression. The JDRF-funded nPOD [9] program was started to address these issues through analysis of human pancreata obtained from nondiabetic individuals with islet autoantibody positivity and recent-onset T1D. In addition, examination of pancreata from cases with gestational diabetes or post-transplant pancreatic transplantation for T1D will provide additional critical information regarding β-cell regenerative capacity in different clinical settings. Furthermore, efforts like the Type 1 Diabetes Genetics Consortium [45] are expected to lead to the identification of additional genes including possibly those outside the MHC locus. Discovery of genes related to β-cell function could explain how environmental factors, as opposed to high-risk HLA, could play a greater role in T1D incidence in recent years [11, 24]. Ideally, information from projects such as these will clarify the complex interaction between autoimmunity, environment, and host genome that combine to present the clinical heterogeneity seen in T1D.
References
Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group (2003) Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA 290:2159–2167
Aizawa T, Kaneko T, Yamauchi K, Yajima H, Nishizawa T, Yada T, Matsukawa H, Nagai M, Yamada S, Sato Y, Komatsu M, Itoh N, Hidaka H, Kajimoto Y, Hashizume K (2001) Size-related and size-unrelated functional heterogeneity among pancreatic islets. Life Sci 69:2627–2639
Akerblom HK, Savilahti E, Saukkonen TT, Paganus A, Virtanen SM, Teramo K, Knip M, Ilonen J, Reijonen H, Karjalainen J et al (1993) The case for elimination of cow's milk in early infancy in the prevention of type 1 diabetes: the Finnish experience. Diab Metab Rev 9:269–278
Argoud GM, Schade DS, Eaton RP (1987) Insulin suppresses its own secretion in vivo. Diabetes 36:959–962
Atkinson MA, Maclaren NK, Luchetta R (1990) Insulitis and diabetes in NOD mice reduced by prophylactic insulin therapy. Diabetes 39:933–937
Atkinson MA, Kaufman DL, Newman D, Tobin AJ, Maclaren NK (1993) Islet cell cytoplasmic autoantibody reactivity to glutamate decarboxylase in insulin-dependent diabetes. J Clin Invest 91:350–356
Atkinson MA (1997) Molecular mimicry and the pathogenesis of insulin-dependent diabetes mellitus: still just an attractive hypothesis. Ann Med 29:393–399
Atkinson MA, Leiter EH (1999) The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med 5:601–604
Atkinson MA, Gianani R (2009) The pancreas in human type 1 diabetes: providing new answers to age-old questions. Curr Opin Endocrinol Diab Obes 16:279–285
Bjork E, Berne C, Kampe O, Wibell L, Oskarsson P, Karlsson FA (1996) Diazoxide treatment at onset preserves residual insulin secretion in adults with autoimmune diabetes. Diabetes 45:1427–1430
Borchers AT, Uibo R, Gershwin ME (2010) The geoepidemiology of type 1 diabetes. Autoimmun Rev 9:A355–A365
Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG, Gamble DR (1985) In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med 313:353–360
Bowman MA, Campbell L, Darrow BL, Ellis TM, Suresh A, Atkinson MA (1996) Immunological and metabolic effects of prophylactic insulin therapy in the NOD-scid/scid adoptive transfer model of IDDM. Diabetes 45:205–208
Brown RJ, Rother KI (2008) Effects of beta-cell rest on beta-cell function: a review of clinical and preclinical data. Pediatr Diab 9:14–22
Brugman S, Klatter FA, Visser JT, Wildeboer-Veloo AC, Harmsen HJ, Rozing J, Bos NA (2006) Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia 49:2105–2108
Buschard K, Brogren CH, Ropke C, Rygaard J (1988) Antigen expression of the pancreatic beta-cells is dependent on their functional state, as shown by a specific, BB rat monoclonal autoantibody IC2. APMIS 96:342–346
Butler AE, Galasso R, Meier JJ, Basu R, Rizza RA, Butler PC (2007) Modestly increased beta cell apoptosis but no increased beta cell replication in recent-onset type 1 diabetic patients who died of diabetic ketoacidosis. Diabetologia 50:2323–2331
Dahlquist G (2006) Can we slow the rising incidence of childhood-onset autoimmune diabetes? The overload hypothesis. Diabetologia 49:20–24
Fan Y, Rudert WA, Grupillo M, He J, Sisino G, Trucco M (2009) Thymus-specific deletion of insulin induces autoimmune diabetes. EMBO J 28:2812–2824
Ferrannini E, Mari A, Nofrate V, Sosenko JM, Skyler JS (2010) Progression to diabetes in relatives of type 1 diabetic patients: mechanisms and mode of onset. Diabetes 59:679–685
Foulis AK, Stewart JA (1984) The pancreas in recent-onset type 1 (insulin-dependent) diabetes mellitus: insulin content of islets, insulitis and associated changes in the exocrine acinar tissue. Diabetologia 26:456–461
Foulis AK, Liddle CN, Farquharson MA, Richmond JA, Weir RS (1986) The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: a 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia 29:267–274
Foulis AK (1996) The pathology of the endocrine pancreas in type 1 (insulin-dependent) diabetes mellitus. APMIS 104:161–167
Fourlanos S, Varney MD, Tait BD, Morahan G, Honeyman MC, Colman PG, Harrison LC (2008) The rising incidence of type 1 diabetes is accounted for by cases with lower-risk human leukocyte antigen genotypes. Diab Care 31:1546–1549
Fousteri G, von Herrath M, Bresson D (2007) Mucosal exposure to antigen: cause or cure of type 1 diabetes? Curr Diab Rep 7:91–98
Fox CS, Coady S, Sorlie PD, D'Agostino RB Sr, Pencina MJ, Vasan RS, Meigs JB, Levy D, Savage PJ (2007) Increasing cardiovascular disease burden due to diabetes mellitus: the Framingham Heart Study. Circulation 115:1544–1550
Fronczak CM, Baron AE, Chase HP, Ross C, Brady HL, Hoffman M, Eisenbarth GS, Rewers M, Norris JM (2003) In utero dietary exposures and risk of islet autoimmunity in children. Diab Care 26:3237–3242
Gepts W (1965) Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14:619–633
Gepts W, De Mey J (1978) Islet cell survival determined by morphology. An immunocytochemical study of the islets of Langerhans in juvenile diabetes mellitus. Diabetes 27(Suppl 1):251–261
Gersell DJ, Gingerich RL, Greider MH (1979) Regional distribution and concentration of pancreatic polypeptide in the human and canine pancreas. Diabetes 28:11–15
Gianani R, Putnam A, Still T, Yu L, Miao D, Gill RG, Beilke J, Supon P, Valentine A, Iveson A, Dunn S, Eisenbarth GS, Hutton J, Gottlieb P, Wiseman A (2006) Initial results of screening of nondiabetic organ donors for expression of islet autoantibodies. J Clin Endocrinol Metab 91:1855–1861
Gianani R, Campbell-Thompson M, Sarkar SA, Wasserfall C, Pugliese A, Solis JM, Kent SC, Hering BJ, West E, Steck A, Bonner-Weir S, Atkinson MA, Coppieters K, von Herrath M, Eisenbarth GS (2010) Dimorphic histopathology of long-standing childhood-onset diabetes. Diabetologia 53:690–698
Hagopian WA, Karlsen AE, Petersen JS, Teague J, Gervassi A, Jiang J, Fujimoto W, Lernmark A (1993) Regulation of glutamic acid decarboxylase diabetes autoantigen expression in highly purified isolated islets from Macaca nemestrina. Endocrinology 132:2674–2681
Hanafusa T, Imagawa A (2008) Insulitis in human type 1 diabetes. Ann N Y Acad Sci 1150:297–299
Hasel C, Bhanot UK, Heydrich R, Strater J, Moller P (2005) Parenchymal regression in chronic pancreatitis spares islets reprogrammed for the expression of NFkappaB and IAPs. Lab Invest 85:1263–1275
Hayek A, Woodside W (1979) Correlation between morphology and function in isolated islets of the Zucker rat. Diabetes 28:565–569
Huang Y, Parker M, Xia C, Peng R, Wasserfall C, Clarke T, Wu L, Chowdhry T, Campbell-Thompson M, Williams J, Clare-Salzler M, Atkinson MA, Womer KL (2009) Rabbit polyclonal mouse antithymocyte globulin administration alters dendritic cell profile and function in NOD mice to suppress diabetogenic responses. J Immunol 182:4608–4615
Imagawa A, Hanafusa T, Miyagawa J, Matsuzawa Y (2000) A novel subtype of type 1 diabetes mellitus characterized by a rapid onset and an absence of diabetes-related antibodies. Osaka IDDM Study Group. N Engl J Med 342:301–307
Imagawa A, Hanafusa T, Makino H, Miyagawa JI, Juto P (2005) High titres of IgA antibodies to enterovirus in fulminant type-1 diabetes. Diabetologia 48:290–293
In't Veld P, Lievens D, De Grijse J, Ling Z, Van der Auwera B, Pipeleers-Marichal M, Gorus F, Pipeleers D (2007) Screening for insulitis in adult autoantibody-positive organ donors. Diabetes 56:2400–2404
Itoh N, Hanafusa T, Miyazaki A, Miyagawa J, Yamagata K, Yamamoto K, Waguri M, Imagawa A, Tamura S, Inada M et al (1993) Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest 92:2313–2322
Ize-Ludlow D, Sperling MA (2005) The classification of diabetes mellitus: a conceptual framework. Pediatr Clin N Am 52:1533–1552
Jansen A, Homo-Delarche F, Hooijkaas H, Leenen PJ, Dardenne M, Drexhage HA (1994) Immunohistochemical characterization of monocytes-macrophages and dendritic cells involved in the initiation of the insulitis and beta-cell destruction in NOD mice. Diabetes 43:667–675
Jansen A, Rosmalen JG, Homo-Delarche F, Dardenne M, Drexhage HA (1996) Effect of prophylactic insulin treatment on the number of ER-MP23+ macrophages in the pancreas of NOD mice. Is the prevention of diabetes based on beta-cell rest? J Autoimmun 9:341–348
Julier C, Akolkar B, Concannon P, Morahan G, Nierras C, Pugliese A (2009) The Type I Diabetes Genetics Consortium ‘Rapid Response’ family-based candidate gene study: strategy, genes selection, and main outcome. Genes Immun 10(Suppl 1):S121–S127
Junker K, Egeberg J, Kromann H, Nerup J (1977) An autopsy study of the islets of Langerhans in acute-onset juvenile diabetes mellitus. Acta Pathol Microbiol Scand A 85:699–706
Kaufman DL, Clare-Salzler M, Tian J, Forsthuber T, Ting GS, Robinson P, Atkinson MA, Sercarz EE, Tobin AJ, Lehmann PV (1993) Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature 366:69–72
Kida K, Mimura G, Kobayashi T, Matsuura N, Toyota T, Kitagawa T, Hibi I, Ikeda Y, Tuchida I, Kuzuya H et al (1994) ICA and organ-specific autoantibodies among Japanese patients with early-onset insulin-dependent diabetes mellitus—the JDS study. Diab Res Clin Pract 23:187–193
Klinke DJ 2nd (2008) Extent of beta cell destruction is important but insufficient to predict the onset of type 1 diabetes mellitus. PLoS ONE 3:e1374
Korhonen S, Knip MM, Kulmala P, Savola K, Akerblom HK, Knip M (2002) Autoantibodies to GAD, IA-2 and insulin in ICA-positive first-degree relatives of children with type 1 diabetes: a comparison between parents and siblings. Diabetes Metab Res Rev 18:43–48
Lee CH, Chen YG, Chen J, Reifsnyder PC, Serreze DV, Clare-Salzler M, Rodriguez M, Wasserfall C, Atkinson MA, Leiter EH (2006) Novel leptin receptor mutation in NOD/LtJ mice suppresses type 1 diabetes progression: II. Immunologic analysis. Diabetes 55:171–178
Lee YS, Ng WY, Thai AC, Lui KF, Loke KY (2001) Prevalence of ICA and GAD antibodies at initial presentation of type 1 diabetes mellitus in Singapore children. J Pediatr Endocrinol Metab 14:767–772
Lehmann R, Zuellig RA, Kugelmeier P, Baenninger PB, Moritz W, Perren A, Clavien PA, Weber M, Spinas GA (2007) Superiority of small islets in human islet transplantation. Diabetes 56:594–603
Leiter EH, Prochazka M, Coleman DL (1987) The non-obese diabetic (NOD) mouse. Am J Pathol 128:380–383
Lempainen J, Vaarala O, Makela M, Veijola R, Simell O, Knip M, Hermann R, Ilonen J (2009) Interplay between PTPN22 C1858T polymorphism and cow's milk formula exposure in type 1 diabetes. J Autoimmun 33:155–164
Liggins C, Orlicky DJ, Bloomquist LA, Gianani R (2003) Developmentally regulated expression of Survivin in human pancreatic islets. Pediatr Dev Pathol 6:392–397
Luopajarvi K, Savilahti E, Virtanen SM, Ilonen J, Knip M, Akerblom HK, Vaarala O (2008) Enhanced levels of cow's milk antibodies in infancy in children who develop type 1 diabetes later in childhood. Pediatr Diab 9:434–441
MacDonald PE, Joseph JW, Yau D, Diao J, Asghar Z, Dai F, Oudit GY, Patel MM, Backx PH, Wheeler MB (2004) Impaired glucose-stimulated insulin secretion, enhanced intraperitoneal insulin tolerance, and increased beta-cell mass in mice lacking the p110gamma isoform of phosphoinositide 3-kinase. Endocrinology 145:4078–4083
MacFarlane AJ, Strom A, Scott FW (2009) Epigenetics: deciphering how environmental factors may modify autoimmune type 1 diabetes. Mamm Genome 20:624–632
MacGregor RR, Williams SJ, Tong PY, Kover K, Moore WV, Stehno-Bittel L (2006) Small rat islets are superior to large islets in in vitro function and in transplantation outcomes. Am J Physiol Endocrinol Metab 290:E771–E779
Maclaren N, Lan M, Coutant R, Schatz D, Silverstein J, Muir A, Clare-Salzer M, She JX, Malone J, Crockett S, Schwartz S, Quattrin T, DeSilva M, Vander Vegt P, Notkins A, Krischer J (1999) Only multiple autoantibodies to islet cells (ICA), insulin, GAD65, IA-2 and IA-2beta predict immune-mediated (Type 1) diabetes in relatives. J Autoimmun 12:279–287
Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y (1980) Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu 29:1–13
Martin CL, Albers J, Herman WH, Cleary P, Waberski B, Greene DA, Stevens MJ, Feldman EL (2006) Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diab Care 29:340–344
McEvoy RC, Madson KL (1980) Pancreatic insulin-, glucagon-, and somatostatin-positive islet cell populations during the perinatal development of the rat. II. Changes in hormone content and concentration. Biol Neonate 38:255–259
Mehers KL, Gillespie KM (2008) The genetic basis for type 1 diabetes. Br Med Bull 88:115–129
Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC (2008) Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 57:1584–1594
Muntoni S (2006) Epidemiological association between some dietary habits and the increasing incidence of type 1 diabetes worldwide. Ann Nutr Metab 50:11–19
Neu A, Ehehalt S, Willasch A, Kehrer M, Hub R, Ranke MB (2001) Rising incidence of type 1 diabetes in Germany: 12-year trend analysis in children 0–14 years of age. Diab Care 24:785–786
Ortqvist E, Bjork E, Wallensteen M, Ludvigsson J, Aman J, Johansson C, Forsander G, Lindgren F, Berglund L, Bengtsson M, Berne C, Persson B, Karlsson FA (2004) Temporary preservation of beta-cell function by diazoxide treatment in childhood type 1 diabetes. Diab Care 27:2191–2197
Ouyang Q, Standifer NE, Qin H, Gottlieb P, Verchere CB, Nepom GT, Tan R, Panagiotopoulos C (2006) Recognition of HLA class I-restricted beta-cell epitopes in type 1 diabetes. Diabetes 55:3068–3074
Parker MJ, Xue S, Alexander JJ, Wasserfall CH, Campbell-Thompson ML, Battaglia M, Gregori S, Mathews CE, Song S, Troutt M, Eisenbeis S, Williams J, Schatz DA, Haller MJ, Atkinson MA (2009) Immune depletion with cellular mobilization imparts immunoregulation and reverses autoimmune diabetes in nonobese diabetic mice. Diabetes 58:2277–2284
Pelegri C, Rosmalen JG, Durant S, Throsby M, Alves V, Coulaud J, Esling A, Pleau JM, Drexhage HA, Homo-Delarche F (2001) Islet endocrine-cell behavior from birth onward in mice with the nonobese diabetic genetic background. Mol Med 7:311–319
Pundziute-Lycka A, Persson LA, Cedermark G, Jansson-Roth A, Nilsson U, Westin V, Dahlquist G (2004) Diet, growth, and the risk for type 1 diabetes in childhood: a matched case-referent study. Diab Care 27:2784–2789
Rangasami JJ, Greenwood DC, McSporran B, Smail PJ, Patterson CC, Waugh NR (1997) Rising incidence of type 1 diabetes in Scottish children, 1984–93. The Scottish Study Group for the Care of Young Diabetics. Arch Dis Child 77:210–213
Rasmussen SB, Sorensen TS, Hansen JB, Mandrup-Poulsen T, Hornum L, Markholst H (2000) Functional rest through intensive treatment with insulin and potassium channel openers preserves residual beta-cell function and mass in acutely diabetic BB rats. Horm Metab Res 32:294–300
Rodriguez-Villar C, Conget I, Casamitjana R, Ercilla G, Gomis R (1999) Effects of insulin administration in a group of high-risk, non-diabetic, first-degree relatives of Type 1 diabetic patients: an open pilot trial. Diabet Med 16:160–163
Roep BO, Hiemstra HS, Schloot NC, De Vries RR, Chaudhuri A, Behan PO, Drijfhout JW (2002) Molecular mimicry in type 1 diabetes: immune cross-reactivity between islet autoantigen and human cytomegalovirus but not Coxsackie virus. Ann N Y Acad Sci 958:163–165
Rosmalen JG, Homo-Delarche F, Durant S, Kap M, Leenen PJ, Drexhage HA (2000) Islet abnormalities associated with an early influx of dendritic cells and macrophages in NOD and NODscid mice. Lab Invest 80:769–777
Sakaguchi Y, Inaba M, Kusafuka K, Okazaki K, Ikehara S (2006) Establishment of animal models for three types of pancreatitis and analyses of regeneration mechanisms. Pancreas 33:371–381
Samuelsson U, Ludvigsson J (2004) The concentrations of short-chain fatty acids and other microflora-associated characteristics in faeces from children with newly diagnosed Type 1 diabetes and control children and their family members. Diabet Med 21:64–67
Savage MO, Smith CP, Dunger DB, Gale EA, Holly JM, Preece MA (1992) Insulin and growth factors adaptation to normal puberty. Horm Res 37(Suppl 3):70–73
Shaltout AA, Moussa MA, Qabazard M, Abdella N, Karvonen M, Al-Khawari M, Al-Arouj M, Al-Nakhi A, Tuomilehto J, El-Gammal A (2002) Further evidence for the rising incidence of childhood Type 1 diabetes in Kuwait. Diabet Med 19:522–525
Siljander HT, Simell S, Hekkala A, Lahde J, Simell T, Vahasalo P, Veijola R, Ilonen J, Simell O, Knip M (2009) Predictive characteristics of diabetes-associated autoantibodies among children with HLA-conferred disease susceptibility in the general population. Diabetes 58:2835–2842
Simon G, Parker M, Ramiya V, Wasserfall C, Huang Y, Bresson D, Schwartz RF, Campbell-Thompson M, Tenace L, Brusko T, Xue S, Scaria A, Lukason M, Eisenbeis S, Williams J, Clare-Salzler M, Schatz D, Kaplan B, Von Herrath M, Womer K, Atkinson MA (2008) Murine antithymocyte globulin therapy alters disease progression in NOD mice by a time-dependent induction of immunoregulation. Diabetes 57:405–414
Skak K, Gotfredsen CF, Lundsgaard D, Hansen JB, Sturis J, Markholst H (2004) Improved beta-cell survival and reduced insulitis in a type 1 diabetic rat model after treatment with a beta-cell-selective K(ATP) channel opener. Diabetes 53:1089–1095
Sobel-Maruniak A, Grzywa M, Orlowska-Florek R, Staniszewski A (2006) The rising incidence of type 1 diabetes in south-eastern Poland. A study of the 0–29 year-old age group, 1980–1999. Endokrynol Pol 57:127–130
Solimena M, Dirkx R Jr, Hermel JM, Pleasic-Williams S, Shapiro JA, Caron L, Rabin DU (1996) ICA 512, an autoantigen of type I diabetes, is an intrinsic membrane protein of neurosecretory granules. EMBO J 15:2102–2114
Suarez-Pinzon WL, Lakey JR, Brand SJ, Rabinovitch A (2005) Combination therapy with epidermal growth factor and gastrin induces neogenesis of human islet {beta}-cells from pancreatic duct cells and an increase in functional {beta}-cell mass. J Clin Endocrinol Metab 90:3401–3409
Taplin CE, Craig ME, Lloyd M, Taylor C, Crock P, Silink M, Howard NJ (2005) The rising incidence of childhood type 1 diabetes in New South Wales, 1990–2002. Med J Aust 183:243–246
Toumba M, Savva SC, Bacopoulou I, Apsiotou T, Georgiou T, Stavrou S, Skordis N (2007) Rising incidence of type 1 diabetes mellitus in children and adolescents in Cyprus in 2000–2004. Pediatr Diab 8:374–376
Von Meyenburg H (1940) Über ‘insulitis’ bei diabetes. Schweiz Med Wochenschr 21:554
Vreugdenhil GR, Geluk A, Ottenhoff TH, Melchers WJ, Roep BO, Galama JM (1998) Molecular mimicry in diabetes mellitus: the homologous domain in coxsackie B virus protein 2C and islet autoantigen GAD65 is highly conserved in the coxsackie B-like enteroviruses and binds to the diabetes associated HLA-DR3 molecule. Diabetologia 41:40–46
Wagner R, McNally JM, Bonifacio E, Genovese S, Foulis A, McGill M, Christie MR, Betterle C, Bosi E, Bottazzo GF (1994) Lack of immunohistological changes in the islets of nondiabetic, autoimmune, polyendocrine patients with beta-selective GAD-specific islet cell antibodies. Diabetes 43:851–856
Weichselbaum A (1910) Über der Veränderungen des Pankreas bei Diabetes Mellitus. Sitzungsberichte der Kaiserlichen Akademie der Wissenschaften 119:73–281
Weir GC, Bonner-Weir S (2004) Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 53(Suppl 3):S16–S21
Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG (2009) Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 155:173–181
Xue S, Wasserfall CH, Parker M, Brusko TM, McGrail S, McGrail K, Moore M, Campbell-Thompson M, Schatz DA, Atkinson MA, Haller MJ (2008) Exendin-4 therapy in NOD mice with new-onset diabetes increases regulatory T cell frequency. Ann N Y Acad Sci 1150:152–156
Xue S, Wasserfall C, Parker M, McGrail S, McGrail K, Campbell-Thompson M, Schatz DA, Atkinson MA, Haller MJ (2009) Exendin-4 treatment of nonobese diabetic mice increases beta-cell proliferation and fractional insulin reactive area. J Diabetes Complications
Yip L, Su L, Sheng D, Chang P, Atkinson M, Czesak M, Albert PR, Collier AR, Turley SJ, Fathman CG, Creusot RJ (2009) Deaf1 isoforms control the expression of genes encoding peripheral tissue antigens in the pancreatic lymph nodes during type 1 diabetes. Nat Immunol 10:1026–1033
Ziegler AG, Baumgartl HJ, Standl E, Mehnert H (1990) Risk of progression to diabetes of low titer ICA-positive first-degree relatives of type I diabetics in southern Germany. J Autoimmun 3:619–624
Acknowledgments
This study was supported, in part, by funds obtained from the Juvenile Diabetes Research Foundation (nPOD program), the National Institutes of Health, the Keene Professorship, and the Brehm Coalition for Type 1 Diabetes.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Rowe, P.A., Campbell-Thompson, M.L., Schatz, D.A. et al. The pancreas in human type 1 diabetes. Semin Immunopathol 33, 29–43 (2011). https://doi.org/10.1007/s00281-010-0208-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-010-0208-x