
Abstract
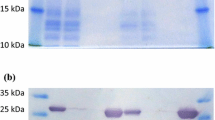
In this study, we have investigated a propionate CoA-transferase (Pct) homologue encoded in the genome of Ralstonia eutropha H16. The corresponding gene has been cloned into the vector pET-19b to yield a histidine-tagged enzyme which was expressed in Escherichia coli BL21 (DE3). After purification, high-performance liquid chromatography/mass spectrometry (HPLC/MS) analyses revealed that the enzyme exhibits a broad substrate specificity for carboxylic acids. The formation of the corresponding CoA-thioesters of acetate using propionyl-CoA as CoA donor, and of propionate, butyrate, 3-hydroxybutyrate, 3-hydroxypropionate, crotonate, acrylate, lactate, succinate and 4-hydroxybutyrate using acetyl-CoA as CoA donor could be shown. According to the substrate specificity, the enzyme can be allocated in the family I of CoA-transferases. The apparent molecular masses as determined by gel filtration and detected by SDS polyacrylamide gel electrophoresis were 228 and 64 kDa, respectively, and point to a quaternary structure of the native enzyme (α4). The enzyme exhibited similarities in sequence and structure to the well investigated Pct of Clostridium propionicum. It does not contain the typical conserved (S)ENG motif, but the derived motif sequence EXG with glutamate 342 to be, most likely, the catalytic residue. Due to the homo-oligomeric structure and the sequence differences with the subclasses IA–C of family I CoA-transferases, a fourth subclass of family I is proposed, comprising — amongst others — the Pcts of R. eutropha H16 and C. propionicum. A markerless precise-deletion mutant R. eutropha H16∆pct was generated. The growth and accumulation behaviour of this mutant on gluconate, gluconate plus 3,3′-dithiodipropionic acid (DTDP), acetate and propionate was investigated but resulted in no observable phenotype. Both, the wild type and the mutant showed the same growth and storage behaviour with these carbon sources. It is probable that R. eutropha H16 is upregulating other CoA-transferase(s) or CoA-synthetase(s), thereby compensating for the lacking Pct. The ability of R. eutropha H16 to substitute absent enzymes by isoenzymes has been already shown in different other studies in the past.



Similar content being viewed by others

References
Altschul FA, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Anderson AJ, Dawes EA (1990) Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol Rev 54:450–472
Auras R, Harte B, Selke S (2004) An overview of polylactides as packaging materials. Macromol Biosci 4:835–864
Bradford MM (1976) Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Brandl H, Gross RA, Lenz RW, Fuller RC (1988) Pseudomonas oleovorans as a source of poly(β-hydroxyalkanoates) for potential applications as biodegradable polyesters. Appl Environ Microbiol 66:2117–2124
Brigham CJ, Budde CF, Holder JW, Zeng Q, Mahan AE, Rha C, Sinskey AJ (2010) Elucidation of β-oxidation pathways in Ralstonia eutropha H16 by examination of global gene expression. J Bacteriol 192:5454–5464
Buckel W, Dorn U, Semmler R (1981) Glutaconate CoA-transferase from Acidaminococcus fermentans. Eur J Biochem 118:315–321
Corthésy-Theulaz IE, Bergonzelli GE, Henry H, Bachmann D, Schorderet DF, Ornston N (1997) Cloning and characterization of Helicobacter pylori succinyl-CoA:acetoacetate CoA-transfease, a novel prokaryotic member of the CoA-transferase family. J Biol Chem 272:25659–25667
Han X, Satoh Y, Satoh T, Matsumoto K, Kakuchi T, Taguchi S, Dairi T, Munekata M, Tajima K (2011) Chemo-enzymatic synthesis of polyhydroxyalkanoate (PHA) incorporating 2-hydroxybutyrate by wild-type class I PHA synthase from Ralstonia eutropha. Appl Microbiol Biotechnol 92:509–517
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580
Heider J (2001) A new family of CoA-transferases. FEBS Lett 509:345–349
Hogrefe C, Römermann D, Friedrich B (1981) Alcaligenes eutrophus hydrogenase genes (Hox). J Bacteriol 158:43–48
Jacob U, Mack T, Clausen T, Huber R, Buckel W, Messerschmidt A (1997) Glutaconate CoA-transferase from Acidaminococcus fermentans: the crystal structure reveals homology with other CoA-transferases. Structure 5:415–426
Jung YK, Kim TY, Park SJ, Lee SY (2010) Metabolic engineering of Escherichia coli for the production of polylactic acid and its copolymers. Biotechnol Bioeng 105:161–171
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lindenkamp N, Peplinski K, Volodina E, Ehrenreich A, Steinbüchel A (2010) Impact of multipe β-ketothiolase deletion mutations in Ralstonia eutropha H16 on the composition of 3-mercaptopropionic acid-containing copolymers. Appl Environ Microbiol 76:5373–5382
Mack M, Buckel W (1995) Identification of glutamate beta 54 as the covalent-catalytic residue in the active site of glutaconate CoA-transferase from Acidaminococcus fermentans. FEBS Lett 357:145–148
Mack M, Bendrat K, Zelder O, Eckel E, Linder D, Buckel W (1994) Location of the two genes encoding glutaconate coenzyme A-transferase at the beginning of the hydroxyglutarate operon in Acidaminococcus fermentans. Eur J Biochem 226:41–51
Marmur J (1961) A procedure for the isolation of desoxyribonucleic acids from microorganisms. J Mol Biol 3:208–218
Matsumoto K, Taguchi S (2010) Enzymatic and whole-cell synthesis of lactate-containing polyesters: toward the complete biological production of polylactate. Appl Microbiol Biotechnol 85:921–932
Matsumoto K, Okei T, Honma I, Ooi T, Aoki H, Taguchi S (2012) Efficient (R)-3-hydroxybutyrate production using acetyl CoA-regenerating pathway catalyzed by coenzyme A transferase. Appl Microbiol Biotechnol. doi:10.1007/s00253-012-4104-2
Parales RE, Harwood CS (1992) Characterization of the genes encoding β-ketoadipate succinyl-coenzyme A transferase in Pseudomonas putida. J Bacteriol 174:4657–4666
Peplinski K, Ehrenreich A, Döring C, Bömeke M, Steinbüchel A (2010) Investigations on the microbial catabolism of the organic sulfur compounds TDP and DTDP in Ralstonia eutropha H16 employing DNA microarrays. Appl Microbiol Biotechnol 88:1145–1159
Pohlmann A, Fricke WF, Reinecke F, Kusian B, Liesegang H, Cramm R, Eitinger T, Ewering C, Pötter M, Schwartz E, Strittmatter A, Voss I, Gottschalk G, Steinbüchel A, Friedrich B, Bowien B (2006) Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat Biotechnol 24:1257–1262
Pötter M, Müller H, Steinbüchel A (2005) Influence of homologous phasins (PhaP) on PHA accumulation and regulation of their expression by the transcriptional repressor PhaR in Ralstonia eutropha H16. Microbiol (SGM) 151:825–833
Quandt J, Hynes MF (1993) Versatile suicide vectors which allow direct selection for gene replacement in Gram-negative bacteria. Gene 127:15–21
Rangarajan ES, Li Y, Ajamian E, Iannuzzi P, Kernaghan SD, Fraser ME, Cygler M, Matte A (2005) Crystallographic trapping of the glutamyl-CoA thioesters intermediate of family I CoA transferases. J Biol Chem 280:42919–42928
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor
Schlegel HG, Kaltwasser H, Gottschalk G (1961) Ein Submersverfahren zur Kultur wasserstoffoxidierender Bakterien: Wachstumsphysiologische Untersuchungen. Arch Mikrobiol 38:209–222
Schubert PA, Steinbüchel A, Schlegel HG (1988) Cloning of the Alcaligenes eutrophus genes for synthesis of poly-β-hydroxybutyrate. J Bacteriol 170:5837–5847
Schürmann M, Wübbeler JH, Grote J, Steinbüchel A (2011) Novel reaction of succinyl-CoA synthetase: Activation of 3-sulfinopropionate to 3-sulfinopropionyl-CoA in Advenella mimigardefordensis strain DPN7T during degradation of 3,3′-dithiodipropionic acid. J Bacteriol 193:3078–3089
Schwartz E, Henne A, Cramm R, Eitinger T, Friedrich B, Gottschalk G (2003) Complete nucleotide sequence of pHG1: a Ralstonia eutropha H16 megaplasmid encoding key enzymes of H2-based lithoautotrophy and anaerobiosis. J Mol Biol 332:369–383
Schweiger G, Buckel W (1984) On the dehydration of (R)-lactate in the fermentation of alanine to propionate by Clostridium propionicum. FEBS Lett 171:79–84
Selmer T, Buckel W (1999) Oxygen exchange between acetate and the catalytic glutamate residue in glutaconate CoA-transferase from Acidaminococcus fermentans: implications for the mechanism of CoA-ester hydrolysis. J Biol Chem 274:20772–20778
Selmer T, Willanzheimer A, Hetzel M (2002) Propionate CoA-transferase from Clostridium propionicum — cloning of the gene and identification of glutamate 324 at the active site. Eur J Biochem 269:372–380
Simon R, Priefer U, Pühler A (1983) Vector plasmids for in vivo and in vitro manipulations of Gram-negative bacteria. In: Pühler A (ed) Molecular genetics of the bacteria-plant interaction. Springer, Berlin, Germany, pp 98–106
Solomon F, Jencks WP (1968) Identification of an enzyme-γ-glutamyl coenzyme A intermediate from coenzyme A transferase. J Biol Chem 244:1079–1081
Steinbüchel A, Schlegel HG (1989) Excretion of pyruvate by mutants of Alcaligenes eutrophus, which are impaired in the accumulation of poly(β-hydroxybutyric acid) (PHB), under conditions permissive for synthesis of PHB. Appl Microbiol Biotechnol 31:168–175
Taguchi S, Yamada M, Matsumoto K, Tajima K, SatohY MM, Ohno K, Kohda K, Shimamura T, Kambe H, Obata S (2008) A microbial factory for lactate-based polyesters using a lactate-polymerizing enzyme. Proc Natl Acad Sci U S A 105:17323–17327
Tajima K, Han X, Satoh Y, Ishii A, Araki Y, Munekata M, Taguchi S (2012) In vitro synthesis of polyhydroxyalkanoate (PHA) incorporating lactate (LA) with a block sequence by using a newly engineered thermostable PHA synthase from Pseudomonas sp. SG4502 with acquired LA-polymerizing activity. Appl Microbiol Biotechnol 94:365–376
Tielens AGM, van Grinsven KWA, Henze K, van Hellemond JJ, Martin W (2010) Acetate formation in the energy metabolism of parasitic helminthes and protists. Int J Parasitol 40:387–397
Timm A, Steinbüchel A (1990) Formation of polyesters consisting of medium-chain-length 3-hydroxyalkanoic acids from gluconate by Pseudomonas aeruginosa and other fluorescent pseudomonads. Appl Environ Microbiol 56:3360–3367
Valentin HE, Steinbüchel A (1994) Application of enzymatically synthesized short-chain-length hydroxyl fatty acid coenzyme A thioesters for assay of polyhydroxyalkanoic acid synthases. Appl Microbiol Biotechnol 40:699–709
Weber K, Osborn M (1969) The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J Biol Chem 244:4406–4412
Wübbeler JH, Raberg M, Brandt U, Steinbüchel A (2010) Dihydrolipoamide dehydrogenase of Advenella mimigardefordensis and Ralstonia eutropha catalyze cleavage of 3,3′dithiodipropionic acid into 3-mercaptopropionic acid. Appl Environ Microbiol 21:7023–7028
Yamada M, Matsumoto K, Shimizu K, Uramoto S, Nakai T, Shozui F, Taguchi S (2010) Adjustable mutations in lactate (LA)-polymerizing enzyme for the microbial production of LA-based polyesters with tailor-made monomer composition. Biomacromolecules 11:815–819
Yang TH, Kim TW, Kang HO, Lee S-H, Lee EJ, Lim S-C, Oh SO, Song A-J, Park SJ, Lee SY (2010) Biosynthesis of polylactic acid and its copolymers using evolved propionate coA transferase and PHA synthase. Biotechnol Bioeng 105:150–160
Yang Y-H, Brigham CJ, Song E, Jeon J-M, Rha CK, Sinskey AJ (2012) Biosynthesis of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) containing a predominant amount of 3-hydroxyvalerate by engineered Escherichia coli expressing propionate-CoA transferase. doi:10.1111/j.1365-2672.2012.05391.x
Acknowledgements
This study was supported by a grant provided by the Bundesministerium für Bildung und Forschung (BMBF; Förderkennzeichen 0313751E).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lindenkamp, N., Schürmann, M. & Steinbüchel, A. A propionate CoA-transferase of Ralstonia eutropha H16 with broad substrate specificity catalyzing the CoA thioester formation of various carboxylic acids. Appl Microbiol Biotechnol 97, 7699–7709 (2013). https://doi.org/10.1007/s00253-012-4624-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4624-9