
Abstract
Objectives
Pantoprazole is metabolized by cytochrome P450 2 C19, which shows genetic polymorphism. The effect of CYP2C19 polymorphism on single-dose pharmacokinetics of oral pantoprazole in healthy volunteers was evaluated.
Methods
Pantoprazole pharmacokinetics was determined in 32 healthy volunteers after a 40-mg single oral dose of the drug.
Results
Carriers of CYP2C19*2/*2 (n = 2) were characterized by higher, starting from 3.5 h post dose, plasma concentrations of pantoprazole in comparison to wild-type (CYP2C19*1/*1, n = 6) volunteers. In subjects with CYP2C19*17/*17 genotype (n = 6) significantly lower plasma concentrations of the drug vs CYP2C19*1/*1 carriers, were observed from 3.0 h after oral pantoprazole administration. Carriers of CYP2C19*1/*17 (n = 6) and CYP2C19*2/*17 (n = 6) displayed concentration–time profiles comparable to wild-type subjects. CYP2C19*2/*2 volunteers showed a decrease in terminal elimination rate constant (λz) by 83.3%, prolongation of terminal half-life (t½) by 572%, a rise in area under the concentration–time curve (AUC) and mean residence time (MRT) by 506% and 259% respectively. Heterozygotes, i.e.. CYP2C19*1/*2 vs CYP2C19*1/*1 were characterized by higher AUC (4.38 ± 1.00 mg⋅h/L vs 3.00 ± 1.02 mg⋅h/L, p < 0.05) and Cmax (2.13 ± 0.42 mg/L vs 1.61 ± 0.35 mg/L, p < 0.05) respectively. A significant reduction in MRT (3.83 ± 0.82 h vs 2.73 ± 0.23 h, p < 0.05) in carriers of CYP2C19*17/*17 vs CYP2C19*1/*1 genotypes was observed. Population modeling confirmed the influence of *1/*2, *2/*2, and *17/*17 genotypes on the pharmacokinetics of pantoprazole. The lowest population oral clearance was assessed in the carriers of genotype *2/*2 (3.68 L/h) and the highest value in subjects with genotype *17/*17 (31.13 L/h).
Conclusion
These data suggest that CYP2C19 polymorphism is an important determinant of pantoprazole pharmacokinetics.
Similar content being viewed by others


Introduction
CYP2C19 is an important drug-metabolizing enzyme encoded by a highly polymorphic gene, with the two most frequent allelic variants in Caucasian populations, CYP2C19*2 and CYP2C19*17. Population studies have demonstrated that subjects homozygous for the inactivating allele CYP2C19*2 associated with a splicing defect are phenotypically poor metabolizers of CYP2C19 substrates [1, 2]. Poor metabolizers are found in 3–5% of Caucasians, and much more frequently in Asian populations, i.e., 12–23% [3]. The CYP2C19*17 allele was documented to correlate with high CYP2C19 activity [4]. In European populations the CYP2C19*17 allele is found in 18–28% of subjects, in 17–18% of Africans and 0.3–4% in Asian populations [5].
Proton pump inhibitors (PPI), among them pantoprazole, are the mainstays in the treatment of gastric acid-related disorders. It was documented that the pharmacokinetics of PPI as well as the clinical outcome of treatment are associated with CYP2C19 polymorphism, being their main metabolizing enzyme. However, the effect of CYP2C19 on pantoprazole metabolism was not as extensively studied as in the case of omeprazole. Tanaka et al. reported that in poor CYP2C19 metabolizers metabolism of R(+)-pantoprazole was impaired to a greater extent than that of S(-)-pantoprazole [6]. Some studies on the effects of the CYP2C19 polymorphism on pantoprazole kinetics revealed an association of drug pharmacokinetics and genotypes in adults and children [7–9]. However, these studies were carried out on a limited number of cases and did not involve CYP2C19*17/*17 homozygotes, which seems to be an important determinant in pantoprazole metabolism.
Our previous preliminary study also suggested an impact of the CYP2C19 polymorphism on pantoprazole metabolism. It was found that concentrations of the drug at 3 h post-oral dose were the highest in heterozygous subjects, carriers of CYP2C19*1/*2 genotypes (unfortunately, we were not able to measure pantoprazole concentrations in CYP2C19*2/*2 patients), and the lowest in the case of CYP2C19*17/*17 subjects [10]. Therefore, it was decided to study an association between CYP2C19 polymorphism and single-dose pharmacokinetics of oral pantoprazole in healthy volunteers.
Materials and methods
Subjects
In the first part of the study 120 healthy, unrelated volunteers of Caucasian origin, Polish nationality (52 males and 68 females; age rank 20–27 years) were genotyped for CYP2C19 polymorphism. Informed consent was obtained from all participants. The study was approved by the Ethics Committee of Pomeranian Medical University, Szczecin, Poland. Within the genotyped group, 46 subjects were homozygous: 38 for the CYP2C19*1 allele, 6 for the CYP2C19*17 allele, and 2 for the CYP2C19*2 allele. Seventy-four were heterozygous with the following CYP2C19 genotypes: 20 participants with *1/*2, 38 with *1/*17, and 16 with *2/*17 genotypes.
In the second part of the study pharmacokinetics of pantoprazole after a single oral dose of 40 mg in 32 healthy volunteers defined for specific CYP2C19 genotypes was determined. Subject characteristics and genotypes are shown in Table 1.
Genotyping
CYP2C19 genotyping was performed in all study subjects. Genomic DNA was extracted from 200 μL of whole blood samples using GeneMATRIX Quick Blood DNA Purification Kit (EURx, Poland). Each individual was genotyped for a presence of SNPs marking CYP2C19 variant alleles: rs4244285 (681 G > A) for allele *2, and rs12248560 (−806 C > T) for allele *17. The allelic discrimination TaqMan real-time PCR assays (Assay IDs: C_25986767_70, C_469857_10, Applied Biosystems, USA) were used for detection. Fluorescence data were captured using an ABI PRISM 7500 FAST Real-Time PCR System (Applied Biosystems), after 40 cycles of PCR.
Drug concentration analysis
Pantoprazole concentration in plasma was determined by a validated high-performance liquid chromatography method (HPLC) with ultraviolet detection following solid phase extraction [11]. The chromatographic system consisted of a pump HP 1100, autosampler HP 1100, an ultraviolet detector HP 1100 and DAD detector Agilent 1100. The isocratic mobile phase was composed of a buffer solution consisting of 25 mM potassium phosphate monobasic in water containing 0.25% triethylamine and acetonitrile (35:65% v/v). The pH = 6.5 was adjusted with phosphoric acid. The mobile phase was pumped at an isocratic flow rate of 1 mL/min at a temperature of 25°C. The wavelength of UV detection was set at 290 nm. Chromatographic separations were achieved on Supelcosil LC-8-DB column (5 μm, 15 × 4.6 mm). Solid-phase extraction was used to extract the pantoprazole and tinidazole as an internal standard. Recoveries from human plasma were ≥ 90% for pantoprazole. The lower limit of detection (LOD) at a signal:noise ratio 3:1 was 0.012 mg/L and the lower limit of quantitation for pantoprazole was 0.025 mg/L. Intra- and interassay coefficients of variation were consistently ≤10% for plasma pantoprazole concentrations within the range of linearity. No interfering peaks from metabolites of pantoprazole and endogenous compounds were observed at the retention time of analytes. All study samples were analyzed within 3 months of their collection.
Pharmacokinetic study
Pharmacokinetic parameters of pantoprazole such as clearance/F (CL/F, where F is the systemically available fraction of a dose), volume of distribution (Vz/F), terminal half-life (t½), mean residence time (MRT), time to maximum concentration (tmax), and the maximum concentration (Cmax) were derived by noncompartmental analysis using Phoenix WinNonlin 6.2 (Pharsight Corporation, Mountain View, CA, USA). For each individual the terminal elimination rate constant (λz) was determined by log-linear regression of the terminal phase of the plasma concentration–time curve. The area under the concentration–time curve (AUC) was determined by the linear trapezoidal rule from time zero to the time of the last observed concentration [12].
Differences between genotypes were evaluated using t test for independent samples. The level of significance was set at 0.05.
Population pharmacokinetic modeling
Because it was impossible to assess statistically the influence of rare genotypes, e.g., CYP2C19*2/*2 on pharmacokinetics of pantoprazole using standard statistical tests, an attempt was made to evaluate the impact of CYP2C19*2 and CYP2C19*17 genotypes when included in the population pharmacokinetic model as covariates.
To assess the influence of CYP2C19 genotypes on pantoprazole pharmacokinetics, drug concentrations were analyzed using the nonlinear mixed effects modeling program, NONMEM (version V, level 1.1). The one- and two-compartment models with first-order absorption from the central compartment and a lag time were tested. These models were implemented in the PREDPP library subroutine ADVAN 2 TRANS 2 or ADVAN4 TRANS4 in NONMEM respectively. Pharmacokinetic analysis was performed using the first-order (FO) estimation method with post-hoc estimation of individual parameters.
The intersubject variability in pharmacokinetic parameters was estimated using an exponential model:
where Pi is the jth pharmacokinetic parameter for the ith subject, TVP is the typical value of jth parameter, ηij is a random variable for the ith individual in the jth parameter. It is assumed that the values of η i are normally distributed with a mean of zero and a variance of ω2.
Residual variability was described by a combined proportional and additive error model:
where COBS and CPRED are observed and predicted pantoprazole concentrations, and ε1 and ε2 are the residual intrasubject variability with means of zero and variances of σ 21 and σ 22 respectively.
The possible relationship between the typical value of the oral clearance (CL/F) and CYP2C19 genetic polymorphisms was modeled according to the following equation:
where θ 1 is the population value of oral clearance for the wild-type group, θ 2 - θ 6 are the fractional changes in the oral clearance for different genotypes, and G1 – G5 represent CYP2C19 genotypes: *1/*2, *1/*17, *2/*17, *2/*2, and*17/*17 respectively.
Model building was performed using a stepwise approach, adding an additional covariate at each step. The effect of each covariate was evaluated based on changes in the objective function value (OFV). The results were considered statistically significant if the decreases in OFV were 3.84 units (p < 0.05), 6.63 units (p < 0.01), and 10.83 units (p < 0.001). To assess the significance of each covariate a backward elimination procedure was used. An increase in OFV greater than 6.63 (P < 0.01) was required to retain the covariate in the final model.
The goodness-of-fit of each analysis was assessed by the percentage relative standard error (%RSE) of the mean of parameters estimates, changes in the estimates of intersubject and residual variability, and visual inspection of scatterplots representing population (PRED) and individual (IPRED) predicted concentrations versus observed concentrations and weighted residuals (WRES) versus predicted pantoprazole concentrations. In addition, epsilon shrinkage defined as 1-SD(IWRES) was calculated in order to ensure validity of the diagnostics. Graphical diagnostics were obtained using Visual-NM, v. V [13].
Results
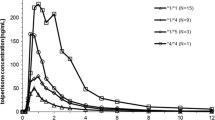
Plasma concentration versus time profiles of pantoprazole in healthy volunteers stratified according to the CYP2C19 genotypes are presented in Fig. 1. Carriers of CYP2C19*2/*2 (n = 2) were characterized by considerably higher, starting from 3.5 h post dose, plasma concentrations of pantoprazole in comparison to wild-type (CYP2C19*1/*1; n = 6) volunteers. In subjects with the CYP2C19*17/*17 genotype (n = 6) significantly lower plasma concentrations of the drug vs CYP2C19*1/*1 (n = 6) carriers, were observed from 3.0 h after oral pantoprazole administration. Carriers of CYP2C19*1/*17 (n = 6) and CYP2C19*2/*17 (n = 6) displayed concentration–time curves comparable to wild-type subjects.

Mean plasma concentration–time profiles in subjects with different CYP2C19 genotypes after administration of 40 mg pantoprazole as a single oral dose. Concentrations of CYP2C19*17/* were significantly different from CYP2C19*1/*1 at each time point starting from 3.0 h post-dose. Number of subjects per group n = 6, except for CYP2C19*2/*2 where n = 2
Changes in pantoprazole pharmacokinetics in subjects with different CYP2C19 genotypes are also reflected in altered pharmacokinetic parameters (Table 2). As in the case of the drug concentrations, most prominent deviations from wild-type genotype carriers were observed in CYP2C19*2/*2 (n = 2) and CYP2C19*17/*17 (n = 6) subjects. In CYP2C19*2/*2 (n = 2) volunteers a marked decrease in λz by 83.3%, prolongation of t½ by 572% and MRT by 259%, a rise of AUC by 506% and Cmax by 32.3% respectively were noted. In carriers of CYP2C19*17/*17 (n = 6) significant reduction in MRT by 24.8% (p < 0.05) was revealed. Subjects with CYP2C19*1/*2 (n = 6) genotype were characterized by markedly higher Cmax by 32.3% (p < 0.05) and a significant increase in AUC by 146% (p < 0.05).
Population modeling confirmed a strong influence of *1/*2, *2/*2 and *17/*17 genotypes on the pharmacokinetics of pantoprazole. Based on the goodness-of-fit criteria, serum pantoprazole concentration versus time profiles were best described by a two-compartment model with first-order absorption from the central compartment and a lag time. Following preliminary analyses, the random effects were included on oral clearance (CL/F), volume of the central compartment (Vc/F), first order absorption rate constant (ka), and the absorption lag time (tlag). Volume of peripheral compartment (Vp/F) and intercompartmental clearance (Q/F) were the remaining model parameters. As shown in Table 3, genetic polymorphism of CYP2C19 significantly affected pantoprazole oral clearance. Based on the changes in OFV, the strongest impact on this parameter was observed in subjects with genotype *1/*2 and in both homozygotes *2/*2, *17/*17, whereas in heterozygotes CYP2C19*17 the effect was significant but less pronounced. In addition, their inclusion in the model did not lead to a decrease in interindividual variability in CL/F. When backward deletion was performed, for genotypes *1/*17 and *2/*17 differences in OFV were less than 6.63 and, in consequence, they were removed from the model (Table 3). The estimates of final population model parameters are presented in Table 4. When comparing the basic and the final model, the interindividual variability in CL was reduced from 92.95% to 35.92% after adding CYP2C19*1/*2, *2/*2, and *17/*17 genotypes as model covariates. As expected, the lowest population oral clearance was assessed in the carriers of genotype 2*/2* (3.68 L/h) and the highest value in subjects with genotype *17/*17 (31.13 L/h). The value of this parameter for *1/*2 genotype was 8 L/h. None of other covariates, such as age, gender, and body weight significantly influenced any of the pharmacokinetic parameters of pantoprazole (data not shown).
Figures 2 and 3 represent the relationship between the population (PRED) and the individual (IPRED) model predicted pantoprazole concentrations versus observed concentrations and weighted residuals (WRES) as a function of PRED plots respectively. From Figs. 2 and 3, it seems that the final model where CYP2C19 genotypes were included as covariates describes well concentration versus time profiles of pantoprazole. This can be confirmed both by the plot representing IPRED versus observed concentrations where the data points are aligned on the identity line (Fig. 2), and by the WRES versus the PRED relationship, which shows random scatter around zero (Fig. 3). Moreover, calculated epsilon shrinkage was low (12.6%), indicating that the diagnostic plot involving IPRED is relevant.

Population (upper panel) and individual (lower panel) models predicted pantoprazole concentrations versus observed concentrations. The solid line represents the line of unity

Weighted residuals versus population model predicted pantoprazole concentrations
Discussion
Definition of CYP2C19 polymorphism is important for clinical practice as it was documented that the polymorphism is associated with the efficacy of the treatment of gastric acid-related disorders, with better clinical outcome in poor metabolizers, i.e., carriers of the CYP2C19*2 and CYP2C19*3 allele [14–17]. However, the pharmacokinetic studies did not involve subjects homozygous for the CYP2C19*17 allele, which seems to have a clinically significant impact, as it was proven for antiplatelet action of clopidogrel [18]. Sim et al. in the first report on the CYP2C19*17 allele in reporter vector experiments found an increased transcriptional activity of CYP2C19*17 allele in vivo in mice [4]. The clinical part of the study revealed up to 50–60% higher metabolic rates for omeprazole and mephenytoin in homozygotes for CYP2C19*17 allele than subjects homozygous for the CYP2C19*1 allele after a single dose of the drug.
Pantoprazole undergoes O-demethylation via CYP2C19, followed by sulfate conjugation and sulfone/sulfide formation [17]. Having in mind widespread clinical application of the drug it is very important to define the impact of CYP2C19 polymorphism on its pharmacokinetics. The completed studies recruited healthy volunteers and patients being carriers of CYP2C19*1, CYP2C19*2, CYP2C19*3, and CYP2C19*17. However, the CYP2C19*17 allele was solely evaluated in a heterozygous setting. The study of Sim et al. [4], as well as our preliminary report [10], suggest that the CYP2C19*17/*17 state?? might significantly affect PPI pharmacokinetics. Sim et al. [4] demonstrated a higher omeprazole/5-hydroxyomeprazole metabolic ratio and Gawrońska-Szklarz et al. [10] higher blood concentrations of pantoprazole attained 3 h after oral drug administration in CYP2C19*17/*17 subjects in comparison to wild-type CYP2C19*1/*1 homozygotes. Our former study [10] evaluated pantoprazole concentration at a single time point as stated above, giving only preliminary information on CYP2C19 polymorphism effects on the drug concentrations in patients stratified according to genotype (however, CYP2C19*2/*2 patients were not available for drug concentration studies). The present study analyzes pharmacokinetic data that enable better definition of the role of the CYP2C19 polymorphism in pantoprazole pharmacokinetics, and also involves CYP2C19*2/*2 volunteers. It is demonstrated that polymorphism of CYP2C19 affects pharmacokinetics of pantoprazole. The impact was documented for CYP2C19*1/*2, CYP2C19*2/*2, and CYP2C19*17/*17 genotypes; however, statistical analysis did not reveal any prominent effects of CYP2C19*1/17* and CYP2C19*2/*17 genotypes. Carriers of the genotype CYP2C19*2/*2 were characterized by higher drug concentrations beginning from 3.5 h after oral pantoprazole administration, and an increase in AUC and MRT were seen in CYP2C19*1/*2 and CYP2C19*2/*2 subjects compared with CYP2C19 wild-type individuals. Also, a significantly higher Cmax was observed in CYP2C19*1/*2 volunteers. These findings are in line with the reports of Hunfeld et al. [7, 8], who documented higher, although not significantly different AUC, in CYP2C19*1/*2 patients in 2008 [7], and significant differences between CYP2C19*1/*2 and CYP2C19*1/*1 in 2010 [8]. Similar findings were reported by Kearns et al. [9] from the pediatric population, where AUC values for pantoprazole were significantly elevated in CYP2C19*1/*2 children. Results from homozygous CYP2C19*2 patients were also reported in the aforementioned studies, i.e. Hunfeld et al. [7] found a higher AUC of pantoprazole in those subjects, although the differences did not reach statistical significance. Our observations do confirm this finding, but at a statistically significant level, and the results are in keeping with pediatric observations by Kearns et al. [9].
According to the results of the statistical analysis performed in the present study heterozygosity for the CYP2C19*17 allele does not affect the pharmacokinetics of pantoprazole. Both subjects with CYP2C19*1/*17 and CYP2C19*2/*17 were characterized with similar concentration–time curves and values of the calculated pharmacokinetic parameters as CYP2C19*1/*1 carriers. These observations are in line with the report by Hunfeld et al. [7], who did not find an effect of CYP2C19*1/*17 on the AUC of the drug, and by Kearns et al. [9] for CYP2C19*1/*17 and CYP2C19*2/*17 genotypes and AUC of pantoprazole. Based on these reports and our own results it can be concluded that heterozygosity for the CYP2C19*17 allele do not affect considerably pantoprazole kinetics.
Our population modeling confirmed the influence of the *1/*2, *2/*2, and *17/*17 genotypes on the pharmacokinetics of pantoprazole. Based on the changes in objective function value, a strong impact on pantoprazole oral clearance was observed in both homozygotes *2/*2 and *17/*17, whereas in genotype *1/*2 the effect was significant, but less evident.
Our study is the first one evaluating an effect of CYP2C19*17/*17 on pantoprazole plasma levels. It is revealed that the drug concentrations in carriers of the CYP2C19*17/*17 genotype are significantly higher than in the CYP2C19 wild-type genotype beginning from 3 h after single oral administration of 40 mg of the drug. The calculated pharmacokinetic parameters also demonstrate lower drug accumulation and accelerated elimination in CYP2C19*17/*17 vs. CYP2C19*1/*1 subjects. An effect of CYP2C19*17 polymorphism was also evaluated for another PPIs, i.e., omeprazole. In CYP2C19*17/*17 subjects, it was found to influence the metabolic ratio of omeprazole/5-hydroxyomeprazole [4] and had no effects on esomeprazole [19] and omeprazole [20] pharmacokinetics, whereas for CYP2C19*17 heterozygosity no impact on omeprazole pharmacokinetics was documented [7, 9].
Like the CYP2C19*17/*17 genotype, a prominent effect of the CYP2C19*2/*2 genotype was observed in the present study. Homozygous carriers of the CYP2C19*2 allele were characterized by higher plasma levels of pantoprazole along with a decreased λz, prolongation of MRT and t½, and a rise in the AUC in comparison to the CYP2C19*1/*1 genotype. Effects of poor CYP2C19 status on the pharmacokinetics of other PPIs were also documented for omeprazole [7, 19, 20], esomeprazole [8], rabeprazole [21], and lansoprazole [7, 21, 22].
These observations indicate that the CYP2C19 status affects the pharmacokinetics of pantoprazole. Moreover, our findings document that 57% of the intersubject variability in pantoprazole clearance can be explained by the CYP2C19 genotype status. The results also support clinical observations indicating the variability of cure rates of gastric acid-related disorders treated with pantoprazole. It was found that the CYP2C19*2/*2 genotype was associated with higher Helicobacter pylori eradication rates, whereas no associations were noted for CYP2C19*1/*17 and CYP2C19*17/*17 patients [23]. Our previous study also indicated an influence of the CYP2C19 polymorphism on Helicobacter pylori eradication, since carriers of *1/*1, *1/*17, and *17/*17 were at a higher risk of treatment failure in comparison to *2/*2, *1/*2, and *2/*17 patients [10].
The results of the present study suggest that CYP2C19 genotyping should be used as a predictor of pantoprazole concentrations, and thus it may be useful as a dose adjustment tool. However, it should be stated that the results of the present study refer to single-dose pantoprazole pharmacokinetics, and an effect of CYP2C19 polymorphism on multiple dosing should be addressed in further investigations.
References
Wilkinson GR, Guengerich FP, Branch RA (1995) Genetic polymorphism of S-mephenytoin hydroxylation. Pharmacol Ther 43:53–76
De Morais SM, Wilkinson GR, Blaisdell J, Nakamura K, Meyer U, Goldstein JA (1994) The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem 269:15419–15422
Goldstein JA, de Morais SM (1994) Biochemistry and molecular biology of the human CYP2C subfamily. Pharmacogenetics 4:285–299
Sim SC, Risinger C, Dahl ML, Aklillu E, Christensen M, Bertilsson L, Ingelman-Sundberg M (2006) A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther 79:103–113
Pedersen RS, Brasch-Andersen C, Sim SC, Bergmann TH, Halling J, Petersen MS, Weihe P, Edvardsen H, Kristensen VN, Brosen K, Ingelman-Sundberg M (2010) Linkage disequilibrium between the CYP2C19*17 allele and wildtype CYP2C8 and CYP2C9 alleles: identification of CYP2C haplotypes in healthy Nordic populations. Eur J Clin Pharmacol 66:1199–1205
Tanaka M, Ohkubo T, Otani K, Tanaka M, Ohkubo T, Otani K, Suzuki A, Kaneko S, Sugawara K, Ryokawa Y, Ishizaki T (2001) Stereoselective pharmacokinetics of pantoprazole, a proton pump inhibitor, in extensive and poor metabolizers of S-mephenytoin. Clin Pharmacol Ther 69:108–113
Hunfeld NG, Mathot RA, Touw DJ, van Schaik RH, Mulder PG, Franck PF, Kuipers EJ, Geus WP (2008) Effect of CYP2C19*2 and *17 mutations on pharmacodynamics and kinetics of proton pump inhibitors in Caucasians. Br J Clin Pharmacol 65:752–760
Hunfeld NG, Touw DJ, Mathot RA, Mulder PG, VAN Schaik RH, Kuipers EJ, Kooiman JC, Geus WP (2010) A comparison of the acid-inhibitory effects of esomeprazole and pantoprazole in relation to pharmacokinetics and CYP2C19 polymorphism. Aliment Pharmacol Ther 31:150–159
Kearns GL, Leeder JS, Gaedigk A (2010) Impact of the CYP2C19*17 allele on the pharmacokinetics of omeprazole and pantoprazole in children: evidence for a differential effect. Drug Metab Dispos 38:894–897
Gawrońska-Szklarz B, Siuda A, Kurzawski M, Bielicki D, Marlicz W, Droździk M (2010) Effects of CYP2C19, MDR1, and interleukin 1-B gene variants on the eradication rate of Helicobacter pylori infection by triple therapy with pantoprazole, amoxicillin, and metronidazole. Eur J Clin Pharmacol 66:681–687
Storms ML, Stewart JT (2002) Development of a reversed-phase liquid chromatographic method for the analysis of amoxicillin, metronidazole and pantoprazole in human plasma using solid-phase extraction. J Liquid Chromatogr Rel Tech 25:2433–2443
Rowland M, Tozer TN (1989) Clinical pharmacokinetics: concepts and applications, 2nd edn. Lea and Febiger, Philadelphia
NONMEM Users Guides, (1989–98). Beal SL, Sheiner LB (eds) Icon Development Solutions, Ellicott City, MD
Zhao F, Wang J, Yang Y, Wang X, Shi R, Xu Z, Huang Z, Zhang G (2008) Effect of CYP2C19 genetic polymorphisms on the efficacy of proton pump inhibitor-based triple therapy for Helicobacter pylori eradication: a meta-analysis. Helicobacter 13:532–541
Shi S, Klotz U (2008) Proton pump inhibitors: an update of their clinical use and pharmacokinetics. Eur J Clin Pharmacol 64:935–951
Furuta T, Shirai N, Takashima M, Xiao F, Hanai H, Sugimura H, Ohashi K, Ishizaki T, Kaneko E (2001) Effect of genotypic differences in CYP2C19 on cure rates for Helicobacter pylori infection by triple therapy with a proton pump inhibitor, amoxicillin, and clarithromycin. Clin Pharmacol Ther 69:158–168
Cheer SM, Prakash A, Faulds D, Lamb HM (2003) Pantoprazole: an update of its pharmacological properties and therapeutic use in the management of acid-related disorders. Drugs 63:101–133
Sibbing D, Koch W, Gebhard D, Schuster T, Braun S, Stegherr J, Morath T, Schömig A, von Beckerath N, Kastrati A (2010) Cytochrome 2 C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation 121:512–518
Ohlsson Rosenborg S, Mwinyi J, Andersson M, Baldwin RM, Pedersen RS, Sim SC, Bertilsson L, Ingelman-Sundberg M, Eliasson E (2008) Kinetics of omeprazole and escitalopram in relation to the CYP2C19*17 allele in healthy subjects. Eur J Clin Pharmacol 64:1175–1179
Baldwin RM, Ohlsson S, Pedersen RS, Mwinyi J, Ingelman-Sundberg M, Eliasson E, Bertilsson L (2006) Increased omeprazole metabolism in carriers of the CYP2C19*17 allele; a pharmacokinetic study in healthy volunteers. Br J Clin Pharmacol 65:767–774
Qiao HL, Hu YR, Tian X, Jia LJ, Gao N, Zhang LR, Guo YZ (2006) Pharmacokinetics of three proton pump inhibitors in Chinese subjects in relation to the CYP2C19 genotype. Eur J Clin Pharmacol 62:107–112
Ieiri I, Kishimoto Y, Okochi H, Momiyama K, Morita T, Kitano M, Morisawa T, Fukushima Y, Nakagawa K, Hasegawa J, Otsubo K, Ishizaki T (2001) Comparison of the kinetic disposition of and serum gastrin change by lansoprazole versus rabeprazole during an 8-day dosing scheme in relation to CYP2C19 polymorphism. Eur J Clin Pharmacol 57:485–492
Kurzawski M, Gawrońska-Szklarz B, Wrześniewska J, Siuda A, Starzyńska T, Droździk M (2006) Effect of CYP2C19*17 gene variant on Helicobacter pylori eradication in peptic ulcer patients. Eur J Clin Pharmacol 62:877–880
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gawrońska-Szklarz, B., Adamiak-Giera, U., Wyska, E. et al. CYP2C19 polymorphism affects single-dose pharmacokinetics of oral pantoprazole in healthy volunteers. Eur J Clin Pharmacol 68, 1267–1274 (2012). https://doi.org/10.1007/s00228-012-1252-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-012-1252-3