
Abstract
Purpose
Although the definition of septic shock has been standardized, some variation in mortality rates among clinical trials is expected. Insights into the sources of heterogeneity may influence the design and interpretation of septic shock studies. We set out to identify inclusion criteria and baseline characteristics associated with between-trial differences in control group mortality rates.
Methods
We conducted a systematic review of RCTs published between 2006 and 2018 that included patients with septic shock. The percentage of variance in control-group mortality attributable to study heterogeneity rather than chance was measured by I2. The association between control-group mortality and population characteristics was estimated using linear mixed models and a recursive partitioning algorithm.
Results
Sixty-five septic shock RCTs were included. Overall control-group mortality was 38.6%, with significant heterogeneity (I2 = 93%, P < 0.0001) and a 95% prediction interval of 13.5–71.7%. The mean mortality rate did not differ between trials with different definitions of hypotension, infection or vasopressor or mechanical ventilation inclusion criteria. Population characteristics univariately associated with mortality rates were mean Sequential Organ Failure Assessment score (standardized regression coefficient (β) = 0.57, P = 0.007), mean serum creatinine (β = 0.48, P = 0.007), the proportion of patients on mechanical ventilation (β = 0.61, P < 0.001), and the proportion with vasopressors (β = 0.57, P = 0.002). Combinations of population characteristics selected with a linear model and recursive partitioning explained 41 and 42%, respectively, of the heterogeneity in mortality rates.
Conclusions
Among 65 septic shock trials, there was a clinically relevant amount of heterogeneity in control group mortality rates which was explained only partly by differences in inclusion criteria and reported baseline characteristics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The fundamental criteria from the consensus definitions of septic shock are used to select patients for inclusion in clinical studies [1,2,3,4]. While the mortality rate of septic shock was found to be 46% (95% confidence interval (CI) 43–50%) in a meta-analysis of observational cohorts [5], randomized controlled trials report more diverse numbers. For example, two high-profile septic shock trials published a year apart reported control group mortality rates as disparate as 16% [6] and 80% [7]. Despite the seemingly wide range of mortality rates there has not yet been a systematic inquiry into its patterns and possible causes.
Identifying the correct patient population to benefit from a specific therapy has been recognized as an essential condition for improving critical care research [8,9,10]. Yet large unexplained mortality differences among trials that all aim to include septic shock patients may hamper reproducibility and generalizability. Insights into the magnitude and sources of between-trial heterogeneity are therefore valuable in the design, reporting, and interpretation of septic shock trials. For example, incorrect prediction of baseline mortality rates has been identified as a major reason for negative critical care trials, as a discrepancy between expected and observed event rates often leads to underpowered studies [11].
We sought to quantify between-trial heterogeneity and identify inclusion criteria and population characteristics associated with differences in control group mortality rates.
Methods
After a systematic search to identify all trials published in the past decade that aimed to include patients with septic shock, we used linear mixed models to estimate the total heterogeneity in control group mortality rates and its association with reported baseline characteristics. Using both a multivariate linear model and a machine learning algorithm, we estimated the proportion of heterogeneity that can be explained by population characteristics.
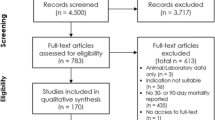
The review protocol was prospectively registered [12] and adheres to the PRISMA checklist [13], which is included in the electronic supplementary material (ESM). Study screening, application of the inclusion- and exclusion criteria and data-extraction were performed independently by two reviewers (HJdG and JP). Conflicting entries were resolved by consensus.
Inclusion criteria and search strategy
PubMed, Embase, and the Cochrane Central Register of Controlled Trials were queried using the search term [“septic shock” AND (random* or rct)]. Embase was additionally queried using the search term “septic shock” with the randomized controlled trial filter activated. The queries were limited to publications from 1 January 2006 and the queries were last performed on 20 January 2018.
We limited the search to trials published between 2006 and 2018 as a compromise between the number of eligible studies and secular trends in clinical practice, research practice, and reporting standards. Publications from 2006 and later had sufficient lead time to incorporate the 2004 update of the Surviving Sepsis Campaign guidelines [4].
Eligible for inclusion were parallel-group randomized controlled trials with adult patients in septic shock according to the published consensus definitions or Surviving Sepsis Campaign guidelines [1, 2, 4]. Trials were excluded if the report was not written in English, if it was only available in abstract, if no baseline characteristics were reported, or if no mortality outcome was reported. Trials that aimed to include a specific subcategory of septic shock patients (e.g. “septic shock patients requiring renal replacement therapy”) were also excluded, as these would be a major source of between-trial heterogeneity.
Identification of the control group and variables of interest
Because the nature of the randomized intervention could contribute to heterogeneity, we focused on the control groups. For each trial, we identified the control group as defined by the authors as ‘control group’, ‘usual care group’, or a variation thereof. When no control group could be identified (in a comparison of two usual care therapies) we defined the control group as the means of the two groups in terms of sample size, mortality, and baseline characteristics. A sensitivity analysis was performed towards this construct by analyzing whether trials with and without specifically defined control groups differed in terms of mean mortality or the amount of between-trial heterogeneity.
For each trial, we recorded the type of intervention, single- or multicenter design, and the primary endpoint. Trials were graded according to the Jadad scale [14]. For the control group in each trial, we recorded the sample size, the reported baseline characteristics, and the mortality rates.
Estimation of heterogeneity in mortality rates and associations with population characteristics
We used 28-day mortality throughout all analyses. For trials that did not report this outcome, we estimated 28-day mortality based on reported hospital, ICU, or 90-day mortality using linear regression with data from trials that reported both 28-day and another mortality measure.
To analyze mortality rates across trials we used a random-effects meta-regression model with the log odds of mortality as dependent variable and a random intercept for each study. Each trial was weighted by the inverse of the sampling variance of the mortality rates. A maximum likelihood estimator was used to estimate the mean mortality (random effects pooled estimate), the between-study standard deviation due to heterogeneity (τ), and the percentage of variation due to heterogeneity rather than change (I2). To quantify between-trial heterogeneity, we report the 95% prediction interval (mean mortality ± 1.96 τ), which represents the distribution of estimated future mortality rates based on observed mortalities weighted by sampling variance (trial size) and corrected for random chance [15]. In the absence of between-study heterogeneity, the 95% prediction interval is equal to the 95% confidence interval, but when significant heterogeneity is present the prediction interval estimates the bandwidth of expected mortality rates from similar studies [15, 16]. In other words, the 95% prediction interval can be thought of as the estimate of true between-study distribution of mortality rates. The prediction interval can therefore be used to guide power calculations for future studies [16].
The between-trial heterogeneity in mortality rates was calculated for subcategories of trials employing different inclusion criteria: confirmed or suspected infection; confirmed infection only; different definitions of hypotension; mandatory hyperlactatemia; mandatory vasopressor therapy; and mandatory mechanical ventilation. Differences in mortality rates between subcategories were calculated by addition of dummy variables to the mixed-effects model.
To estimate the association between study and population characteristics and mortality, these variables were added to the model as covariates. Residuals were checked for normality with Q–Q plots, and the goodness of fit of the log‐linear model was compared with quadratic and power models by selecting the model with the lowest Akaike information criterion (AIC). To facilitate comparisons between variables, we report standardized regression coefficients (β) and the proportion of between-trial variability in mortality explained by the population variable (unadjusted R2) for all univariate analyses.
Predicting mortality rates using a linear model and recursive partitioning
We then constructed a comprehensive model to predict between-study differences in mortality. Population variables that were reported by at least 25% of the included trials with a univariate regression R2 ≥ 0.10 were included as regressors in a multivariate model and removed in a stepwise manner for P values ≥ 0.05. The threshold R2 of 0.10 was a compromise between the number of variables and the limited number of observations. This model selection process was not prospectively protocolized as the number of eligible variables could not be estimated a priori. Multiple imputation (generating 20 datasets) with predictive mean matching was used for missing observations (i.e., missing population characteristics). The imputation methods are further described in section 7 of the ESM.
As a complementary approach to predict 28-day mortality rates from population characteristics, we constructed a regression tree model based on recursive partitioning (a machine learning algorithm) [17, 18] for its ability to handle partially missing observations (obviating the need for imputation) and its robustness to nonlinear relations. We set up the model to predict 28-day mortality based on all inclusion criteria and population characteristics. In short, the recursive partitioning algorithm selected the most informative variable, which was then ‘split’ at the value that best differentiates low from high mortality. The algorithm then selected the most informative variable for each of the two resulting subgroups, and split it again. When a splitting variable was missing for a specific trial, a surrogate variable (the variable most closely correlated to the splitting variable) was used. After multiple splits, this recursive partitioning resulted in a regression tree (similar to a decision tree) with subgroups of trials ranked from low to high expected mortality. R2 represents the variance in mortality explained by the decision tree. Overfitting was examined using the cross-validated error.
For all analyses, P < 0.05 was considered significant. The analyses were performed in R version 3.4.2 using the metafor, mice and rpart packages [19,20,21].
Results
Characteristics of the included trials
The search resulted in 65 trials that met all inclusion and exclusion criteria (eFigure 1 in the ESM), representing a total of 8634 control group patients [6, 7, 22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84]. A list of excluded trials is available in the ESM. The trial characteristics are presented in Table 1.
Twenty trials (31%) did not report 28-day mortality but only hospital mortality, ICU mortality, or 90-day mortality. Using trials that reported multiple mortality measures, 28-day mortality was estimated as a linear function of hospital mortality, ICU mortality, or 90-day mortality (R2 values 0.99, 0.98, and 0.98, respectively). The estimates and validation plots are presented in eTable 1 and eFigure 2 of the ESM.
In 14 trials (21%) the control group could not be identified because two usual care therapies were compared. For these trials, the control group characteristics and mortality rates were defined as the means of the two treatment groups. None of these 14 trials reported significant mortality differences between the treatment groups.
The distribution of mortality rates
The control group mortality rates ranged between 13.8 and 84.6%, with a random-effects estimated mean mortality rate of 38.6%. There was significant heterogeneity among trials (I2 = 93%, τ = 0.710, p < 0.0001), and the 95% prediction interval was 13.5–71.7%.
Figure 1 shows the mortality rates of trials categorized by inclusion criteria. The mean mortality rate did not differ between trials with different definitions of hypotension, infection (confirmed vs. suspected), or vasopressor or mechanical ventilation inclusion criteria. There were no significant differences in mean mortality rate or in heterogeneity between large vs. small trials, monocenter vs. multicenter trials, unblinded vs blinded trials, high-quality trials vs. low-quality trials, or trials with vs. without a specifically defined control group (eTable 2 in the ESM).

Control-group mortality rates categorized by trial inclusion criteria. The diamonds represent the mean mortality rates and 95% confidence intervals. The 95% prediction intervals (dashed lines) represents the estimated between-trial variability in mortality rates after adjusting for random chance and sample size. I2 represents the proportion of between-trial variability that cannot be explained by chance. There were no significant differences in mean mortality rates between inclusion criteria. MAP mean arterial pressure, SBP systolic blood pressure
The exclusion criteria employed in the trials were too diverse for statistical analysis, but the total number of exclusion criteria (ranging from 0 to 30) was inversely associated with the mortality rate (β = − 0.375, R2 = 0.14, P = 0.007).
The heatmap in Fig. 2 provides an overview of the between-trial differences in mortality rates and population characteristics. The log-linear associations between the mortality rate and reported control group baseline characteristics are presented in Table 2 (goodness-of-fit statistics are reported in eTable 3 in the ESM). There was no significant decrease in mortality over the period 2006–2018, with only (R2) 4% of heterogeneity explained by the year of publication (Table 2, eFigure 3). Baseline variables that were univariately associated with mortality were: mean Sequential Organ Failure Assessment (SOFA) score, the proportion of patients on mechanical ventilation, the proportion of patients on vasopressors, and mean serum creatinine. Regression plots of selected associations are shown in eFigure 3 of the ESM.

Heatmap of included trials (n = 65) and associated baseline characteristics, ranked by decreasing mortality rates. White tiles represent the mean value across trials, while red and blue tiles are indicative of higher and lower than average values, respectively. Gray tiles (N/A) are variables that were not reported. The 28-day mortality rate ranged between 13.8 and 84.6%, with a mean of 38.6%. APACHE Acute Physiology and Chronic Health Evaluation, SAPS Simplified Acute Physiology Score, SOFA Sequential Organ Failure Assessment score, MAP mean arterial pressure, CVP central venous pressure, CNS central nervous system. (Asterisk) Variables with a significant univariate association with 28-day mortality
Predicting mortality rates from population characteristics
Details of the variable selection process for the multivariate model are available in section 7 of the ESM. Significant independent variables in the final multivariate model were: baseline mean SOFA score (β = 0.39, standardized standard error (SSE) = 0.17, P = 0.019), the proportion of patients on mechanical ventilation (β = 0.42, SSE = 0.18, P = 0.019), and mean serum creatinine (β = 0.31, SSE = 0.10, P = 0.0015). The multivariate model R2 was 0.41 with significant residual heterogeneity (I2 = 82%, τ = 0.544, P < 0.0001). Figure 3 shows the predicted and actual mortality rates of the included trials.

Included trials ordered by predicted control group mortality rate (diamonds). The predicted mortality rates were based on a multivariate weighted random-effects regression model with baseline mean Sequential Organ Failure Assessment (SOFA) score, the proportion of patients on mechanical ventilation, and mean serum creatinine as significant independent variables. The squares and brackets are the observed control-group mortality rates with 95% confidence interval. The figure illustrates that the model explained (R2) 41% of the variability in mortality rates, with significant residual heterogeneity (P < 0.0001). The red dots are the reported a-priori expected mortality rates used for sample size calculations
The recursive partitioning algorithm resulted in a regression tree with the following variables as informative determinants of the mortality rate: mean age (split at 64.8 years); the proportion of patients with a respiratory infection (split at 54.5%); the proportion of patients on mechanical ventilation (split at 74.3%); and the proportion of male patients (splits at 63.8 and 53.8%). The R2 value of the regression tree was 0.42. The cross-validated relative error decreases to below the root (split 0) value, which indicates that the tree was not overfitted. The results from the regression tree analysis are further described in eFigures 4 and 5 of the ESM (section 7).
Discussion
In this analysis of 65 septic shock trials published in the past decade, we found a statistically significant and clinically relevant amount of heterogeneity in control group mortality rates. The mean mortality rate was 38.6% with estimated 95% prediction limits of 13.5–71.7%, revealing a wide range in underlying mortality rates after discounting the effects of random change and small trials.
In contrast to findings from large observational studies that the mortality of sepsis has decreased in the past decade, we found only a small nonsignificant decline in the period 2006–2018 [85, 86]. Different inclusion definitions of septic shock did not affect mean mortality rates, but a higher total number of exclusion criteria was associated with lower mortality. We used three statistical methods to analyze the association between population characteristics and mortality.
The univariate associations reflect how the reader of a trial report could interpret the population characteristics in relation to the mortality rate, and shows that the proportion of ventilated patients, mean SOFA score, and the proportion of patients on vasopressor support were most informative (i.e. have highest standardized regression coefficients).
The multivariate linear model (with missing observations imputed) shows which combinations of characteristics were predictive of mortality if all trials hypothetically reported the same variables. A combination of three independently significant characteristics (mean SOFA score, proportion of ventilated patients, and mean creatinine) explained only 41% of the heterogeneity in mortality rates across trials.
The recursive partitioning algorithm, which is not limited by dependence on multiple imputation and the assumption of linearity, shows which characteristics were most informative, given that different trials report different characteristics. The resulting regression tree explained only 42% of the heterogeneity in mortality.
The linear model and the regression tree arrived at different predictor variables because the linear model is biased towards more informative linear associations, while the regression tree allows for nonlinear relations and is biased towards variables with less missing data.
In all, these results indicate that there are clinically significant between-trial differences in control group mortality rates, and that these differences are not associated with differences in inclusion criteria and only weakly associated with reported baseline characteristics. Visual inspection of the heatmap (Fig. 2) shows that there are no unambiguous patterns in the relation between population characteristics and mortality rates. This heterogeneity is reflected in our finding that different statistical methods result in different predictive variables.
Possible sources of residual heterogeneity
Residual heterogeneity among trials may be caused by population differences in nutrition and socio-economic status, heterogenous exclusion criteria, incomplete reporting, between-trial differences in variable definitions, the timing of randomization, and differences in post-randomization co-interventions and standards of care.
We found that no single measure of chronic comorbidity was reported in more than 40% of the included trials and that characteristics of causative pathogens were reported in only 28–39% of trials. This compromised the power of our analysis to detect associations across all trials, but, more importantly, it also prevents readers of trial reports from evaluating and comparing populations among trials and from judging to what extent a trial population corresponds to the population under their care.
Another source of heterogeneity is the imprecise definition of many variables. It is unclear whether a variable like ‘pre-existing kidney disease’ in one trial has the same meaning as ‘chronic renal insufficiency’ in another trial. Minor variations in variable definitions and data capture methods have been shown to lead to significantly different septic shock populations and to inter-observer variability in severity-of-illness scoring systems [5, 87, 88]. The importance of this ‘fine print’ in defining a population does not receive due attention in the methods section of most trials.
The time of inclusion may be an additional source of heterogeneity. Patients recruited later after the diagnosis of septic shock have not responded to treatment in an earlier phase and are therefore likely to have a worse prognosis. Only 13 trials reported the time from diagnosis to randomization, and for those trials it explained 22% of the heterogeneity.
While we have focused on inclusion criteria and baseline characteristics, the prognosis of septic shock may be largely influenced by post-randomization standards of care and co-interventions. Unfortunately, co-interventions and (control group) treatment standards are often described as ‘according to the Surviving Sepsis Campaign guidelines’ or not discussed at all in trial reports. Variables describing important post-randomization interventions, such as red blood cell transfusions, vasopressor dose, or fluid balance were recently found to be reported in only 33, 17, and 13% of large septic shock trials, respectively [89].
We did not analyze the association between trial countries and the mortality rate because many countries are represented by a single trial in the present sample. Nevertheless, between-country differences in standards of care or access to early healthcare may account for part of the residual heterogeneity. Large international observational studies are a more appropriate instrument for the investigation of differences in mortality rates among countries.
Implications for investigators and clinicians
Clinicians demand of clinical trials that they are relevant, reproducible, and generalizable to a clearly defined patient population. The results of this study indicate that many of the baseline characteristics upon which clinicians rely to gauge the applicability of trial results to their practice are in fact only weakly or not at all associated with mortality outcomes across trials.
The association between the number of exclusion criteria and mortality suggests that many seemingly inconsequential criteria together may have a significant effect on the composition of a trial population. Investigators should therefore be aware of this phenomenon in the design phase of a trial, as it affects the generalizability and external validity of trial results.
The wide prediction limits of control-group mortality have consequences for sample size calculations. Detecting a relative risk reduction of 25% with 80% power requires 245 patients if mortality is estimated to be 71.7%, while it requires 795 patients if control group mortality is 38.6% or 2980 patients if mortality is 13.5%. In practice, misestimation of the mortality rate by more than 7.5% occurred in 65% of critical care trials [11]. We therefore suggest that sample size calculations should not be based on the mean of reported control-group mortality rates in the literature but should be robust towards a wider range of expected event rates.
Reproducibility and generalizability also require a common phenomenological structure with respect to diagnostic definitions, inclusion criteria, patient characteristics, concomitant treatment, and outcomes. A recent review of large septic shock trials found that only half of the information deemed necessary for evaluation of the control group was reported in the investigated trials [89]. In the present study, we now find that many of the reported characteristics are not associated with control-group mortality rates, possibly due to variations in variable definitions.
The third consensus definitions for sepsis and septic shock were partly developed to harmonize the inclusion criteria for clinical studies [3]. We were unable to analyze a subset of trials with populations that might fit the Sepsis-3 septic shock definition, as none of the included trials employed both delta SOFA score and vasopressor inclusion criteria. We do note that SOFA score is independently associated with mortality rates, although baseline SOFA explains only 33% (R2) of the variation in mortality rates in the 37 trials that report it. Furthermore, we found significant heterogeneity within subsets of trials employing similar inclusion criteria (Fig. 2).
We suggest that an international consensus is necessary to standardize variable definitions, data collection, and reporting of patient characteristics and outcomes for sepsis trials, as has been proposed before [89,90,91,92]. The feasibility of harmonizing study protocols has been demonstrated in three large trials investigating early goal-directed therapy [93]. The present results indicate that SOFA score, the proportion of ventilated patients, and creatinine independently reflect baseline risk across trials and should therefore be reported for each trial.
The results from this study also support the practice of data sharing, as we have shown that aggregated population characteristics are less informative than expected. Sharing individual patient data will not only increase the power to detect treatment effects across multiple studies but can also be used to test the generalizability of trial results vis-à-vis large cohorts with septic shock.
Strengths and limitations
This study was performed with a prospectively registered protocol and analysis plan. We chose to include only trials published between 2006 and 2018 to minimize the influence of long-term secular trends in septic shock diagnosis, treatment, and mortality [94, 95]. The search strategy was broad and comprehensive, but we excluded 40 trial reports not written in English, which compromised power and generalizability. We excluded trials that recruited only septic shock patients with specific organ dysfunction (such as kidney or liver failure) to rule out this source of between-trial heterogeneity.
For 20 trials, 28-day mortality was estimated using another reported mortality rate. Although the prediction equations were very precise (R2 values ≥ 0.98), we cannot rule out the possibility that this influenced the results. Excluding these 20 trials would have eroded the power of the study.
Importantly, using study-level data means that, to avoid the ecological fallacy, we cannot make inferences about predictive characteristics at the individual patient level, although several predictor variables are known to be individually associated with mortality (e.g. high SOFA score as a risk factor [96, 97]).The fact that there was substantial variation in the reporting of baseline variables was an important finding in itself, but also limited our power to detect associations across trials. A more in-depth investigation into the heterogeneity among trial populations would require individual patient data, but we think that obtaining such data would lead to significant selection bias.
Conclusion
Septic shock is a syndrome with various etiologies, biochemical characteristics, and phenotypes [9, 98]. Onto this inherently heterogeneous syndrome, a layer of investigator-induced heterogeneity is added when trials employ different inclusion criteria, report different variables, and use different variable definitions. This compounded complexity causes heterogeneity among trial populations that may go unnoticed. We have shown that control-group mortality rates are very dissimilar across trials, and that the majority of this heterogeneity remains unexplained after accounting for reported population characteristics. The lack of standardized reporting limits the usefulness of the variables explaining the mortality differences found in this study. In all, the substantial between-trial heterogeneity limits the reproducibility and generalizability of septic shock research and may inhibit the discovery of beneficial therapies for specific (sub)populations. The findings of this study therefore strongly support the argument for profound standardization and harmonization of septic shock trial reporting as well as data-sharing policies to test the external validity of trial populations.
References
Bone RC, Balk RA, Cerra FB et al (1992) Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 101:1644–1655
Levy MM, Fink MP, Marshall JC et al (2003) 2001 SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Intensive Care Med 29:530–538. https://doi.org/10.1007/s00134-003-1662-x
Singer M, Deutschman CS, Seymour CW et al (2016) The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 315:801–810. https://doi.org/10.1001/jama.2016.0287
Dellinger RP, Carlet JM, Masur H et al (2004) Surviving sepsis campaign guidelines for management of severe sepsis and septic shock. Intensive Care Med 30:536–555. https://doi.org/10.1007/s00134-004-2210-z
Shankar-Hari M, Phillips GS, Levy ML et al (2016) Developing a new definition and assessing new clinical criteria for septic shock: for the third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 315:775–787. https://doi.org/10.1001/jama.2016.0289
ARISE Investigators, ANZICS Clinical Trials Group, Peake SL et al (2014) Goal-directed resuscitation for patients with early septic shock. N Engl J Med 371:1496–1506. https://doi.org/10.1056/nejmoa1404380
Morelli A, Ertmer C, Westphal M et al (2013) Effect of heart rate control with esmolol on hemodynamic and clinical outcomes in patients with septic shock: a randomized clinical trial. JAMA 310:1683–1691. https://doi.org/10.1001/jama.2013.278477
Vincent J-L, Hall JB, Slutsky AS (2015) Ten big mistakes in intensive care medicine. Intensive Care Med 41:505–507. https://doi.org/10.1007/s00134-014-3570-7
Marshall JC (2014) Why have clinical trials in sepsis failed? Trends Mol Med 20:195–203. https://doi.org/10.1016/j.molmed.2014.01.007
Opal SM, Dellinger RP, Vincent J-L et al (2014) The next generation of sepsis clinical trial designs: what is next after the demise of recombinant human activated protein C? Crit Care Med 42:1714–1721. https://doi.org/10.1097/CCM.0000000000000325
Harhay MO, Wagner J, Ratcliffe SJ et al (2014) Outcomes and statistical power in adult critical care randomized trials. Am J Respir Crit Care Med 189:1469–1478. https://doi.org/10.1164/rccm.201401-0056CP
de Grooth HJ, Oudemans-van Straaten HM, Girbes AR (2016) Heterogeneity in a decade of septic shock RCTs: a systematic analysis of mortality outcomes. PROSPERO 2016 CRD42016049330. https://www.crd.york.ac.uk/PROSPERO/display_record.php?RecordID=49330. Accessed 3 Nov 2016
Moher D, Liberati A, Tetzlaff J et al (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ 339:b2535
Jadad AR, Moore RA, Carroll D et al (1996) Assessing the quality of reports of randomized clinical trials: is blinding necessary? Control Clin Trials 17:1–12. https://doi.org/10.1016/0197-2456(95)00134-4
Deeks JJ, Higgins JPT, Altman DG (2011) Section 9.5: heterogeneity. Cochrane handbook for systematic reviews of interventions version 5.1.0 (updated March 2011)
IntHout J, Ioannidis JPA, Rovers MM, Goeman JJ (2016) Plea for routinely presenting prediction intervals in meta-analysis. BMJ Open 6:e010247. https://doi.org/10.1136/bmjopen-2015-010247
Breiman L, Friedman J, Olshen R, Stone C (1984) Classification and regression trees. CRC Press, Boca Raton, Florida, USA
Atkinson EJ, Therneau TM (2017) An introduction to recursive partitioning using the RPART routines. Mayo Found, Rochester, Minnesota, USA
Viechtbauer W (2010) Conducting meta-analyses in R with the metafor package. J Stat Softw. https://doi.org/10.18637/jss.v036.i03
Van Buuren S, Groothuis-Oudshoorn K (2011) mice: Multivariate imputation by chained equations in R. J Stat Softw 45:1–67. https://doi.org/10.18637/jss.v045.i03
Therneau T, Atkinson B, Ripley B (2017) rpart: recursive partitioning and regression trees. R Packag. version 4.1-11
Annane D, Vignon P, Renault A et al (2007) Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet (London, England) 370:676–684. https://doi.org/10.1016/s0140-6736(07)61344-0
Annane D, Timsit J-F, Megarbane B et al (2013) Recombinant human activated protein C for adults with septic shock: a randomized controlled trial. Am J Respir Crit Care Med 187:1091–1097. https://doi.org/10.1164/rccm.201211-2020OC
Asfar P, Meziani F, Hamel J-F et al (2014) High versus low blood-pressure target in patients with septic shock. N Engl J Med 370:1583–1593. https://doi.org/10.1056/NEJMoa1312173
Bahloul M, Chaari A, Ben Mbarek MN et al (2012) Use of heptaminol hydrochloride for catecholamine weaning in septic shock. Am J Ther 19:e8–e17. https://doi.org/10.1097/MJT.0b013e3181e9b630
Chakraborty A, Mathur S, Dhunna R (2007) Comparison of norepinephrine and dopamine in the management of septic shock using impedance cardiography. Indian J Crit Care Med 11:186. https://doi.org/10.4103/0972-5229.37713
Chen C, Kollef MH (2015) Targeted fluid minimization following initial resuscitation in septic shock: a pilot study. Chest 148:1462–1469. https://doi.org/10.1378/chest.15-1525
Cicarelli DD, Vieira JE, Benseñor FEM (2007) Early dexamethasone treatment for septic shock patients: a prospective randomized clinical trial. São Paulo Med J (Rev Paul Med) 125:237–241
COIITSS Study Investigators, Annane D, Cariou A et al (2010) Corticosteroid treatment and intensive insulin therapy for septic shock in adults: a randomized controlled trial. JAMA 303:341–348. https://doi.org/10.1001/jama.2010.2
Dong W, Zhang G, Qu F (2015) Effects of Ringer’s sodium pyruvate solution on serum tumor necrosis factor-α and interleukin-6 upon septic shock. Pak J Med Sci 31:672–677. https://doi.org/10.12669/pjms.313.7170
Donnino MW, Andersen LW, Chase M et al (2016) Randomized, double-blind, placebo-controlled trial of thiamine as a metabolic resuscitator in septic shock: a pilot study. Crit Care Med 44:360–367. https://doi.org/10.1097/CCM.0000000000001572
Dubin A, Pozo MO, Casabella CA et al (2010) Comparison of 6% hydroxyethyl starch 130/0.4 and saline solution for resuscitation of the microcirculation during the early goal-directed therapy of septic patients. J Crit Care 25:659.e1–659.e8. https://doi.org/10.1016/j.jcrc.2010.04.007
Forceville X, Laviolle B, Annane D et al (2007) Effects of high doses of selenium, as sodium selenite, in septic shock: a placebo-controlled, randomized, double-blind, phase II study. Crit Care 11:R73. https://doi.org/10.1186/cc5960
Garg M, Sen J, Goyal S, Chaudhry D (2016) Comparative evaluation of central venous pressure and sonographic inferior vena cava variability in assessing fluid responsiveness in septic shock. Indian J Crit Care Med 20:708–713. https://doi.org/10.4103/0972-5229.195706
Gordon AC, Mason AJ, Perkins GD et al (2014) The interaction of vasopressin and corticosteroids in septic shock: a pilot randomized controlled trial. Crit Care Med 42:1325–1333. https://doi.org/10.1097/CCM.0000000000000212
Gordon AC, Mason AJ, Thirunavukkarasu N et al (2016) Effect of early vasopressin vs norepinephrine on kidney failure in patients with septic shock: the VANISH randomized clinical trial. JAMA 316:509–518. https://doi.org/10.1001/jama.2016.10485
Hajjej Z, Meddeb B, Sellami W et al (2017) Effects of levosimendan on cellular metabolic alterations in patients with septic shock: a randomized controlled pilot study. Shock 48:307–312. https://doi.org/10.1097/SHK.0000000000000851
Hjortrup PB, Haase N, Bundgaard H et al (2016) Restricting volumes of resuscitation fluid in adults with septic shock after initial management: the CLASSIC randomised, parallel-group, multicentre feasibility trial. Intensive Care Med 42:1695–1705. https://doi.org/10.1007/s00134-016-4500-7
Huh JW, Choi H-S, Lim C-M et al (2011) Low-dose hydrocortisone treatment for patients with septic shock: a pilot study comparing 3 days with 7days. Respirology 16:1088–1095. https://doi.org/10.1111/j.1440-1843.2011.02018.x
Hyvernat H, Barel R, Gentilhomme A et al (2016) Effects of increasing hydrocortisone to 300 mg per day in the treatment of septic shock: a pilot study. Shock 46:498–505. https://doi.org/10.1097/SHK.0000000000000665
Juffermans NP, Vervloet MG, Daemen-Gubbels CRG et al (2010) A dose-finding study of methylene blue to inhibit nitric oxide actions in the hemodynamics of human septic shock. Nitric Oxide 22:275–280. https://doi.org/10.1016/j.niox.2010.01.006
Li M-Q, Pan C-G, Wang X-M et al (2015) Effect of the shenfu injection combined with early goal-directed therapy on organ functions and outcomes of septic shock patients. Cell Biochem Biophys 72:807–812. https://doi.org/10.1007/s12013-015-0537-4
Li Y, Zhang X, Lin P et al (2016) Effects of shenfu injection in the treatment of septic shock patients: a multicenter, controlled, randomized, open-label trial. Evid Based Complement Alternat Med 2016:2565169. https://doi.org/10.1155/2016/2565169
Lin S-M, Huang C-D, Lin H-C et al (2006) A modified goal-directed protocol improves clinical outcomes in intensive care unit patients with septic shock: a randomized controlled trial. Shock 26:551–557. https://doi.org/10.1097/01.shk.0000232271.09440.8f
Livigni S, Bertolini G, Rossi C et al (2014) Efficacy of coupled plasma filtration adsorption (CPFA) in patients with septic shock: a multicenter randomised controlled clinical trial. BMJ Open 4:e003536. https://doi.org/10.1136/bmjopen-2013-003536
Loisa P, Parviainen I, Tenhunen J et al (2007) Effect of mode of hydrocortisone administration on glycemic control in patients with septic shock: a prospective randomized trial. Crit Care 11:R21. https://doi.org/10.1186/cc5696
Lu N-F, Zheng R-Q, Lin H et al (2015) Improved sepsis bundles in the treatment of septic shock: a prospective clinical study. Am J Emerg Med 33:1045–1049. https://doi.org/10.1016/j.ajem.2015.04.031
Lv Q, Gu X, Chen Q et al (2017) Early initiation of low-dose hydrocortisone treatment for septic shock in adults: a randomized clinical trial. Am J Emerg Med 35:1810–1814. https://doi.org/10.1016/j.ajem.2017.06.004
McIntyre LA, Fergusson D, Cook DJ et al (2008) Fluid resuscitation in the management of early septic shock (FINESS): a randomized controlled feasibility trial. Can J Anaesth (J Can d’anesthésie) 55:819–826. https://doi.org/10.1007/bf03034053
Memiş D, Kargi M, Sut N (2009) Effects of propofol and dexmedetomidine on indocyanine green elimination assessed with LIMON to patients with early septic shock: a pilot study. J Crit Care 24:603–608. https://doi.org/10.1016/j.jcrc.2008.10.005
Merouani M, Guignard B, Vincent F et al (2008) Norepinephrine weaning in septic shock patients by closed loop control based on fuzzy logic. Crit Care 12:R155. https://doi.org/10.1186/cc7149
Morelli A, Lange M, Ertmer C et al (2007) Glibenclamide dose response in patients with septic shock: effects on norepinephrine requirements, cardiopulmonary performance, and global oxygen transport. Shock 28:530–535. https://doi.org/10.1097/shk.0b013e3180556a3c
Morelli A, Ertmer C, Rehberg S et al (2009) Continuous terlipressin versus vasopressin infusion in septic shock (TERLIVAP): a randomized, controlled pilot study. Crit Care 13:R130. https://doi.org/10.1186/cc7990
Morelli A, Ertmer C, Rehberg S et al (2008) Phenylephrine versus norepinephrine for initial hemodynamic support of patients with septic shock: a randomized, controlled trial. Crit Care 12:R143. https://doi.org/10.1186/cc7121
Morelli A, Ertmer C, Lange M et al (2008) Effects of short-term simultaneous infusion of dobutamine and terlipressin in patients with septic shock: the DOBUPRESS study. Br J Anaesth 100:494–503. https://doi.org/10.1093/bja/aen017
Morelli A, Donati A, Ertmer C et al (2010) Levosimendan for resuscitating the microcirculation in patients with septic shock: a randomized controlled study. Crit Care 14:R232. https://doi.org/10.1186/cc9387
Mouncey PR, Osborn TM, Power GS et al (2015) Trial of early, goal-directed resuscitation for septic shock. N Engl J Med 372:1301–1311. https://doi.org/10.1056/NEJMoa1500896
Ngaosuwan K, Ounchokdee K, Chalermchai T (2017) Clinical outcomes of minimized hydrocortisone dosage of 100 mg/day on lower occurrence of hyperglycemia in septic shock patients. Shock. https://doi.org/10.1097/SHK.0000000000001061
Palizas F, Dubin A, Regueira T et al (2009) Gastric tonometry versus cardiac index as resuscitation goals in septic shock: a multicenter, randomized, controlled trial. Crit Care 13:R44. https://doi.org/10.1186/cc7767
Patel GP, Grahe JS, Sperry M et al (2010) Efficacy and safety of dopamine versus norepinephrine in the management of septic shock. Shock 33:375–380. https://doi.org/10.1097/SHK.0b013e3181c6ba6f
Payen DM, Guilhot J, Launey Y et al (2015) Early use of polymyxin B hemoperfusion in patients with septic shock due to peritonitis: a multicenter randomized control trial. Intensive Care Med 41:975–984. https://doi.org/10.1007/s00134-015-3751-z
Investigators ProCESS, Yealy DM, Kellum JA et al (2014) A randomized trial of protocol-based care for early septic shock. N Engl J Med 370:1683–1693. https://doi.org/10.1056/NEJMoa1401602
Puskarich MA, Kline JA, Krabill V et al (2014) Preliminary safety and efficacy of l-carnitine infusion for the treatment of vasopressor-dependent septic shock: a randomized control trial. JPEN J Parenter Enter Nutr 38:736–743. https://doi.org/10.1177/0148607113495414
Quenot J-P, Binquet C, Vinsonneau C et al (2015) Very high volume hemofiltration with the Cascade system in septic shock patients. Intensive Care Med 41:2111–2120. https://doi.org/10.1007/s00134-015-4056-y
Ranieri VM, Thompson BT, Barie PS et al (2012) Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 366:2055–2064. https://doi.org/10.1056/NEJMoa1202290
Richard J-C, Bayle F, Bourdin G et al (2015) Preload dependence indices to titrate volume expansion during septic shock: a randomized controlled trial. Crit Care 19:5. https://doi.org/10.1186/s13054-014-0734-3
Russell JA, Vincent J-L, Kjølbye AL et al (2017) Selepressin, a novel selective vasopressin V1A agonist, is an effective substitute for norepinephrine in a phase IIa randomized, placebo-controlled trial in septic shock patients. Crit Care 21:213. https://doi.org/10.1186/s13054-017-1798-7
Russell JA, Walley KR, Singer J et al (2008) Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 358:877–887. https://doi.org/10.1056/NEJMoa067373
Schmoelz M, Schelling G, Dunker M, Irlbeck M (2006) Comparison of systemic and renal effects of dopexamine and dopamine in norepinephrine-treated septic shock. J Cardiothorac Vasc Anesth 20:173–178. https://doi.org/10.1053/j.jvca.2005.10.016
Schortgen F, Clabault K, Katsahian S et al (2012) Fever control using external cooling in septic shock: a randomized controlled trial. Am J Respir Crit Care Med 185:1088–1095. https://doi.org/10.1164/rccm.201110-1820OC
Seguin P, Laviolle B, Guinet P et al (2006) Dopexamine and norepinephrine versus epinephrine on gastric perfusion in patients with septic shock: a randomized study [NCT00134212]. Crit Care 10:R32. https://doi.org/10.1186/cc4827
Shum HP, Leung YW, Lam SM et al (2014) Alteco endotoxin hemoadsorption in Gram-negative septic shock patients. Indian J Crit Care Med 18:783–788. https://doi.org/10.4103/0972-5229.146305
Singh RK, Agarwal V, Baronia AK et al (2017) The effects of atorvastatin on inflammatory responses and mortality in septic shock: a single-center, randomized controlled trial. Indian J Crit Care Med 21:646–654. https://doi.org/10.4103/ijccm.IJCCM_474_16
Sprung CL, Annane D, Keh D et al (2008) Hydrocortisone therapy for patients with septic shock. N Engl J Med 358:111–124. https://doi.org/10.1056/NEJMoa071366
Stephens DP, Thomas JH, Higgins A et al (2008) Randomized, double-blind, placebo-controlled trial of granulocyte colony-stimulating factor in patients with septic shock. Crit Care Med 36:448–454. https://doi.org/10.1097/01.CCM.0B013E318161E480
Torraco A, Carrozzo R, Piemonte F et al (2014) Effects of levosimendan on mitochondrial function in patients with septic shock: a randomized trial. Biochimie 102:166–173. https://doi.org/10.1016/j.biochi.2014.03.006
Trof RJ, Beishuizen A, Cornet AD et al (2012) Volume-limited versus pressure-limited hemodynamic management in septic and nonseptic shock. Crit Care Med 40:1177–1185. https://doi.org/10.1097/CCM.0b013e31823bc5f9
Trzeciak S, Glaspey LJ, Dellinger RP et al (2014) Randomized controlled trial of inhaled nitric oxide for the treatment of microcirculatory dysfunction in patients with sepsis. Crit Care Med 42:2482–2492. https://doi.org/10.1097/CCM.0000000000000549
van Genderen ME, Engels N, van der Valk RJP et al (2015) Early peripheral perfusion-guided fluid therapy in patients with septic shock. Am J Respir Crit Care Med 191:477–480. https://doi.org/10.1164/rccm.201408-1575LE
van Haren FMP, Sleigh J, Boerma EC et al (2012) Hypertonic fluid administration in patients with septic shock: a prospective randomized controlled pilot study. Shock 37:268–275. https://doi.org/10.1097/SHK.0b013e31823f152f
Venkatesh B, Finfer S, Cohen J et al (2018) Adjunctive glucocorticoid therapy in patients with septic shock. N Engl J Med NEJMoa1705835. https://doi.org/10.1056/nejmoa1705835
Xu Q, Yan J, Cai G et al (2014) Effect of two volume responsiveness evaluation methods on fluid resuscitation and prognosis in septic shock patients. Chin Med J (Engl) 127:483–487. https://doi.org/10.3760/cma.j.issn.0366-6999.20131830
Yu T, Peng X, Liu L et al (2015) Propofol increases preload dependency in septic shock patients. J Surg Res 193:849–855. https://doi.org/10.1016/j.jss.2014.08.050
Zhou X, Liu D, Su L et al (2017) Use of stepwise lactate kinetics-oriented hemodynamic therapy could improve the clinical outcomes of patients with sepsis-associated hyperlactatemia. Crit Care 21:33. https://doi.org/10.1186/s13054-017-1617-1
Kaukonen K-M, Bailey M, Suzuki S et al (2014) Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA 311:1308–1316. https://doi.org/10.1001/jama.2014.2637
Gaieski DF, Edwards JM, Kallan MJ, Carr BG (2013) Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med 41:1167–1174. https://doi.org/10.1097/CCM.0b013e31827c09f8
Klein Klouwenberg PMC, Ong DSY, Bonten MJM, Cremer OL (2012) Classification of sepsis, severe sepsis and septic shock: the impact of minor variations in data capture and definition of SIRS criteria. Intensive Care Med 38:811–819. https://doi.org/10.1007/s00134-012-2549-5
Féry-Lemonnier E, Landais P, Loirat P et al (1995) Evaluation of severity scoring systems in ICUs—translation, conversion and definition ambiguities as a source of inter-observer variability in Apache II, SAPS and OSF. Intensive Care Med 21:356–360. https://doi.org/10.1007/BF01705416
Pettilä V, Hjortrup PB, Jakob SM et al (2016) Control groups in recent septic shock trials: a systematic review. Intensive Care Med 42:1912–1921. https://doi.org/10.1007/s00134-016-4444-y
Porter ME, Larsson S, Lee TH (2016) Standardizing patient outcomes measurement. N Engl J Med 374:504–506. https://doi.org/10.1056/NEJMp1511701
Myles PS, Grocott MPW, Boney O et al (2016) Standardizing end points in perioperative trials: towards a core and extended outcome set. Br J Anaesth 116:586–589. https://doi.org/10.1093/bja/aew066
Vincent J, Martin-Loeches I, Annane D (2016) What patient data should be collected in this randomized controlled trial in sepsis? Intensive Care Med 42:2011–2013. https://doi.org/10.1007/s00134-016-4560-8
Investigators PRISM, Rowan KM, Angus DC et al (2017) Early, goal-directed therapy for septic shock—a patient-level meta-analysis. N Engl J Med 376:2223–2234. https://doi.org/10.1056/NEJMoa1701380
Shankar-Hari M, Harrison DA, Rowan KM (2016) Differences in impact of definitional elements on mortality precludes international comparisons of sepsis epidemiology—a cohort study illustrating the need for standardized reporting. Crit Care Med 44:2223–2230. https://doi.org/10.1097/CCM.0000000000001876
Martin GS, Mannino DM, Eaton S, Moss M (2003) The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 348:1546–1554. https://doi.org/10.1056/NEJMoa022139
Ferreira FL, Bota DP, Bross A et al (2001) Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA 286:1754–1758. https://doi.org/10.1001/jama.286.14.1754
Minne L, Abu-Hanna A, de Jonge E (2008) Evaluation of SOFA-based models for predicting mortality in the ICU: a systematic review. Crit Care 12:R161. https://doi.org/10.1186/cc7160
Kempker JA, Martin GS (2016) Does sepsis case mix heterogeneity prevent outcome comparisons? Crit Care Med 44:2288–2289. https://doi.org/10.1097/CCM.0000000000001933
Author information
Authors and Affiliations
Contributions
HJdG and AG designed the study. HJdG and JP performed the study screening and extracted the data. HJdG performed the statistical analyses. SL, HO, and AG provided technical, material, and administrative support. HJdG drafted the manuscript. JP, SL, JJP, HO and AG revised the manuscript for important intellectual content.
Corresponding author
Ethics declarations
Conflicts of interest
All authors declare that they have no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
de Grooth, HJ., Postema, J., Loer, S.A. et al. Unexplained mortality differences between septic shock trials: a systematic analysis of population characteristics and control-group mortality rates. Intensive Care Med 44, 311–322 (2018). https://doi.org/10.1007/s00134-018-5134-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-018-5134-8