
Abstract
Aims/hypothesis
Development of type 2 diabetes depends on environmental and genetic factors. We investigated the epigenome-wide association of prevalent diabetes with DNA methylation (DNAm) in peripheral blood.
Methods
DNAm was measured in whole blood with the Illumina Infinium HumanMethylation450 BeadChip in two subsamples of participants from the ESTHER cohort study. Cohort 1 included 988 participants, who were consecutively recruited between July and October 2000 and cohort 2 included 527 randomly selected participants. The association of DNAm with prevalent type 2 diabetes at recruitment was estimated using median regression analysis adjusting for sex, age, BMI, smoking behaviour, cell composition and batch at 361,922 CpG sites.
Results
Type 2 diabetes was prevalent in 16% of the participants, and diabetes was poorly controlled in 45% of the diabetic patients. In cohort 1 (discovery) DNAm at 39 CpGs was significantly associated with prevalent diabetes after correction for multiple testing. In cohort 2 (replication) at one of these CpGs, DNAm was still significantly associated. Decreasing methylation levels at cg19693031 with increasing fasting glucose and HbA1c concentrations were observed using restricted cubic spline analysis. In diabetic patients with poorly controlled diabetes, the decrease in estimated DNAm levels was approximately 5% in comparison with participants free of diagnosed diabetes.
Conclusions/interpretation
Cg19693031, which is located within the 3′-untranslated region of TXNIP, might play a role in the pathophysiology of type 2 diabetes. This result appears biologically plausible given that thioredoxin-interacting protein is overexpressed in diabetic animals and humans and 3′-untranslated regions are known to play a regulatory role in gene expression.
Similar content being viewed by others

Introduction
The escalating prevalence of type 2 diabetes is a major health problem in industrial as well as developing countries. The numerous associated complications such as diabetic retinopathy, nephropathy, neuropathy, atherosclerosis, stroke and hypertension lead to reduced quality of life and increased mortality. Since type 2 diabetes is a heterogeneous disease whose onset and progression depends on genetic and environmental factors, epigenetic mechanisms may also play a key role in the pathology of diabetes and its complications. DNA methylation (DNAm), the most studied epigenetic mechanism, has been associated with stable alterations of gene expression and implicated in the pathogenesis of type 2 diabetes and other age-related diseases [1–4].
The study of epigenetic mechanisms can provide novel insights into the pathophysiology of diabetes and its complications, which may result in the identification of new drug targets. In addition, the investigation of associations with DNAm in peripheral blood may offer the identification of novel biomarkers for noninvasive early disease detection, since peripheral blood is the most widely available DNA source in large-scale epidemiological studies [5].
So far, the association of DNAm with type 2 diabetes or insulin has been investigated in human pancreatic islets [6], CD4+ T cells [7] and human adipose tissue [8, 9]. Owing to the potential use of peripheral blood as a biomarker, we aimed to identify type 2 diabetes-associated CpG sites in whole blood DNA. In our population-based cohort of 1,515 older adults with a type 2 diabetes prevalence of 16% (n = 240), we investigated the association of whole blood DNAm with type 2 diabetes at more than 360,000 CpG sites measured with the Illumina Infinium HumanMethylation450 BeadChip.
Methods
Study population
The cross-sectional data used for this analysis are from the baseline examination of the prospective ESTHER cohort study. The ESTHER study (n = 9,949) is a general population-based epidemiological study of older adults (aged 50−75 years at baseline) who were recruited by their general practitioners during a routine health check-up between 2000 and 2002 in Saarland, a state in southwest Germany (n = 9,949), and has been described in detail previously [10]. From all participants, two non-overlapping subcohorts were selected. Cohort 1 included 988 participants of the ESTHER cohort study, who were consecutively recruited at the start of the study between July and October 2000. Cohort 2 included 527 participants randomly selected from 3,499 ESTHER participants (with DNA available) recruited between October 2000 and March 2001. Baseline sociodemographic, lifestyle, health and diet information was obtained by a comprehensive questionnaire. Height, weight and history of several diseases, such as hypertension, stroke, diabetes mellitus and cardiovascular disease, were either obtained from a comprehensive participant questionnaire or from the general practitioner’s health check-up report at baseline. Only information obtained at baseline was used for the presented analyses. The study was approved by the ethics committees of the University of Heidelberg and of the physicians’ board of Saarland. Informed consent was obtained from all participants.
Laboratory methods
Blood samples were taken at recruitment. DNA was extracted from whole blood samples using a salting out procedure [11]. DNAm profile was measured in 2012 for subcohort 1 and in 2014 for subcohort 2 using the Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA), which enables the simultaneous quantitative measurement of the methylation status at 485,577 CpG sites [12]. The laboratory work was done in the Genomics and Proteomics Core Facility at the German Cancer Research Center, Heidelberg, Germany (DKFZ) as previously described [13]. Methylation levels at each CpG quantified by average beta values, where 1 corresponds to complete methylation and 0 to no methylation, were calculated with Illumina’s GenomeStudio 2011.1 (Modul M Version 1.9.0). Since the different normalisation approaches like β-mixture quantile normalisation (BMIQ), Illumina normalisation and preprocessing method implemented in Illumina’s GenomeStudio software and subset-quantile within array normalisation (SWAN) were all comparable in both the number of discovered CpGs and the number that were validated [14], data were processed according to the manufacturer’s protocol, no background correction was done and data were normalised to internal controls provided by Illumina. All controls were checked for inconsistencies in each measured plate. Each batch contained samples of participants with and without type 2 diabetes. Samples on the same batch were measured on the same day, with the same BeadChip and located on the same plate. Signals of probes with a detection p value >0.01 were excluded from analysis. We also excluded CpG sites associated with single nucleotide polymorphisms (SNPs), cross-reactive probes and CpG sites located on sex chromosomes, leaving 361,922 CpG sites to analyse.
Plasma glucose levels were measured by general practitioners during the preventive health check-up offered to people older than 35 years in the German healthcare system. Information on whether the participants had fasted overnight was documented on a standardised report sheet together with the measured values. HbA1c levels were measured in EDTA blood at the central laboratory of the University Clinic Heidelberg using standard high-performance liquid chromatography methods.
Statistical analysis
Prevalent diabetes was defined based on a physician’s diagnosis obtained from a preventive health check-up report or from the validation questionnaire at follow-up and by use of glucose-lowering drugs. If the prevalent diabetes value was zero and the HbA1c concentration was ≥6.5% (48 mmol/mol) the participant was considered to have undiagnosed diabetes [15]. Poorly controlled diabetes was defined as an HbA1c concentration ≥7% (53 mmol/mol) in participants with prevalent diabetes [16].
The following potential confounders of the association of DNAm with type 2 diabetes were considered: age, sex, BMI, smoking status (never, former, current) and batch. Restricted cubic spline regression was used to model the shape of dose–response relationships between whole blood DNAm and fasting glucose or HbA1c concentration, controlling for potential confounding factors (sex, age, BMI, smoking status and batch) for CpG sites at which the association of DNAm with type 2 diabetes was significant in both cohorts [17, 18]. Knots were set at 5th, 35th, 65th and 95th percentiles [17]. Since DNAm was measured in whole blood and methylation may vary between different leucocyte subtypes [19], we additionally adjusted for white blood cell composition using the method proposed by Houseman and colleagues [20–23].
No missing values were observed for the variables age, sex and HbA1c. The number of missing values was less than 2% for BMI and smoking status. Nonfasting blood samples were taken in 8% of the participants, which were excluded to determine fasting glucose levels. Overall, overnight fasting glucose levels were missing for 13% of participants and dose–response relationships were calculated for nonmissing values only. Less than 1% of the DNAm data were missing. Although neighbouring CpG sites are often highly correlated with each other, the methylation levels in general might also be quite different at neighbouring CpG sites being either hypomethylated or hypermethylated. Since multiple imputation is based on functional relationships between variables, we replaced missing values by the median for each CpG site. The same approach was used for replacement of missing values for BMI. Participants with missing smoking status were considered as nonsmokers.
In subcohort 1 (discovery cohort) we investigated the association of whole blood DNAm with type 2 diabetes at 361,922 CpG sites. Since DNAm levels quantified by beta values at most of the CpG sites are not normally distributed, we used nonparametric median regression models, in which the dependent variables were the beta values at ~362,000 CpG sites and the independent variable was ‘type 2 diabetes at recruitment yes or no’, adjusted for sex, age, BMI, smoking status and batch. We corrected for 361,922 multiple tests using the Benjamini–Hochberg approach with a false discovery rate (FDR) of 5%. The q values were calculated according to Storey and colleagues [24, 25]. In subcohort 2 (replication cohort) we examined the association of whole blood DNAm with type 2 diabetes at CpG sites at which the association in subcohort 1 was significant; again we used median regression adjusted for sex, age, BMI, smoking status, batch and cell composition. The conservative method of Bonferroni was used to correct for 39 multiple tests (p < 0.0012).
Median and interquartile range for continuous data or number and percentage for categorical data were calculated for the description of population characteristics. Group differences between diabetic and nondiabetic individuals were compared using the Mann–Whitney U (Wilcoxon) test for continuous variables and the χ 2 test for categorical variables. A two-sided p value of 0.05 was considered significant. Data were analysed with the SAS software package (Version 9.2 and Enterprise Guide 4.2; SAS Institute, Cary, NC, USA) and using the procedures PROC CORR, PROC FREQ, PROC UNIVARIATE, PROC NPAR1WAY, PROC QUANTREG, PROC MULTTEST and PROC GPLOT.
Results
In the discovery cohort, type 2 diabetes was prevalent in 153 of 988 individuals (15%), 2% of the participants had undiagnosed type 2 diabetes (n = 23 with HbA1c ≥6.5% [48 mmol/mol]) and 45% (n = 69/153) of the diabetic patients had poorly controlled diabetes (HbA1c ≥7% [53 mmol/mol]). In the replication cohort, the prevalence of diabetes was 16% (n = 87/527), 4% of the participants had undiagnosed diabetes (n = 24) and 45% of diabetic patients had poorly controlled diabetes (n = 39/87). Characteristics of the study population are presented in Table 1. Diabetic patients were on average older, had higher levels of HbA1c and fasting glucose, and higher BMI. Smoking behaviour was not significantly different between individuals with and without diagnosed type 2 diabetes.
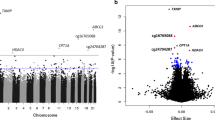
In the discovery cohort, we analysed the association of whole blood DNAm at 361,922 CpG sites with prevalent type 2 diabetes at recruitment. The distribution of p values in the discovery cohort calculated using median regression models for each site is shown in Fig. 1 (Manhattan plot). After correction for multiple testing using the Benjamini–Hochberg approach with an FDR of 5%, we found 39 CpG sites where DNAm was significantly associated with type 2 diabetes. The regression coefficients and the p values for this association at these 39 type 2 diabetes-associated CpG sites are shown in Table 2. The median methylation level in diabetic patients in comparison to individuals free of type 2 diabetes was decreased at 20 CpGs and increased at 19 CpGs.

Manhattan plot of p values for the association of DNAm with prevalent diabetes at 361,922 CpG sites in the discovery cohort of 988 older German adults
We examined the association of DNAm with prevalent type 2 diabetes at these 39 CpGs in the replication cohort. After correction by Bonferroni for 39 multiple tests, DNAm was significantly associated with type 2 diabetes at cg19693031 located within the 3′-untranslated region (3′-UTR) of the gene TXNIP (Table 2). After additional adjustment for leucocyte composition, only marginal changes of the estimates and p values were observed and the association of DNAm with type 2 diabetes at cg19693031 was still significant (Table 2).
Dose–response relationships between DNAm levels at cg19693031 and fasting glucose or HbA1c concentrations adjusted for sex, BMI, age, smoking status, leucocyte composition and batch based on restricted cubic spline models are shown in Fig. 2. We observed a decrease in DNAm levels with increasing glucose and HbA1c concentrations in both subcohorts. Since poorly controlled diabetes was observed in 45% of the diabetic patients and DNAm levels decreased with increasing HbA1c, we estimated the difference in DNAm at cg19693031 in patients with controlled and poorly controlled type 2 diabetes in comparison with individuals free of diagnosed type 2 diabetes by median regression models adjusted for sex, BMI, age, smoking status, leucocyte composition and batch (Table 3). The decrease in DNAm was around 5% in patients with poorly controlled type 2 diabetes in comparison with individuals free of diagnosed type 2 diabetes in both cohorts.

Dose–response relationships between DNAm at cg19693031 (mapping to the gene TXNIP) and fasting glucose and HbA1c concentrations. Dose–response relationship between DNAm and fasting glucose concentration where the reference value for fasting glucose is (a) a median value of 5.2 mmol/l in the discovery cohort (n = 832) and (b) a median value of 5.1 mmol/l in the replication cohort (n = 451). Dose–response relationship between DNAm and HbA1c concentration where the reference value for HbA1c is (c) a median value of 5.6% (38 mmol/mol) in the discovery cohort and (d) a median value of 5.7% (39 mmol/mol) in the replication cohort. Solid line, estimation; grey dashed lines, confidence interval limits; points, knots. Ref., reference value. To convert values for HbA1c in DCCT % into mmol/mol, subtract 2.15 and multiply by 10.929
Discussion
In two independent subcohorts that were drawn from a population-based cohort of elderly German adults, we identified and replicated an association of type 2 diabetes with methylation within the 3′-UTR of TXNIP (cg19693031). Methylation at this CpG site significantly decreased with increasing fasting glucose and HbA1c concentrations. In patients with poorly controlled diabetes, DNAm was decreased by 5% in comparison with individuals free of diagnosed type 2 diabetes.
Thioredoxin-interacting protein (TXNIP), also known as vitamin D-upregulated protein or thioredoxin-binding-protein-2, has been linked to diabetes in multiple previous studies [26–30]. It plays a particularly critical role in pancreatic beta cell biology and glucose homeostasis [26–30]. TXNIP is activated in both hyperglycaemic animals and human adipose tissue treated with 25 mmol/l glucose [29] and regulates insulin-dependent and insulin-independent pathways of glucose uptake in human skeletal muscle [30].
The CpG site cg19693031 is located within the 3′-UTR, a region that contains regulatory regions that post-transcriptionally influence gene expression [31, 32]. It therefore appears biologically plausible that methylation within the 3′-UTR of TXNIP might be involved in the regulation of TXNIP expression and might play a role in defective glucose homeostasis preceding type 2 diabetes. Although we discovered and replicated an association of DNAm with type 2 diabetes at only one CpG site (cg19693031) this finding is plausible since of the 19 CpGs located within the gene region of TXNIP measured with the Illumina Infinium HumanMethylation450 BeadChip, cg19693031 is the only one that is located within the 3′-UTR (one CpG is located in the gene body and 17 CpGs are located in the promoter regions).
An epigenome-wide study using the Illumina Infinium HumanMethylation450 BeadChip reported the association of DNAm in CD4+ T cells with insulin and HOMA-IR in individuals free of type 2 diabetes at two CpG sites, cg01881899 and cg06500161, located within the gene ABCG1 [7]. In our study in whole blood, the p value for the association of DNAm with type 2 diabetes at cg01881899 was 0.36. Cg06500161 was not analysed because we excluded all probes with SNPs according to the annotation files since probe signals may be biased by the presence of SNPs in target CpG with the Illumina Infinium HumanMethylation450 BeadChip [33]. However, these apparent differences should be interpreted with caution because quite different study designs were used. While finalising this manuscript, two studies were published that found an association of DNAm within the gene region of TXNIP with type 2 diabetes in whole blood [34, 35]. Kulkarni et al reported an association of DNAm at cg19693031 with prevalent type 2 diabetes, fasting blood glucose and insulin resistance in 850 pedigreed Mexican American individuals using the 450k BeadChip, which was validated by pyrosequencing [34]. Chambers et al observed an association of DNAm at five loci, including TXNIP, with incident type 2 diabetes. Epigenome-wide association analyses using blood samples from Indian Asian individuals with incident type 2 diabetes and age-matched and sex-matched Indian Asian controls, followed by replication of top-ranking signals in Europeans were performed [35]. TXNIP was identified as one of seven loci at which methylome–metabotype associations independent of genetic variation and potentially driven by common environmental and lifestyle-dependent factors were observed [36].
A genome-wide study in skeletal muscle tissue from 28 men with or without a family history of type 2 diabetes and from nine monozygotic twin pairs discordant for type 2 diabetes observed differential DNAm at 26 CpG sites [37]. Differential non-CpG methylation of the PGC-1a (also known as PPARGC1A) promoter had already been found in skeletal muscle tissue of nondiabetic and diabetic individuals in 2009 [38]. In human pancreatic islets from 15 type 2 diabetic and 34 nondiabetic donors, 1,649 CpGs were identified with differential DNAm after correction for multiple testing by FDR [6]. In human adipose tissue from 28 type 2 diabetic donors and 28 age- and sex-matched controls, 15,627 CpG sites (a large number due to an FDR of 15%) with differential DNAm were identified [8]. In adipose tissue of 96 nondiabetic men, HbA1c level correlated significantly with DNAm at 711 CpG sites after correction for multiple testing (FDR of 5%), but in the female validation cohort (consisting of 94 nondiabetic women) these finding were not replicated [9]. Apart from the use of different tissues, discrepancies in the results described above can be explained by differences in study design. We replicated the findings of the current study in an independent sample for CpG sites that were identified in the discovery cohort and we additionally adjusted for smoking behaviour and BMI, as both factors have been associated with DNAm previously [39–41].
Our study has specific strengths and limitations. Strengths include the large study population of more than 1,500 individuals, the replication of the significant associations in an independent sample and adjustment of the effect estimates for potential confounders including smoking behaviour and BMI. Besides the limitations resulting from the use of the Illumina Infinium HumanMethylation450 BeadChip (e.g. selection bias due to selection of available probes defined by the company and a consortium of experts), a potential limitation is the measurement of DNAm in whole blood rather than specific cell types. To adjust for the different leucocyte subtype composition we used the method introduced by Houseman and colleagues [20–23].
In summary, in this large cohort of older adults from Germany, we found a novel association of DNAm within the gene region of TXNIP, which has been confirmed by others recently while we finalised this manuscript. Given that overexpression of TXNIP in diabetic animals and humans has been reported and the regulatory role of 3′-UTRs in gene expression is well known, it appears biologically plausible that methylation at cg19693031 might play a role in the pathophysiology of type 2 diabetes.
Abbreviations
- 3′-UTR:
-
3′-Untranslated region
- DNAm:
-
DNA methylation
- ESTHER:
-
Epidemiologische Studie zu Chancen der Verhütung, Früherkennung und optimierten THerapie chronischer ERkrankungen in der älteren Bevölkerung
- FDR:
-
False discovery rate
- SNP:
-
Single nucleotide polymorphism
- TXNIP:
-
Thioredoxin-interacting protein
References
Reddy MA, Zhang E, Natarajan R (2015) Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 58:443–455
Bell JT, Tsai P-C, Yang T-P et al (2012) Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy aging population. PLoS Genet 8, e1002629
Xu Z, Taylor JA (2014) Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer. Carcinogenesis 35:356–364
Roessler J, Ammerpohl O, Gutwein J et al (2014) The CpG island methylator phenotype in breast cancer is associated with the lobular subtype. Epigenomics 27:1–13
Gillberg L, Ling C (2015) The potential use of DNA methylation biomarkers to identify risk and progression of type 2 diabetes. Front Endocrinol (Lausanne) 30:43
Dayeh T, Volkov P, Salö S et al (2014) Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet 10, e1004160
Hidalgo B, Irvin MR, Sha J et al (2014) Epigenome-wide association study of fasting measures of glucose, insulin, and HOMA-IR in the Genetics of Lipid Lowering Drugs and Diet Network Study. Diabetes 63:801–807
Nilsson E, Jansson PA, Perfilyev A et al (2014) Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 63:2962–2976
Rönn T, Volkov P, Gillberg L et al (2015) Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet 24:3792–3813
Raum E, Rothenbacher D, Löw M et al (2007) Changes of cardiovascular risk factors and their implications in subsequent birth cohorts of older adults in Germany: a life course approach. Eur J Cardiovasc Prev Rehabil 14:809–814
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Bibikova M, Barnes B, Tsan C et al (2011) High density DNA methylation array with single CpG site resolution. Genomics 98:288–295
Florath I, Butterbach K, Müller H et al (2014) Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum Mol Genet 25:51–56
Wu MC, Joubert BR, Kuan P-F et al (2014) A systematic assessment of normalization approaches for the Infinium 450K methylation platform. Epigenetics 9:318–329
American Diabetes Association (2010) Diagnosis and classification of diabetes mellitus. Diabetes Care 33(Suppl 1):S62–S69
Standards of medical care in diabetes-2012. Diabetes Care 35(Suppl 1):S11–S63
Harrell FE Jr (2001) Regression modeling strategies: with applications to linear models, logistic regression, and survival analysis (Springer Series in Statistics). Springer, New York
Desquilbet L, Mariotti F (2010) Dose–response analyses using restricted cubic spline functions in public health research. Stat Med 29:1037–1057
Reinius LE, Acevedo N, Joerink M et al (2012) Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 7, e41361
Houseman EA, Accomando WP, Koestler DC et al (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13:86
Koestler DC, Christensen B, Karagas MR et al (2013) Blood-based profiles of DNA methylation predict the underlying distribution of cell types: a validation analysis. Epigenetics 8:816–826
Jaffe AE, Irizarry RA (2014) Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 15:R31
Accomando WP, Wiencke JK, Houseman EA et al (2014) Quantitative reconstruction of leukocyte subsets using DNA methylation. Genome Biol 15:R50
Storey JD (2002) A direct approach to false discovery rates. J R Stat Soc Ser B 64:479–498
Storey JD, Taylor JE, Siegmund D (2004) Strong control, conservative point estimation, and simultaneous conservative consistency of false discovery rates: a unified approach. J R Stat Soc Ser B 66:187–205
Chen J, Jing G, Xu G, Shalev A (2014) Thioredoxin-interacting protein stimulates its own expression via a positive feedback loop. Mol Endocrinol 28:674–680
Jo SH, Kim MY, Park JM, Kim TH, Ahn YH (2013) Txnip contributes to impaired glucose tolerance by upregulating the expression of genes involved in hepatic gluconeogenesis in mice. Diabetologia 56:2723–2732
Yoshihara E, Fujimoto S, Inagaki N et al (2010) Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity. Nat Commun 23:127
Koenen TB, Stienstra R, van Tits LJ et al (2011) Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1ß transcription in human adipose tissue. Diabetes 60:517–524
Parikh H, Carlsson E, Chutkow WA et al (2007) TXNIP regulates peripheral glucose metabolism in humans. PLoS Med 4, e158
Misquitta CM, Iyer VR, Werstiuk ES, Grover AK (2001) The role of 3′-untranslated region (3′-UTR) mediated mRNA stability in cardiovascular pathophysiology. Mol Cell Biochem 224:53–67
Merritt C, Rasoloson D, Ko D, Seydoux G (2008) 3′ UTRs are the primary regulators of gene expression in the C. elegans germline. Curr Biol 18:1476–1482
Price ME, Cotton AM, Lam LL et al (2013) Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin 6:4
Kulkarni H, Kos MZ, Neary J et al (2015) Novel epigenetic determinants of type 2 diabetes in Mexican-American families. Hum Mol Genet 24:5330–5344
Chambers JC, Loh M, Lehne B et al (2015) Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol 3:526–534
Petersen K-A, Zeilinger S, Kastenmuller G et al (2014) Epigenetics meets metabolomics: an epigenome-wide association study with blood serum metabolic traits. Hum Mol Gen 23:534–545
Nitert MD, Dayeh T, Volkov P et al (2012) Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 61:3322–3332
Barres R, Osler ME, Yan J et al (2009) Non-CpG methylation of the PGC-1a promoter through DNMT3B controls mitochondrial density. Cell Metab 10:189–198
Dick KJ, Nelson CP, Tsaprouni L et al (2014) DNA methylation and body-mass index: a genome-wide analysis. Lancet 383:1990–1998
Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H (2011) Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 88:450–457
Shenker NS, Polidoro S, van Veldhoven K et al (2013) Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet 22:843–851
Acknowledgements
We thank the Microarray Unit of the DKFZ Genomics and Proteomics Core Facility, Heidelberg, Germany, especially M. Schick and R. Fischer for performing the Illumina Human Methylation arrays and related services.
Funding
The ESTHER study was supported in part by grants from the Baden-Württemberg State Ministry of Science, Research and Arts and by the German Federal Ministry of Education and Research.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
HB and IF designed the study. IF analysed the data and wrote the manuscript. KB, JH, MB-H, YZ and BS contributed to the acquisition and analysis of the data. All authors revised the manuscript critically and approved the final version. IF and HB are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Florath, I., Butterbach, K., Heiss, J. et al. Type 2 diabetes and leucocyte DNA methylation: an epigenome-wide association study in over 1,500 older adults. Diabetologia 59, 130–138 (2016). https://doi.org/10.1007/s00125-015-3773-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3773-7