
Abstract
Heparanase is the sole mammalian endoglycosidase that selectively degrades heparan sulfate, the key polysaccharide associated with the cell surface and extracellular matrix of a wide range of tissues. Extensively studied for its capacity to promote cancer progression, heparanase enzyme was recently implicated as an important determinant in several inflammatory disorders as well. Applying immunohistochemical staining, we detected preferential expression of heparanase by epidermal keratinocytes in human psoriatic lesions. To investigate the role of the enzyme in the pathogenesis of psoriasis, we utilized heparanase transgenic mice in a model of 12-O-tetradecanoyl phorbol 12-myristate 13-acetate-induced cutaneous inflammation. We report that over-expression of the enzyme promotes development of mouse skin lesions that strongly recapitulate the human disease in terms of histomorphological appearance and molecular/cellular characteristics. Importantly, heparanase of epidermal origin appears to facilitate abnormal activation of skin-infiltrating macrophages, thus generating psoriasis-like inflammation conditions, characterized by induction of STAT3, enhanced NF-κB signaling, elevated expression of TNF-α and increased vascularization. Taken together, our results reveal, for the first time, involvement of heparanase in the pathogenesis of psoriasis and highlight a role for the enzyme in facilitating abnormal interactions between immune and epithelial cell subsets of the affected skin. Heparanase inhibitors (currently under clinical testing in malignant diseases) could hence turn highly beneficial in psoriatic patients as well.
Similar content being viewed by others


Introduction
Psoriasis is a common chronic inflammatory skin disease of unsolved pathogenesis. Skin of psoriatic patients is characterized by well-demarcated erythematous lesions with specific histopathological features, including epidermal hyperplasia, increased dermal angiogenesis, and dense infiltrates of immunocytes [1, 2]. Psoriatic plaques are dominated by T lymphocytes as well as innate immune cells such as dendritic cells and macrophages [3–6]. Crosstalk between immunocytes and keratinocytes, which results in the production of cytokines, chemokines, and growth factors, is thought to mediate the disease. The infiltrating immune cells generate inflammatory cytokines that can enhance expression of MHC II, adhesion and co-stimulatory molecules, angiogenic and growth promoting factors by keratinocytes, further facilitating pathologic interactions between immune and epithelial compartments of the affected skin [2]. Important advancements have been made in deciphering the role of growth factors (e.g., VEGF, FGF, KGF), inflammatory cytokines (e.g., TNF-α, IFN-γ, IL-12, IL-17, IL-23) and their downstream transcription factors (NF-κB, STAT) in the crosstalk between immune and epithelial cells that underlies the pathogenesis of psoriasis [2, 6]. Much less is known about the role of the extracellular matrix (ECM) and its enzymatic degradation in this crosstalk.
Heparanase is the sole mammalian endoglycosidase that cleaves heparan sulfate (HS), the key polysaccharide of the ECM and basement membranes. Heparan sulfate is a ubiquitous macromolecule associated with the cell surface and ECM of a wide range of tissues and organs. By interacting with ECM protein components, HS contributes to the self-assembly and structural integrity of the ECM and basement membrane [7, 8]. Accumulating evidence indicates that HS chains act to preserve proper tissue organization and inhibit cellular migration by promoting tight cell–cell and cell–ECM interactions [9, 10]. Alterations in HS content are characteristic for several inflammation disorders [11, 12], including psoriasis [13, 14]. HS is believed to coordinate the inflammatory response at a number of levels, interacting with an array of chemokines and cytokines (i.e., macrophage inflammatory protein (MIP)-1, IL-2, -8, -10), growth factors (VEGF, FGF, KGF) and cell-surface molecules (i.e., selectins) [11, 12, 15, 16].
Enzymatic cleavage of HS by heparanase profoundly affects a variety of pathophysiological processes [17, 18], including inflammation, where heparanase activity was often associated with ECM remodeling, immunocyte activation, and release of chemokines anchored within the ECM network and cell surface [12]. Up-regulation of heparanase, locally expressed (i.e., by epithelial cells, vascular endothelium) at the site of inflammation, was demonstrated in mouse models of delayed type hypersensitivity [19], vascular injury [20], chronic colitis [21], sepsis-associated lung injury [22], as well as in several auto-immune and auto-inflammatory human disorders, including rheumatoid arthritis [23], atherosclerosis [24, 25], and inflammatory bowel disease [21, 26, 27]. However, a role for heparanase in the pathogenesis of psoriasis has not been demonstrated.
Based on the involvement of heparanase in several autoimmune disorders mechanistically related to psoriasis (i.e., rheumatoid arthritis, inflammatory bowel disease [21, 23, 26]), we investigated the biological significance of heparanase in the pathogenesis of psoriasis. Here we report that heparanase is preferentially expressed in human psoriatic lesions. Moreover, applying a mouse model of cutaneous inflammation induced by topical applications of 12-O-tetradecanoyl phorbol 12-myristate 13-acetate (TPA, a protein kinase C agonist commonly used as a non-antigen-dependent chemical stimulator of immune cells), we found that heparanase-over-expressing Hpa-tg mice (but not their wild-type littermates) develop chronic skin inflammation with striking similarities to human psoriasis. Our data suggest that in psoriasis heparanase acts through facilitation of pathologic crosstalk between keratinocyte and immunocyte communication circuits. Heparanase over-expression creates psoriasis-like phenotype in the mouse skin via generation of inflammation-preserving conditions, characterized by induction of STAT3, enhanced NF-κB signaling and increased vascularization. Furthermore, our data indicate that heparanase-dependent macrophage activation represents a relevant mechanism in the pathogenesis of psoriasis. This involves a self-sustained inflammatory circle through which heparanase of epidermal origin facilitates abnormal activation of macrophages, which, in turn, preserves chronic inflammatory conditions in the skin and in parallel controls further production/activation of the enzyme by the epithelial compartment.
Materials and methods
Multiple TPA application to mouse skin
Male BALB/c mice were purchased from Harlan Laboratories (Jerusalem, Israel). Hpa-tg mice in which transgenic heparanase is driven by a constitutive ß-actin promoter [28] in a BALB/c genetic background were bred at the animal facility of the Hadassah-Hebrew University Medical Center. All mice were kept under conventional pathogen-free conditions, and all experiments were performed in accordance with the Hebrew University IACUC. The TPA treatment was based on the previously reported methods [29–31]. The dorsal skin of each mouse was shaved 2 days prior to the treatment. For the TPA application, the mice (n = 6/each group) were treated six times at an interval of 48 h with TPA (8.1 nmol in 100 μl acetone; Sigma–Aldrich, St. Louis, MO, USA). The acetone-treated mice were used for control.
Measurement of the epidermis thickness and histopathological analysis
The back skins of the mice were obtained by 8-mm-diameter punch and fixed with 4 % paraformaldehyde, embedded in paraffin, and sectioned at 5-μm thickness. The epidermal thickness of skin was measured at five sites of randomly selected region utilizing an image analysis system with a Zeiss axioscope microscope and Scion Image software. Inflammatory lesions were scored in a blinded fashion by expert pathologist (T.N.). The vertical thickness of the epidermis was defined as the distance from the basal layer to the stratum corneum.
Skin biopsy specimens from healthy individuals and patients with psoriasis were provided by the Department of Pathology, Hadassah Medical Center, Jerusalem. The use of these specimens was exempted from IRB review because the study does not meet Common Rule Section 101(b) criteria for “research involving human subjects”. Tissue samples were deidentified making it impossible to trace back to the patient’s identity.
Antibodies
Immunostaining was carried out with the following antibodies: anti-F4/80 (Serotec); anti-cyclin D1 (Thermo Scientific); anti-cathepsin-L (R&D Systems); anti-COX2 (Cayman); anti-CD31 (Biocare Medical); anti-phospho NF-κB p65, anti-pStat3 (Cell Signaling); anti-PCNA (Santa Cruz); and anti-heparanase monoclonal antibody 01385-126, recognizing both the 50-kDa subunit and the 65-kDa proheparanase [21, 32], kindly provided by Dr. P. Kussie (ImClone Systems Inc., New York, NY, USA).
Immunohistochemistry
Paraffin-embedded slides were deparaffinized and incubated in 3 % H2O2. Antigen unmasking was carried out by heating (20 min) in a microwave oven in 10 mM Tris buffer containing 1 mM EDTA. Slides were incubated with primary antibodies diluted in CAS-Block (Invitrogen) or with CAS-Block alone, as a control. Appropriate secondary antibodies (Nichirei) were then added and slides incubated at room temperature for 30 min. Mousestain kit (Nichirei) was used when primary mouse antibodies were applied to stain mouse tissues. Color was developed using the DAB substrate kit (Thermo Scientific) or Zymed AEC substrate kit (Zymed Laboratories), followed by counter staining with Mayer’s hematoxylin. Controls without addition of primary antibody showed low or no background staining in all cases.
Immunofluorescence
For immunofluorescence analysis, DyLight 488 donkey anti-rat and DyLight 549 donkey anti-goat and anti-mouse (Jackson Labs) antibodies were used as secondary antibodies. Nuclear staining was performed with 1,5-bis{[2-(di-methylamino) ethyl]amino}-4,8-dihydroxyanthracene-9, 10-dione (DRAQ5) (Cell Signaling). Images were captured using a Zeiss LSM 5 Confocal microscope and analyzed with Zen software (Carl Zeiss).
Determination of vessel density
Blood vessels were visualized by staining with rat anti-mouse CD31 antibody (Biocare Medical). Zeiss LSM 5 software (Carl Zeiss Inc.) was used to calculate the pixel density of labeled cells per 200 μm2 microscopic field, in ten randomly selected microscopic fields of each skin sample (four mice/group) and the mean value ± SD was determined.
Analysis of gene expression by quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from snap-frozen tissue samples using TRIzol (Invitrogen), according to the manufacturer’s instructions, and quantified by spectrophotometry. After oligo (dT)-primed reverse transcription of 500 ng total RNA, the resulting single stranded cDNA was amplified using real-time quantitative PCR analysis with an automated rotor gene system RG-3000A (Corbett Research). The PCR reaction mix (20 μl) was composed of 10 μl QPCR sybr master mix (Takara), 5 μl of diluted cDNA (each sample in a triplicate) and a final concentration of 0.3 μM of each primer. PCR conditions were as follows: an initial denaturation step at 95 °C for 10 min; 40 cycles of denaturation at 94 °C for 15 s, hybridization at 57 °C for 30 s, and elongation at 72 °C for 30 s. Actin primers were used as an internal standard. The following primers were used:
- Mouse β-Actin S::
-
5′-ATG CTC CCC GGG CTG TAT-3′
- AS::
-
5′-CAT AGG AGT CCT TCT GAC CCA TTC-3′
- Mouse TNF-α S::
-
5′-CAT CTT CTC AAA ATT CGA GTG ACAA-3′
- AS::
-
5′-TGG GAG TAG ACA AGG TAC AAC CC-3′
- Mouse IL-12/23p40 S::
-
5′-GTT CAA CAT CAA GAG CAG TAG CA-3′
- AS::
-
5′-CTG CAG ACA GAG ACG CCA TT-3′
- Mouse Heparanase S::
-
5′-ACT TGA AGG TAC CGC CTC CG-3′
- AS::
-
5′-GAA GCT CTG GAA CTC GGC AA-3′
TNF-α ELISA
TNF-α levels were assayed with ELISA MAX mouse TNF-α kit (Biolegend).
Isolation of murine peritoneal macrophages
Ten-week-old male C57BL/6 mice were injected i.p. with 1.5 ml of 4 % thioglycollate (BD Biosciences), and killed 3 days later. A total of 2.5 ml of DMEM was injected into the peritoneal cavity and the peritoneal lavage fluid was pooled and centrifuged at 1,000 rpm for 5 min. The pellet was resuspended in DMEM supplemented with 1 mM glutamine, 50 μg/ml streptomycin, 50 U/ml penicillin and 10 % fetal calf serum, and the cells were plated for 1 h and then washed three times with DMEM to remove non-adherent cells. This procedure yielded cells that were >95 % positive for F4/80.
Results
Preferential expression of heparanase in human skin psoriatic lesions
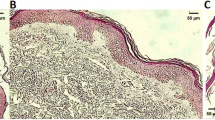
We applied immunohistochemical staining to examine heparanase expression in skin specimens derived from psoriatic patients (n = 29) and in control samples derived from healthy individuals (n = 7). While heparanase expression was practically not detected in healthy skin specimens (negative staining in six biopsies and faint staining in one biopsy, Fig. 1a, top), in the psoriatic lesions a marked over-expression of heparanase was detected in 72 % of specimens (Fig. 1a, middle, bottom; Fig. 1b; Chi-square test p < 0.001). Of note, epidermal rather than inflammatory cells represent the main source of heparanase protein in the involved areas (Fig. 1a).

Preferential expression of heparanase in psoriatic lesions. a Tissue specimens derived from normal skin tissue (top panels), or psoriatic patients (middle and bottom panels) were stained with anti-heparanase antibody (brown staining). Photographs are representative of seven control and 29 psoriatic samples (original magnification ×200). b Specimens were then scored by expert pathologist as negatively (gray bars) or positively (black bar) stained for heparanase. Chi-squared analysis confirmed highly statistically significant (p < 0.001) prevalence of heparanase over-expression in psoriatic skin vs. normal skin tissue
Backing our immunohistochemical findings, several microarray studies available at the Gene Expression Omnibus repository database (i.e., GEO accession numbers GDS2518; GDS3539) clearly demonstrate significant increase in heparanase (Hpse) gene expression in human lesional skin. These findings are also consistent with the previously reported altered content of HS (enzymatic substrate of heparanase), found in involved psoriatic skin [14].
Heparanase over-expression preserves psoriasis-like phenotype in mouse skin following TPA application
We next studied the tempo-spatial pattern of heparanase expression in the mouse model of chemically induced inflammatory reaction in skin. Psoriasis is unique to humans, but multiple topical applications of TPA to mouse skin has traditionally been used to model some aspects of psoriatic disease [29, 30]. Repeated applications of TPA have been reported to induce a chronic cutaneous inflammation, characterized by infiltration of the skin with T cells and macrophages, and by epidermal hyperplasia [31] which returns to the normal values rapidly after the TPA application is ended [33].
In our initial experiments TPA was applied six times over 14 days to BALB/c mouse back skin, as indicated in Fig. 2a, and described in the “Materials and methods” section. Mice were killed on the indicated time points, their back skin samples were collected and heparanase expression was assessed by qRT-PCR and immunostaining. As shown in Fig. 2, markedly increased levels of heparanase mRNA and protein were detected in the skin (primarily epidermis, Fig. 2b) of TPA-treated mice on experimental day 15, concomitant with the development of inflammation, manifested by hyperplasia of the epidermis (>threefold increase in epidermal thickness), hyper- and para-keratosis, and neutrophil accumulation. However, in parallel with the disappearance of inflammatory changes in the skin during the following 7 days, as expected based on previous reports [31, 33], heparanase expression gradually decreased (Fig. 2b, c). We next utilized heparanase transgenic (Hpa-tg) mice, along with their wild-type (wt) counterparts, in multiple TPA topical application model. Hpa-tg mice, overexpressing heparanase in skin, as well as in additional tissue types (but not in splenocytes) [28] have been previously used in studies on the role of heparanase in several autoimmune/autoinflammatory conditions [19–21, 23, 34]. The skin of Hpa-tg mice has normal appearance and is characterized by an enhanced hair re-growth [28, 35]. Of note, analysis of skin tissue sections immunostained with antibodies directed against proliferating cell nuclear antigen (PCNA) revealed no differences in proliferation of interfollicular keratinocytes between healthy Hpa-tg and wt mice (not shown). Applying multiple topical TPA challenges (as shown in Fig. 2a) in both genotypes, we found prolonged skin inflammation with remarkable similarities to human psoriasis in Hpa-tg, but not in wt mice. While in TPA-treated wt mice epidermal hyperplasia and the associated fourfold increase in mean epidermal thickness observed on day 15 gradually returned to the normal levels within 6 days, in TPA-treated Hpa-tg mice epidermal hyperplasia and the ~fivefold increase in mean epidermal thickness detected on day 15 persisted until day 21 (Fig. 3a, b). In vehicle-treated wt and Hpa-tg mice no significant increase in skin thickness was observed during the experiment (not shown). Importantly, topical application of the anti-inflammatory drug betamethasone-17-valerate (BMS), common reference compound against psoriasis, markedly reduced (>threefold) epidermal hyperplasia in TPA-treated Hpa-tg mice on experimental day 21(not shown).

Transient induction of heparanase in mouse model of TPA-induced chronic cutaneous inflammation. a Schematic representation of TPA induced skin inflammation, induced as described in the “Materials and methods” section. Mouse skin tissues were harvested on indicated time points and analyzed as described below (b–c). b Immunostaining (brown) of mouse skin tissue sections with anti-heparanase. Note heparanase staining in the epithelial compartment (original magnification ×200). c Heparanase mRNA expression during the course of TPA skin inflammation, measured by qRT-PCR and normalized to actin mRNA; (n = 4; *p = 0.034)

Heparanase over-expression preserves psoriasis-like phenotype in mouse skin following multiple TPA application. Skin samples were obtained from wt and Hpa-tg mice on experimental days 1 (prior to TPA application), 15 and 21 and processed for H&E staining and morphometric analysis of epidermal thickness (a, b). Vascular density was determined by (c) immunofluorescent staining with antibody directed against CD31 (scale bars 200 μm, cell nuclei were counterstained with DRAQ5) and d quantification of CD31-positive microvessels, based on five sections from four independent mice *p < 0.003. e Histopathological evaluation revealed psoriasis-like phenotype in skin samples harvested from Hpa-tg (right), but not wt (left) mice on experimental day 21. Note the absence of the granular layer (white arrowhead), psoriasiform hyperplasia of the epidermis (white arrow), infiltration of parakeratotic scale by polymorphonuclear leukocytes and formation of Munro micro abscess (black arrow). Six mice per genotype were examined, representative microphotographs are shown (original magnification, ×400). f Immunohistochemical staining with antibody directed against Cyclin D1. Note the increased number of cyclin D1-positive keratinocytes in the epidermis of Hpa-tg (red arrows), but not wt mice on experimental day 21
Microscopic examination of the skin samples collected on day 21 revealed presence of histopathological features characteristic for human psoriatic lesions in Hpa-tg but not in wt skin. These changes included hypervascularity (Fig. 3c, d), psoriasiform hyperplasia of the epidermis, hyperparakeratosis, loss of the granular layer, and transmigration of polymorphonuclear leukocytes through the reactive epidermis into the parakeratotic scale, resembling formation of Munro microabscesses (Fig. 3e). In addition, on day 21, keratinocytes in the Hpa-tg but not wt TPA-treated skin were highly positive for cyclin D1 (Fig. 3f), a key cell-cycle promoting gene, whose induction is characteristic of psoriatic lesions [36].
Cyclin D1 is a well-defined target gene of signal transducer and activator of transcription 3 (STAT3). Importantly, STAT3 signaling emerged as a critical component in the pathogenesis of psoriasis [37, 38]. This notion, taken together with the previous reports on activation of STAT3 in the presence of elevated levels of heparanase [21, 39], prompted us to examine STAT3 activation in TPA-treated wt and Hpa-tg skin samples. As shown in Fig. 4, a high number of cells positive for nuclear-localized pSTAT3 was observed on day 21 in Hpa-tg (Fig. 4 bottom, middle lower panel), as compared to wt epidermis (Fig. 4 top, middle lower panel). Moreover, applying double-immunofluorescent staining with antibody directed against the marker of hyperproliferation PCNA, we demonstrated that STAT3 activation co-localizes with highly proliferating cells in Hpa-tg epidermis (Fig. 4 bottom, right lower panel). These findings are in agreement with the proposed role of STAT3 in enhanced keratinocyte proliferation in psoriatic lesions [38].

Co-localization of phosphoSTAT3 positivity and increased cell proliferation in Hpa-tg epidermis. Skin tissue was obtained from wt (top) and Hpa-tg (bottom) mice on experimental days 1 and 21, and processed for immunofluorescent analysis using the combination of anti-PCNA (green) and anti-phosphoSTAT3 (red) antibodies. Cell nuclei were counterstained with DRAQ5 (blue). Overlay represents proliferating cells positive for nuclear-localized phosphoSTAT3. At least three mice per time point were analyzed and representative photographs are shown; Scale bar 100 μm
In addition, qRT-PCR analysis of the TPA-treated skin taken from Hpa-tg and wt mice on experimental day 21 revealed increased levels of mRNA encoding for IL-12/23p40 (a p40 subunit shared by IL-12 and IL-23) and TNF-α, both central components of psoriasis-driving cytokine network [6, 40–43] in Hpa-tg skin (Supplementary Figure 1A, B). Importantly, IL-12, IL-23, and TNF-α were shown to induce epithelial STAT3 signaling [44–46], providing a mechanistic explanation for the observed STAT3 activation in Hpa-tg mice.
TNF-α and IL-23 are important inducers of NF-κB pathway, and enhanced NF-κB signaling was demonstrated in almost all epidermal keratinocytes in human psoriatic lesions [47]. In agreement with these notions, marked activation of the NF-κB pathway was observed in the epidermal layers of TPA-treated Hpa-tg vs. wt skin on experimental day 21, as manifested by a higher number of cells positive for nuclear-localized phospho-p65 NF-κB (Supplementary Figure 1C).
Role of macrophages in psoriasis-like phenotype of TPA-treated Hpa-tg mice
Activated macrophages have been identified as important producers of TNF-α and IL-12/23p40 in psoriasis and other chronic inflammatory disorders [4, 48–50]. Moreover, experimental and clinical observations suggest that activation of macrophages represents an important pathogenic event in the development and maintenance of psoriatic skin disease [4, 5, 48, 49, 51–53]. This notion, along with the recently described role of heparanase in sustaining continuous activation of macrophages in a mouse model of chronic inflammatory bowel disease, mechanistically related to psoriasis [21], led us to examine the involvement of macrophages in our system. Interestingly, on day 15 (immediately after cessation of TPA treatment) immunostaining with antibody directed against F4/80 (mouse macrophage specific marker [54]) revealed no difference in the extent of skin infiltration of macrophages in both genotypes (not shown). However, on day 21, examination of F4/80-stained skin sections revealed retention of macrophage infiltration in Hpa-tg mice vs. little or no infiltration in wt mice (Fig. 5a, b). Macrophages were mainly detected in the upper portion of Hpa-tg dermis and near the dermo-epidermal junction, resembling the pattern reported for human psoriatic lesion-infiltrating macrophages [49]. To quantify macrophage infiltration, following immunofluorescence staining with F4/80-specific antibody, an average F4/80 pixel density for labeled cells was calculated for Hpa-tg and wt skin samples harvested on day 21. As shown in Fig. 5b, twofold increase in macrophage infiltration was detected in Hpa-tg mice, similar to observations reported in psoriatic patients [49].

a Increased macrophage accumulation in psoriasiform skin lesions in TPA-treated Hpa-tg mice. Skin tissue was obtained from TPA-treated Hpa-tg and wt mice on day 21 and stained with anti-F4/80 antibody. b F4/80-positive pixel density was quantified per 400 μm2 microscopic field, based on five fields from three independent mice of each group. p < 0.05
Supporting the ability of heparanase to facilitate activation of macrophages in the setting of psoriasis is the observation that in vitro pre-treatment with recombinant heparanase strongly sensitized mouse peritoneal macrophages to activation by IFN-γ (which is present in ample amounts in psoriatic lesions [49, 55]), as indicated by a ~ninefold increase in TNF-α secretion and ~twofold increase in IL-12/23p40 expression, compared to macrophages treated with IFN-γ alone (p < 0.01, not shown). This effect of heparanase was dependent on its enzymatic activity, since heat-inactivated heparanase did not affect macrophage response to IFN-γ.
Heparanase enzymatic activity requires proteolytic processing of 65-kDa pro-heparanase into 8- and 50-kDa subunits that form the active enzyme [56, 57]. Cathepsin L (CatL) is the predominant protease responsible for proteolytic activation of pro-heparanase [58]. Of note, upregulation of CatL was reported in several skin inflammatory disorders, with the highest CatL levels found in psoriatic lesions [59]. In agreement, immunohistochemical examination revealed increased number of CatL-positive cells both in human psoriatic skin (Supplementary Figure 2A) and in TPA-induced psoriasiform Hpa-tg skin inflammation (Supplementary Figure 2B), with a distribution pattern similar to that of macrophages (Fig. 5) and [49]. Based on the fact that macrophages represent an important source for secreted mature CatL [60] and on previously reported role of macrophage-derived CatL in activation of pro-heparanase in the mouse model of colitis [21], we assumed that a similar mode of heparanase post-translational activation may operate in the setting of psoriasis as well. Since we were unable to isolate sufficient quantities of macrophages from the mouse lesional skin, we utilized in vitro approach to investigate the heparanase-activating ability of macrophages under psoriatic conditions. As IFN-γ-mediated activation of macrophages emerges as an important event in the pathogenesis of psoriasis [4, 47, 49], we incubated purified 65-kDa heparanase precursor with medium conditioned by either resting or IFN-γ-stimulated mouse peritoneal macrophages and examined the resulting heparanase enzymatic activity. As shown in Supplementary Figure 2C, incubation of the 65-kDa pro-heparanase with medium conditioned by IFN-γ-stimulated macrophages (IFN/Mϕ+pHpa), but not by resting macrophages (Mϕ+pHpa) resulted in a marked increase in heparanase enzymatic activity.
Discussion
Our study demonstrates previously unknown role of heparanase in the pathogenesis of psoriasis. We attested keratinocytes as an important source of heparanase in human psoriatic lesions (Fig. 1) and in the mouse model of TPA-induced cutaneous inflammation (Fig. 2). Moreover, analysis of TPA-treated skin in Hpa-tg mice, characterized by constantly increased levels of heparanase in keratinocytes (among other cell types, but not in splenocytes [28]), suggests that heparanase over-expression promotes development of skin lesions that recapitulate human disease in terms of histomorphological appearance and molecular/cellular characteristics (Figs. 3, 4, 5). In fact, several mouse models (including topical application of imiquimod) are considered superior to TPA model in recapitulating human psoriatic lesions (reviewed in [61]), as TPA application in wt mice does not result in typical psoriasiform phenotype (Fig. 3e, left panel). Thus, induction of psoriasiform lesions in Hpa-tg skin following TPA treatment (Fig. 3e, right panel), along with the immunohistochemistry findings in the clinical samples (Fig. 1), convincingly implicate the enzyme in pathogenesis of psoriasis.
In addition, beneficial effect of betamethasone valerate treatment observed in this system suggests that TPA-induced Hpa-tg skin inflammation can become a useful model in testing new anti-psoriatic compounds, especially when keratinocyte-immunocyte interplay is the target.
Extensively studied for its capacity to promote cancer progression, heparanase enzyme was recently linked to several inflammatory disorders as well [20–27]. In particular, ability of heparanase to stimulate macrophage activation and direct macrophage responses toward chronic inflammatory pattern was highlighted in our studies focusing on inflammatory bowel disease [21, 62], mechanistically related to psoriasis. The exact mechanism underlying heparanase-dependent sensitization of macrophages is not fully understood, however generation of soluble HS degradation fragments (shown to stimulate TLR signaling both in vitro and in vivo [63, 64]) may play a role. This feature of heparanase (further supported by reports describing augmented macrophage activation in the presence of increased heparanase levels in a model of neointimal lesions following vascular injury [20] and in atherosclerotic plaque progression toward vulnerability [65]), could be highly relevant to the enzyme’s role in the pathogenesis of psoriasis, as well.
Although it is largely accepted that psoriasis is primarily mediated by T-cells (i.e., Th1 and, more recently, Th17 and Th22 subsets) [66–68], the cytokine pattern, particularly the dominance of TNF-α, implicates innate immunocytes (i.e., macrophages) in psoriasis pathogenesis [69]. Indeed, considerable evidence from genetic mouse models and clinical observations suggest that activation of macrophages represents an important pathogenic event in the development and maintenance of psoriatic disease [5, 49, 51, 52, 68]. Macrophage infiltration, especially at the epidermal/dermal interface, has been repetitively reported in human psoriatic lesions [5, 49, 51, 70–73]. Moreover, depletion of skin macrophages in both T cell-dependent and epithelial-based mouse models of psoriasis [48, 53], resulted in significant alleviation of the disease, attesting macrophages as important players in the pathogenesis of psoriasis [4]. Activated macrophages, recruited to the psoriatic lesions (i.e., by IFN-γ-producing T cells) represent a major (although not the sole) source of TNF-α, the key inflammation-maintaining cytokine [4, 47, 48, 53]. Interestingly, enzymatically active (but not heat-inactivated) heparanase facilitated macrophages’ response to IFN-γ in vitro (~ninefold increase in TNF-α and ~twofold increase in IL-12/23p40 vs. macrophages treated with IFN-γ alone; p < 0.01, not shown).
Importantly, processing of pro-heparanase [56, 57] by CatL is a prerequisite for its enzymatic activity [58]. CatL is upregulated in psoriasis [59] and it was shown that psoriatic skin lysates contained abundant mature enzyme, while mostly inactive CatL precursors were detected in normal skin [74]. Macrophages are important source for secreted mature CatL due to a specific mechanism that stabilizes active CatL and allows its extracellular accumulation [60]. Notably, in human lesions and in psoriasiform Hpa-tg skin we detected increased number of CatL-positive cells (Supplementary Figure 2A, B), with the spatial distribution pattern closely resembling that of macrophages (Fig. 5) and [49]. Taken together with the previously reported role of macrophage-derived CatL in heparanase activation in the mouse colitis [21], these findings led us to speculate that a similar mode of post-translational activation may be relevant in psoriasis. In support of this idea, a marked increase in heparanase enzymatic activity was observed following incubation of the latent pro-heparanase with medium conditioned by IFN-γ-stimulated (as compared to resting) macrophages (Supplementary Figure 2C).
Collectively our results reveal, for the first time, involvement of heparanase in the pathogenesis of psoriasis and suggest a role for the enzyme in facilitating abnormal interactions between immune (i.e., macrophages) and epithelial (keratinocytes) cellular subsets of the affected skin.
In addition, previously described effects of heparanase enzymatic activity (i.e., promotion of angiogenesis [75], vessel permeability [19], leukocyte extravasation and migration [11, 22], release of HS-bound cytokines anchored within the ECM [7]) can also contribute to the appearance of psoriasiform phenotype in TPA treated Hpa-tg skin.
The emerging link between heparanase activity and psoriasis implies that heparanase-targeting therapeutic approaches (which are now under intensive development/clinical testing in malignant diseases [76]) could turn highly beneficial in psoriatic patients as well. Further studies aimed at detailed understanding of heparanase-driven molecular/cellular events occurring in psoriatic skin, along with systematic analysis of heparanase inhibiting compounds in the setting of skin inflammation, are important toward development of effective therapeutic strategies in psoriasis.
References
Griffiths CE, Barker JN (2007) Pathogenesis and clinical features of psoriasis. Lancet 370:263–271
Nickoloff BJ, Qin JZ, Nestle FO (2007) Immunopathogenesis of psoriasis. Clin Rev Allergy Immunol 33:45–56
Nikaein A, Phillips C, Gilbert SC, Savino D, Silverman A et al (1991) Characterization of skin-infiltrating lymphocytes in patients with psoriasis. J Invest Dermatol 96:3–9
Clark RA, Kupper TS (2006) Misbehaving macrophages in the pathogenesis of psoriasis. J Clin Invest 116:2084–2087
Marble DJ, Gordon KB, Nickoloff BJ (2007) Targeting TNFalpha rapidly reduces density of dendritic cells and macrophages in psoriatic plaques with restoration of epidermal keratinocyte differentiation. J Dermatol Sci 48:87–101
Wagner EF, Schonthaler HB, Guinea-Viniegra J, Tschachler E (2010) Psoriasis: what we have learned from mouse models. Nat Rev Rheumatol 6:704–714
Bishop JR, Schuksz M, Esko JD (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446:1030–1037
Timpl R (1996) Macromolecular organization of basement membranes. Curr Opin Cell Biol 8:618–624
Sanderson RD (2001) Heparan sulfate proteoglycans in invasion and metastasis. Semin Cell Dev Biol 12:89–98
Timar J, Lapis K, Dudas J, Sebestyen A, Kopper L et al (2002) Proteoglycans and tumor progression: Janus-faced molecules with contradictory functions in cancer. Semin Cancer Biol 12:173–186
Parish CR (2006) The role of heparan sulphate in inflammation. Nat Rev Immunol 6:633–643
Li JP, Vlodavsky I (2009) Heparin, heparan sulfate and heparanase in inflammatory reactions. Thromb Haemost 102:823–828
Tomas D, Vucic M, Situm M, Kruslin B (2008) The expression of syndecan-1 in psoriatic epidermis. Arch Dermatol Res 300:393–395
Seyger MM, van den Born J, Schalkwijk J, van de Kerkhof PC, de Jong EM (1997) Altered distribution of heparan sulfate proteoglycans in psoriasis. Acta Derm Venereol 77:105–109
Wang L, Fuster M, Sriramarao P, Esko JD (2005) Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol 6:902–910
Axelsson J, Xu D, Kang BN, Nussbacher JK, Handel TM et al (2012) Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood 120:1742–1751
Vreys V, David G (2007) Mammalian heparanase: what is the message? J Cell Mol Med 11:427–452
Vlodavsky I, Elkin M, Abboud-Jarrous G, Levi-Adam F, Fuks L et al (2008) Heparanase: one molecule with multiple functions in cancer progression. Connect Tissue Res 49:207–210
Edovitsky E, Lerner I, Zcharia E, Peretz T, Vlodavsky I et al (2006) Role of endothelial heparanase in delayed-type hypersensitivity. Blood 107:3609–3616
Baker AB, Groothuis A, Jonas M, Ettenson DS, Shazly T et al (2009) Heparanase alters arterial structure, mechanics, and repair following endovascular stenting in mice. Circ Res 104:380–387
Lerner I, Hermano E, Zcharia E, Rodkin D, Bulvik R et al (2011) Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J Clin Invest 121:1709–1721
Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ et al (2012) The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med 18:1217–1223
Li RW, Freeman C, Yu D, Hindmarsh EJ, Tymms KE et al (2008) Dramatic regulation of heparanase activity and angiogenesis gene expression in synovium from patients with rheumatoid arthritis. Arthritis Rheum 58:1590–1600
Osterholm C, Folkersen L, Lengquist M, Ponten F, Renne T et al (2013) Increased expression of heparanase in symptomatic carotid atherosclerosis. Atherosclerosis 226:67–73
Rao G, Ding HG, Huang W, Le D, Maxhimer JB et al (2011) Reactive oxygen species mediate high glucose-induced heparanase-1 production and heparan sulphate proteoglycan degradation in human and rat endothelial cells: a potential role in the pathogenesis of atherosclerosis. Diabetologia 54:1527–1538
Waterman M, Ben-Izhak O, Eliakim R, Groisman G, Vlodavsky I et al (2007) Heparanase upregulation by colonic epithelium in inflammatory bowel disease. Mod Pathol 20:8–14
Zhang S, Qing Q, Wang Q, Xu J, Zhi F et al (2013) Syndecan-1 and heparanase: potential markers for activity evaluation and differential diagnosis of Crohn’s disease. Inflamm Bowel Dis 19:1025–1033
Zcharia E, Metzger S, Chajek-Shaul T, Aingorn H, Elkin M et al (2004) Transgenic expression of mammalian heparanase uncovers physiological functions of heparan sulfate in tissue morphogenesis, vascularization, and feeding behavior. Faseb J 18:252–263
Stanley PL, Steiner S, Havens M, Tramposch KM (1991) Mouse skin inflammation induced by multiple topical applications of 12-O-tetradecanoylphorbol-13-acetate. Skin Pharmacol 4:262–271
Petersen TK (2006) In vivo pharmacological disease models for psoriasis and atopic dermatitis in drug discovery. Basic Clin Pharmacol Toxicol 99:104–115
Alford JG, Stanley PL, Todderud G, Tramposch KM (1992) Temporal infiltration of leukocyte subsets into mouse skin inflamed with phorbol ester. Agents Actions 37:260–267
Kelly T, Miao HQ, Yang Y, Navarro E, Kussie P et al (2003) High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res 63:8749–8756
Hvid H, Teige I, Kvist PH, Svensson L, Kemp K (2008) TPA induction leads to a Th17-like response in transgenic K14/VEGF mice: a novel in vivo screening model of psoriasis. Int Immunol 20:1097–1106
van den Hoven MJ, Rops AL, Bakker MA, Aten J, Rutjes N et al (2006) Increased expression of heparanase in overt diabetic nephropathy. Kidney Int 70:2100–2108
Zcharia E, Philp D, Edovitsky E, Aingorn H, Metzger S et al (2005) Heparanase regulates murine hair growth. Am J Pathol 166:999–1008
Abou ELEM, Nagui N, Mahgoub D, El-Eishi N, Fawzy M et al (2010) Expression of cyclin D1 and p16 in psoriasis before and after phototherapy. Clin Exp Dermatol 35:781–785
Wagner P, Koch M, Nummer D, Palm S, Galindo L et al (2008) Detection and functional analysis of tumor infiltrating T-lymphocytes (TIL) in liver metastases from colorectal cancer. Ann Surg Oncol 15:2310–2317
Sano S, Chan KS, DiGiovanni J (2008) Impact of Stat3 activation upon skin biology: a dichotomy of its role between homeostasis and diseases. J Dermatol Sci 50:1–14
Cohen-Kaplan V, Jrbashyan J, Yanir Y, Naroditsky I, Ben-Izhak O, et al. (2011) Heparanase induces STAT protein phosphorylation: pre-clinical and clinical significance in head and neck cancer. J Biol Chem 287:6668–6678
Nair RP, Duffin KC, Helms C, Ding J, Stuart PE et al (2009) Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 41:199–204
Nestle FO, Conrad C (2004) The IL-12 family member p40 chain as a master switch and novel therapeutic target in psoriasis. J Invest Dermatol 123: xiv–xv
Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C et al (2007) A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med 356:580–592
Griffiths CE, Strober BE, van de Kerkhof P, Ho V, Fidelus-Gort R et al (2010) Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med 362:118–128
Parham C, Chirica M, Timans J, Vaisberg E, Travis M et al (2002) A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol 168:5699–5708
Guo D, Dunbar JD, Yang CH, Pfeffer LM, Donner DB (1998) Induction of Jak/STAT signaling by activation of the type 1 TNF receptor. J Immunol 160:2742–2750
Jarnicki A, Putoczki T, Ernst M (2010) Stat3: linking inflammation to epithelial cancer—more than a “gut” feeling? Cell Div 5:14
Wang H, Peters T, Sindrilaru A, Scharffetter-Kochanek K (2009) Key role of macrophages in the pathogenesis of CD18 hypomorphic murine model of psoriasis. J Invest Dermatol 129:1100–1114
Wang H, Peters T, Kess D, Sindrilaru A, Oreshkova T et al (2006) Activated macrophages are essential in a murine model for T cell-mediated chronic psoriasiform skin inflammation. J Clin Invest 116:2105–2114
Fuentes-Duculan J, Suarez-Farinas M, Zaba LC, Nograles KE, Pierson KC et al (2010) A subpopulation of CD163-positive macrophages is classically activated in psoriasis. J Invest Dermatol 130:2412–2422
Murray PJ, Wynn TA (2011) Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11:723–737
Gillitzer R, Wolff K, Tong D, Muller C, Yoshimura T et al (1993) MCP-1 mRNA expression in basal keratinocytes of psoriatic lesions. J Invest Dermatol 101:127–131
Nickoloff BJ (2000) Characterization of lymphocyte-dependent angiogenesis using a SCID mouse: human skin model of psoriasis. J Investig Dermatol Symp Proc 5:67–73
Stratis A, Pasparakis M, Rupec RA, Markur D, Hartmann K et al (2006) Pathogenic role for skin macrophages in a mouse model of keratinocyte-induced psoriasis-like skin inflammation. J Clin Invest 116:2094–2104
Starkey PM, Turley L, Gordon S (1987) The mouse macrophage-specific glycoprotein defined by monoclonal antibody F4/80: characterization, biosynthesis and demonstration of a rat analogue. Immunology 60:117–122
Lew W, Bowcock AM, Krueger JG (2004) Psoriasis vulgaris: cutaneous lymphoid tissue supports T-cell activation and “Type 1” inflammatory gene expression. Trends Immunol 25:295–305
Levy-Adam F, Miao HQ, Heinrikson RL, Vlodavsky I, Ilan N (2003) Heterodimer formation is essential for heparanase enzymatic activity. Biochem Biophys Res Commun 308:885–891
McKenzie E, Young K, Hircock M, Bennett J, Bhaman M et al (2003) Biochemical characterization of the active heterodimer form of human heparanase (Hpa1) protein expressed in insect cells. Biochem J 373:423–435
Abboud-Jarrous G, Atzmon R, Peretz T, Palermo C, Gadea BB et al (2008) Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J Biol Chem 283:18167–18176
Bylaite M, Moussali H, Marciukaitiene I, Ruzicka T, Walz M (2006) Expression of cathepsin L and its inhibitor hurpin in inflammatory and neoplastic skin diseases. Exp Dermatol 15:110–118
Fiebiger E, Maehr R, Villadangos J, Weber E, Erickson A et al (2002) Invariant chain controls the activity of extracellular cathepsin L. J Exp Med 196:1263–1269
Swindell WR, Johnston A, Carbajal S, Han G, Wohn C et al (2011) Genome-wide expression profiling of five mouse models identifies similarities and differences with human psoriasis. PLoS One 6:e18266
Hermano E, Lerner I, Elkin M (2012) Heparanase enzyme in chronic inflammatory bowel disease and colon cancer. Cell Mol Life Sci 15:2501–2513
Akbarshahi H, Axelsson JB, Said K, Malmstrom A, Fischer H et al (2011) TLR4 dependent heparan sulphate-induced pancreatic inflammatory response is IRF3-mediated. J Transl Med 9:219
Brunn GJ, Bungum MK, Johnson GB, Platt JL (2005) Conditional signaling by Toll-like receptor 4. Faseb J 19:872–874
Blich M, Golan A, Arvatz G, Sebbag A, Shafat I et al (2013) Macrophage activation by Heparanase is mediated by TLR-2 and TLR-4 and associates with plaque progression. Arterioscler Thromb Vasc Biol 33:56–65
Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC et al (2008) Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol 128:1207–1211
Haider AS, Lowes MA, Suarez-Farinas M, Zaba LC, Cardinale I et al (2008) Identification of cellular pathways of “type 1”, Th17 T cells, and TNF- and inducible nitric oxide synthase-producing dendritic cells in autoimmune inflammation through pharmacogenomic study of cyclosporine A in psoriasis. J Immunol 180:1913–1920
Perera GK, Di Meglio P, Nestle FO (2012) Psoriasis. Annu Rev Pathol 7:385–422
Nickoloff BJ, Karabin GD, Barker JN, Griffiths CE, Sarma V et al (1991) Cellular localization of interleukin-8 and its inducer, tumor necrosis factor-alpha in psoriasis. Am J Pathol 138:129–140
van den Oord JJ, de Wolf-Peeters C (1994) Epithelium-lining macrophages in psoriasis. Br J Dermatol 130:589–594
Djemadji-Oudjiel N, Goerdt S, Kodelja V, Schmuth M, Orfanos CE (1996) Immunohistochemical identification of type II alternatively activated dendritic macrophages (RM 3/1 + 3, MS-1+/−, 25F9-) in psoriatic dermis. Arch Dermatol Res 288:757–764
Vestergaard C, Just H, Baumgartner Nielsen J, Thestrup-Pedersen K, Deleuran M (2004) Expression of CCR2 on monocytes and macrophages in chronically inflamed skin in atopic dermatitis and psoriasis. Acta Derm Venereol 84:353–358
Boehncke WH, Wortmann S, Kaufmann R, Mielke V, Sterry W (1995) A subset of macrophages located along the basement membrane (“lining cells”) is a characteristic histopathological feature of psoriasis. Am J Dermatopathol 17:139–144
Kawada A, Hara K, Kominami E, Hiruma M, Noguchi H et al (1997) Processing of cathepsins L, B and D in psoriatic epidermis. Arch Dermatol Res 289:87–93
Elkin M, Ilan N, Ishai-Michaeli R, Friedmann Y, Papo O et al (2001) Heparanase as mediator of angiogenesis: mode of action. Faseb J 15:1661–1663
Ramani VC, Purushothaman A, Stewart MD, Thompson CA, Vlodavsky I et al (2013) The heparanase/syndecan-1 axis in cancer: mechanisms and therapies. FEBS J 280:2294–2306
Acknowledgments
This work was supported by grants from the Israel Science Foundation (grant 593/10) and the National Cancer Institute, NIH (grant RO1-CA106456). I. Vlodavsky is a Research Professor of the Israel Cancer Research Fund (ICRF).
Author information
Authors and Affiliations
Corresponding author
Additional information
I. Lerner, E. Zcharia and T. Neuman contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Lerner, I., Zcharia, E., Neuman, T. et al. Heparanase is preferentially expressed in human psoriatic lesions and induces development of psoriasiform skin inflammation in mice. Cell. Mol. Life Sci. 71, 2347–2357 (2014). https://doi.org/10.1007/s00018-013-1496-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-013-1496-9