
Abstract
Plants have developed several morphological and physiological strategies to adapt to phosphate stress. We analyzed the inducible transcripts associated with phosphate starvation and over-abundant phosphate supply to characterize the transcriptome in rice seedlings using the mRNA-Seq strategy. Fifty-three million reads obtained from 16 libraries under various phosphate stress and recovery treatments were uniquely mapped to the rice genome. Transcripts identified specifically tagged to 40,574 (root) and 39,748 (shoot) Rice Annotation Project (RAP) transcripts. Additionally, we detected uniquely 10,388 transcripts with no match to any RAP transcript. These transcripts that showed specific response to Pi stress include those without ORFs that may act as non-protein coding transcripts. With an accompanying browser of the transcriptome under Pi stress, a deeper understanding of the structural and functional features of both annotated and unannotated Pi stress-responsive transcripts can provide useful information in improving Pi acquisition and utilization in rice and other cereal crops.
Similar content being viewed by others


Introduction
Phosphorus (P) is a component of key cellular molecules such as nucleic acids, proteins, phospholipids, phytic acid, and ATP in plants. Plants absorb P almost exclusively in an inorganic phosphate form (Pi), primarily as H2PO −4 from the soil. Among the major nutrients necessary for plant growth, P is the most dilute and the least mobile in soil; hence it is often a limiting factor for crop yield. Pi starvation during farming is alleviated by the massive application of fertilizers. However, continuous usage of phosphate fertilizers may have a negative impact on the environment as the rock phosphate in the world is now in short supply and maybe depleted within the next century (Vance et al. 2003). On the other hand, massive Pi fertilization is also known to inhibit plant growth and productivity. A thorough understanding of plant response to stress due to Pi starvation (−P) as well as an over-abundant Pi supply (++P) is therefore indispensable in improving acquisition and utilization of this nutrient.
The molecular mechanisms by which plants respond and acclimate to changes in the nutritional Pi concentration are complex but of great importance and could be useful in developing strategies for elucidating the gene networks involved in plant response to various kinds of abiotic stress. The regulation system for the—P signaling pathway in plants has been proposed using studies in Arabidopsis and involved the sumoylation of the MYB transcription factor PHR1 by a small ubiquitin-like modifier SIZ1 in a process dependent on E3 ligase (Miura et al. 2005; Gojon et al. 2009). Located downstream of PHR1, miR399 and IPS1 (induced by phosphate starvation1) are specifically induced by −P stress. The miR399 reciprocally regulates the expression of PHO2/UBC24 and the resulting protein functions as an ubiquitin-conjugating E2 enzyme (Bari et al. 2006). A loss of function of PHO2/UBC24 could lead to excessive Pi accumulation in the shoot (Fujii et al. 2005). IPS1, a member of the Mt4/TPS1 family, was shown to mimic the target miR399 and offset its function (Franco-Zorrilla et al. 2007). The sequences near the miR399 complementary regions in different plant species, and all known members of the IPS1 family in Arabidopsis, are highly conserved. Of note, if Pi is supplied to plants, IPS1 transcripts rapidly disappear suggesting that they are component riburegulators of the Pi signaling system. PHR1 also upregulates many responsive genes that are expressed under −P stress, including those encoding high-affinity Pi transporters, RNases, and acid phosphatases, by binding to an imperfect palindromic sequence (P1BS, GNATATNC) in the promoter regions of these genes (Rubio et al. 2001). However, although Pi stress due to starvation has been characterized in detail, not much is known on the signaling pathway involved in Pi stress brought about by an over-abundant supply of phosphate in the soil.
Recently, the mRNA-Seq strategy using next generation sequencers has become a useful tool for analyzing genome-wide gene expression and cataloguing of all transcripts including mRNAs, non-coding RNAs and small RNAs. With a high resolution and sensitivity, the mRNA-Seq can provide detailed information on transcriptional structure of genes such as the precise location of transcription boundaries to a single-base resolution, reveal rare transcripts or variants, and identify splicing isoforms of known genes (Wang et al. 2009a). More importantly, it could accurately quantify gene expression levels over a broad dynamic range and detect transcripts expressed at either very low or very high levels including subtle changes which could not be characterized by microarray-based approaches (Li et al. 2009; He et al. 2010; Lu et al. 2010; Mizuno et al. 2010). We therefore used the massive parallel sequencing technology by mRNA-Seq to elucidate the rice transcriptome under stress due to –P and ++P in order to provide a comprehensive overview of the primary molecular events resulting from phosphate stress.
Results
Morphological changes triggered by −P and ++P stress
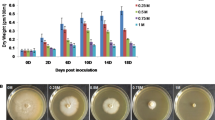
After the first 10 days in −P and ++P stress treatments, the rice seedlings began to show morphological changes in roots and shoots that may be attributed to starvation or over-abundant supply of phosphate. These changes gradually became prominent so that after 30 days of –P stress, shoot growth was retarded with thin and fewer leaves whereas the root became bristlier in texture and more brownish in color as compared with the control (Fig. 1a). Under ++P stress however, the shoot gradually showed signs of wilting with some yellowing of the leaf and relatively inhibited root growth. The total phosphorus content in −P stress-treated plants was lower than the control for both shoots and roots (Fig. 1b). On the other hand, in ++P stress-treated plants, the total P content was 1.3-fold higher in the shoots but slightly lower in the roots than the control. The dry weight of −P and ++P stress-treated plants decreased gradually to 38.4% and 45.3% of the control plants after 30 days, respectively (Fig. 1c). The shoot/root weight ratio differed in the two stress treatments after 10–15 days with a decrease in the −P stress-treated plants and an increase in ++P stress-treated plants as compared with the control (Fig. 1d). Although the roots weight did not decrease much under −P stress, there was a significant decrease in the dry weight of the roots and shoots under ++P stress. These results indicate that rice seedlings respond to stress due Pi starvation or an over-abundant Pi supply with visible changes in root and shoot morphology after 10 days of treatment.

Effect of Pi stress on rice growth. a Phenotypic changes in rice plants after 30 days of growth in culture medium with normal Pi concentration (+P, control), Pi starvation (−P), and over-abundant Pi supply (++P). Both the shoot and root showed growth retardation under Pi stress. b Total phosphorus content in roots and shoots after 30 days in +P, −P, and ++P treatment conditions. The values represent the mean ± SE for three replicates for each treatment. c, d Changes in dry weight and shoot/root weight ratio (in mg) of rice seedlings +P, −P, and ++P treatment conditions. The values represent the mean ± SE for three replicates for each treatment. Changes in dry weight and shoot/root weight ratio became prominent at around 10 days after Pi stress treatment.
Time course expression of OsIPS1 under P stress
We next analyzed the expression of OsIPS1, a Pi starvation responsive non-protein coding regulatory transcript, to confirm the validity of −P stress treatment. OsIPS1 was gradually upregulated from days 1 to 10 in both root and shoot under −P stress, after which the expression remained stable for up to 30 days (Fig. 2a). After transferring the plants to growth conditions with normal Pi concentration (+P) to allow recovery from stress, the expression was immediately downregulated within 1 day. Under ++P stress however, OsIPS1 was not upregulated in both root and shoot (Fig. 2b). This gene was also not upregulated under starvation with other essential nutrients such as nitrogen, calcium and magnesium (data not shown). Based on these results, we focused our gene expression profiling analysis by mRNA-Seq during the early growth stages particularly at 1 (early), 5 (middle), and 10 days (late) after −P and ++P stress treatments. Additionally, the expression profiles at recovery after 10 days of −P and ++P stress treatments were also investigated (Supplementary Fig. S1).

OsIPS1 expression under Pi stress treatments. The expression of OsIPS1 in root and shoot under Pi starvation (a) and over-abundant Pi supply (b) was analyzed by RT-PCR at various time points from 0 to 30 days after stress and at 1 or 2 days after recovery from stress (Re1 and Re2). The amplification of β-tubulin in RT-PCR was used as control.
Sequencing and short-read mapping
A total of 16 libraries were used for mRNA-Seq analysis using the Illumina Genome Analyzer IIx (Illumina Inc., San Diego, CA, USA). Overall, 195 million short-read sequences were produced and used for mapping onto the reference Nipponbare genome sequence (Table 1). Of the 6 to 30 million quality-evaluated reads (Passed Filtered reads) from each library, 12.3% to 39.8% were mapped to single locations (unique) in the genome whereas 23.7% to 45.7% were mapped to multiple locations in the genome. The expression level of all unique transcripts mapped onto the genome were quantified as reads per kilobase of exon model per million mapped (RPKM) values (Mortazavi et al. 2008). On the other hand, 19.0% to 51.1% of the sequence reads from each library had no match in the genome. These unmapped reads may include low-quality reads, sequencing errors, or sequences derived from adaptors and contaminating organisms (Mizuno et al. 2010).
mRNA-Seq vs. microarray analysis
We correlated the mRNA-Seq data with the gene expression profiles derived from analysis using the rice 44 K oligomicroarray platform (Agilent Technologies, Palo Alto, CA, USA). First, we re-mapped the microarray probe sequences into the updated rice genome assembly (Build 5.0 pseudomolecules) to make a valid comparison of the microarray data and the mRNA-Seq data. As a result, we confirmed that 30,026 RAP transcripts (23,113 loci) were represented as probes in the 44 K array (Fig. 3). Sequence comparison with the mRNA-Seq data showed that 28,680 transcripts from the root and 28,650 transcripts from the shoot matched with the microarray probes. This indicates that 95% of the transcripts in the 44 K microarray platform could be validated by mRNA-Seq. A total of 11,888 transcripts from the root and 11,098 transcripts from the shoot were not represented in the 44 K array but showed complete match with the annotated RAP transcripts. These transcripts correspond to gene models without EST and/or full-length cDNA support as well as those predicted based on gene prediction programs. The mRNA-Seq approach could therefore provide a more comprehensive analysis of the transcriptome including about half of RAP transcripts not represented in the microarray platform. We used the Cufflinks program for comparative assembly and estimation of abundance of transcripts in each sample (Trapnell et al. 2010). By excluding the annotated transcripts in RAP and the MSU rice gene models (http://rice.plantbiology.msu.edu/), we were able to detect a total of 8,590 transcripts from the root and 8,193 transcripts from the shoot which have not yet been previously annotated.

Transcripts identified in RAP annotations and 44 K microarray probes. Unique transcripts mapped to the genome were classified as follows: (1) transcripts with match in RAD-DB annotation and microarray, (2) transcripts with match in RAP-DB annotation but no match in microarray, and (3) unannotated transcripts identified by the Cufflinks program. In both root and shoot samples, about 95% of transcripts were supported by the microarray probes. About 50% of transcripts not identified by the array matched with the RAP-DB transcripts. A total of 10,388 unique unannotated transcripts were identified by Cufflinks program.
Gene expression profiling the 44 K microarray platform was performed in root samples at 1, 5, and 10 days after –P and ++P stress treatments. The signal intensities of 28,686 transcripts represented in the microarray were plotted against the RPKM values of corresponding transcripts (Supplementary Fig. S2). For overall gene expression, we observed an average correlation coefficient of >0.8, suggesting a clear validation of the microarray-based gene expression profiling data with the mRNA-Seq data.
Transcripts corresponding to known Pi-related genes
We were able to identify transcripts with match to previously reported Pi-related genes including those involved in sumoylation (SIZ1) and signaling (OsIPS1 and OsIPS2), transcription factors (phosphate starvation response (PHR)1 and PHR2), phosphatases (PAP2 and PAP10), etc. (Supplementary Table S1). As expected, analysis using Cufflinks also revealed the abundance and differential expression of these transcripts. The expression levels of transcripts from root and shoot samples at various Pi starvation (−P_1d, −P_5d, and −P_10d) as well as over-abundant Pi supply (++P_1d, ++P_5d, and ++P_10d) treatments based on RPKM values indicate significant expression of these Pi-related genes (Supplementary Table S2). Moreover, comparison of the fold change in RPKM values from control and treatment conditions with the normalized signal intensity clearly showed a similar pattern of expression profile obtained by mRNA-Seq and microarray-based analysis in all treatments of –P and ++P stress (Supplementary Table S3). These results indicate that the expression profiles for known Pi-related genes can be validated by mRNA-Seq.
Characterization of Pi stress-responsive transcripts
We used the G test (FDR, <0.01) on the RPKM-derived read counts to determine the differences in gene expression at various time points during −P and ++P treatments. A transcript is considered responsive if the expression level after recovery from stress (−P_Re_1d and ++P_Re_1d) reverted to the same level before treatment or the control condition. As a result, we were able to classify the expression patterns into 24 expression patterns based on the statistical significance between treatment conditions (−P and ++P) and the expression levels in root and shoot at each stage of stress designated as early for 1 day, middle for 5 days, and late for 10 days after stress treatment (Table 2). Under stress recovery conditions, the responsive transcripts which were upregulated in roots under −P stress showed similar downregulation as OsIPS1 after recovery from stress starvation. The same pattern of fast and marked recovery of expression after re-addition of Pi was also shown in Arabidopsis with >20-fold increase in AtPht1;4 and AtPAP2 signal intensity as compared with the level during P deprivation (Morcuende et al. 2007). Similarly, upregulated transcripts under ++P stress conditions were also downregulated after 1-day recovery from stress due to over-abundant Pi supply.
The highest number of responsive (upregulated and downregulated) transcripts was observed in root at late stage, which was almost twice the number in root at early stage of –P stress (Table 2; Fig. 4). The highest number of responsive transcripts was found in shoot at early stage and in root at middle stage of ++P stress. Downregulation of transcription in shoot at early ++P treatment was most common among the 24 expression patterns. The expression patterns of representative transcripts such as WRKY (Os03t0321700), Pht1;2 (Os03t0150800), and OsIPS1 (Os03t0146800) clearly show upregulation at early, middle, and late stage of −P stress, respectively (Supplementary Fig. S3).

Distribution of upregulated and downregulated transcripts in response to Pi stress. The total number of upregulated (upper) or downregulated (lower) transcripts identified by mRNA-Seq were determined at various time points of stress (early, middle, and late) during Pi starvation (−P) or phosphate over-abundant Pi supply (++P) treatments. Each bar shows the distribution of transcripts with match in RAP-DB annotation and microarray (gray), transcripts with match in RAP-DB annotation but no match in microarray (white), and unannotated transcripts (black).
The responsive transcripts identified based on G test also included approximately 900 RAP transcripts which were not represented in the microarray as well as 28 unannotated transcripts revealed by the Cufflinks program (Table 2). These unsupported RAP transcripts and unannotated transcripts were classified as novel transcripts induced during –P and ++P stress. Approximately 6.1% to 24.3% (average 13.6%) of all responsive transcripts were classified as novel transcripts in each group.
The Pi stress-responsive genes were also analyzed based on the expression of PHR1. This gene is known to upregulate −P stress-related genes such as high-affinity Pi transporters, RNases, and acid phosphatases by binding to an imperfect palindromic sequence (P1BS, GNATATNC) in the promoter regions (Rubio et al. 2001). The presence of P1BS cis-acting element in the 1-kb upstream region of Pi stress-responsive transcripts was therefore used as a measure of response under stress. Using the signal scan search in the PLACE database (http://www.dna.affrc.go.jp/PLACE/), we investigated the 1-kb upstream region of Pi stress-responsive representative RAP transcripts. The P1BS cis-acting elements were significantly overrepresented in the 1-kb upstream region of transcripts upregulated in root and shoot at late –P stress (Table 2).
Functional characterization of transcripts generated by mRNA-Seq
We used the Gene Ontology (GO) classification to estimate the functional categories of upregulated and downregulated genes in the root and shoot under various stress treatments (−P and ++P) and at different stages of stress (early, middle, and late) based on the different expression patterns (Supplementary Fig. S4). The GO categories showed specific trends for many Pi-related genes in response to –P stress (Supplementary Fig. S5) and ++P stress (Supplementary Fig. S6).
-
1.
Regulatory proteins/riboregulator: during –P stress, we observed specific pattern of response among genes for regulatory proteins/riboregulators such as OsIPS1, WRKY, SPX (SYG/PHO81/XPR1) domain-containing genes, histone, etc. OsIPS1 and OsIPS2 were strongly upregulated in root and shoot but PHO2/UBC24 was downregulated in shoot during late period of –P stress. The expression of OsIPS2 and PHO2/UBC24, which were not supported by the microarray, were confirmed here. The expression patterns of several transcription factors and signaling molecules under −P stress were clarified. In particular, WRKY (Os03t0321700) was upregulated in root during early −P stress. In Arabidopsis, AtWRKY6 and AtWRKY42 are known to modulate PHO1 transcription (Chen et al. 2009) whereas AtWRKY75 modulates Pi acquisition and root development (Devaiah et al. 2007). NAM (NAC) transcription factor (Os03t0624600) was downregulated in root at early stage of −P stress. The NAC gene family shows diverse functions in both plant development and stress responses as in the AtNAC1-mediated auxin signaling to promote lateral root development (Xie et al. 2000). Several SPX (SYG/PHO81/XPR1) domain-containing genes involved in response to environmental cues or internal regulation of nutrition homeostasis such as SPX1, SPX2, and SPX6 were upregulated only in shoot during late –P stress. In rice, SPX1 is partly involved in regulating the induction of some Pi transporters (Wang et al. 2009b). Histone H4 (Os04t0583600) and histone H2B (Os05t0574300) were upregulated in shoot during early –P stress whereas histone H1 (Os03t0799000) and histone H2B (Os01t0152900) were downregulated in shoot during late –P stress thereby supporting the chromatin-level regulation of response to Pi starvation (Smith et al. 2010). Under ++P stress treatments, OsIPS1 and OsIPS2 did not show any specific pattern of expression in either root or shoot. Downregulation of auxin-regulated transcripts such as Os03t0742900 in root at early stage, Os05t0230700 in shoot at middle stage, and Os04t0519700 (auxin response factor) in root at late stage may suggest specific functions of these genes in root and shoot growth under ++P stress. We observed the downregulation of PTF1 in shoot at early ++P stress. Overexpression of this gene however, could enhance tolerance to –P in transgenic rice (Yi et al. 2005). Among histone genes, ++P stress induced the upregulation of histone-like transcription factor (Os03t0413000) in shoot at early stage and histone H3 (Os06t0130900) in shoot at middle stage, and downregulation of histone H2B (Os01t0152900) in root at late stage.
-
2.
Metabolism (energy): many transcripts categorized as tricarboxylic acid (TCA) cycle components were induced during –P stress in both root and shoot, and most were strongly upregulated from the middle to the late stage of –P stress. The gene encoding isocitrate dehydrogenase (ICDH) was downregulated in root at the late stage of –P stress. As a gene involved in the degradation of citrate in the TCA cycle, the repression of ICDH could induce an increase in the internal citrate concentration (Delhaize et al. 2003). The gene encoding malate dehydrogenase (MDH) (Os08t0434300) was upregulated in root at late stage of –P stress. In alfalfa, overexpression of MDH caused an increase in the exudation of various organic acids and subsequently resulted in increased P accumulation in acid soils (Tesfaye et al. 2001). Among the OsA genes, the plasma membrane H+-ATPase encoding OsA7 was strongly expressed under normal Pi concentration but the expression gradually decreased during –P stress (Supplementary Table S2), a response that maybe associated to nutrient uptake and translation during Pi starvation. During ++P stress, OsA2 was upregulated in root and shoot at early stage, OsA3 was upregulated in shoot at middle stage, and OsA7 was downregulated in root at middle stage. Genes encoding glycolysis enzymes were mostly upregulated as in the case of fructose-bisphosphate aldolase (Os11t0171300) in shoot at early stage, phosphofructokinase (Os05t0524400) in shoot at middle stage, Os10t0405600 in shoot at late stage, and enolase (Os06t0136600) in shoot at middle stage of ++P stress. Enzymes involved in TCA cycle and glycolysis may function to produce organic acids, which would permit the recycling of P from phosphorylated intermediates. Organic acids are also known to help release Pi from organic or insoluble inorganic Pi compounds outside the plant.
-
3.
Metabolism (carbohydrate): many genes encoding sucrose metabolism-related proteins showed specific response in root and shoot during –P and ++P stress. This may be associated with the tightly controlled mechanisms that allow the coordination of Pi homeostasis with carbon status and photosynthesis (Wissuwa et al. 2005). Sugars generated in the shoots and transported through the phloem are involved in establishing a physiological, biochemical, and molecular response to −P stress in plants (Hammond and White 2008). Sucrose phosphatase encoding transcript (Os01t0376700) was upregulated in root at late –P stress whereas sucrose synthase encoding transcripts (Os06t0194900 and Os03t0401300) were downregulated in shoot and root at late–P stress and root at middle ++P stress.
-
4.
Metabolism (lipid): we observed specific pattern of response to Pi stress among transcripts related to lipid and fatty-acid metabolism. During the late stage of –P stress, there was prominent upregulation of lipid metabolism-related genes such as UDP-sulfoquinovose synthase 1 (SQD1), SQD2, monogalactosyldiacylglycerol (MGDG) synthases, DGD1 (digalactosyl diacylglycerol 1), DGD2, and glycerophosphoryl diester phosphodiesterase (GDPD), and transcripts encoding lipid transfer proteins (Os07t0174400 and Os03t0794000). In particular, SQD1, SQD2, and MGDG were upregulated in both root and shoot during −P stress. Lipid and fatty-acid metabolism-related genes involved in the synthesis of galacto- and sulfolipids were strongly induced by −P treatment. Lipase encoding transcripts such as Os01t0215000 and Os01t0710700 were upregulated in root at early ++P stress. Induction of these lipid metabolism-related transcripts maybe associated with changes in the lipid composition and fluidity of the membranes that control metabolic and cellular processes.
-
5.
Pi transport: among genes encoding high-affinity Pi transporters, Pht1;2 and Pht1;8 were upregulated in the roots in the middle stage whereas Pht1;3, Pht1;4, Pht1;6, Pht1;9, and Pht1;10 were upregulated in the roots in the late stage of –P stress. Although high Pi concentration is toxic to plants, the Pi transporter encoding genes were mostly upregulated during ++P stress including Pht1;4 and Pht2;1 in shoot at early stage, Pht1;8 in shoot at late stage, and Pht1;1 in root at middle stage. Only Pht1;8 was downregulated in the root at middle stage of ++P stress. These expression patterns may suggest specific functions of these genes in Pi uptake and homeostasis under conditions of over-abundant Pi supply. Among the transcripts for Pi transporters, only Pht1;11 was not detected (Supplementary Table S2) probably because it is specifically activated during mycorrhizal symbiosis (Paszkowski et al. 2002).
-
6.
Pi remobilization: many OsRNS genes showed specific expression patterns during –P and ++P stress. It has been suggested that RNS may have redundant functions or specific functions in different biological processes and tissues under different biotic and abiotic stresses (MacIntosh et al. 2010). Purple acid phosphatase 10 (PAP10) and acid phosphatase 1 (ACP1) were upregulated in both root and shoot at late –P stress, which may suggest a possible role in Pi acquisition and metabolism during –P stress.
-
7.
Morphological changes: From the middle to late stage of –P stress, genes encoding cell wall-related proteins such as pectin methylesterase 8 (Os01t0311800) and cellulose synthase (Os07t0208500) were upregulated suggesting possible roles in the control of cell wall synthesis and extensibility during stress. Upregulation during the initial response to stress suggests that plant growth was affected slightly during the first 10 days of –P stress. An auxin response factor (Os04t0519700) and an Aux/IAA binding protein (Os03t0742900) may be involved in regulating the root architecture in response to −P stress by promoting lateral root elongation and changing auxin distribution within root cells (Nacry et al. 2005). Upregulation of anthocyanin metabolism-related gene such as phenylalanine ammonia-lyase (Os12t0520200) in root at late stage of –P, may be associated with its function in protecting cells from the stress. Many genes encoding auxin-related regulatory proteins, cell wall-related proteins and cytoskeleton proteins were downregulated from the early to late stage without any morphological changes for the first 10 days during ++P stress. The genes encoding protein synthesis-related proteins such as aminoacyl-tRNA synthetase (Os03t0749300, Os05t0150900) translation initiation factor 2 (Os03t0296400) and translation elongation factor EF1B (Os06t0571400) were downregulated, suggesting a repression of protein turnover and recycling of amino acids in roots and shoots. These results also show that ++P stress is a more severe form of stress for rice growth than −P stress.
-
8.
Homeostasis of other ions: Genes encoding transport proteins such as potassium transporter (Os09t0448200), a heavy metal transporter (Os02t0530100) and an iron–sulfur cluster assembly related protein such as NifU-like protein (Os01t0662600) showed specific expression patterns that may be associated to their functions in adjusting the nutrient balance under –P stress. As Pi is important in numerous energy-requiring metabolic and transport processes, genes encoding transport proteins such as heavy metal transporter (Os07t0258400, Os01t0125600, and Os01t0976300) were upregulated during ++P stress.
Characterization of unannotated transcripts induced by −P and ++P stress
The unannotated Pi-responsive transcripts validated by G test a ranged in length from 135 to 1,750 bp with an average of 600 bp (Supplementary Table S4). These transcripts were distributed in the 12 chromosomes. The nearest adjacent transcripts were located from a distance of 1 to more than 3,000 bp. Most transcripts showed specific expression pattern in root and shoot at early, middle or late stage of −P and ++P stress. Two unannotated transcripts, namely, NP_CUFF.172640.1 and NP_CUFF.136416.1, were found to contain an intron which showed their direction based on the junction sequence. Analysis of flanking regions also revealed that most of these transcripts contain a PHR1-binding sequence (P1BS) in the 1-kb upstream or downstream region (Supplementary Table S5). Among10,388 unannotated transcripts, approximately one third (3,006 transcripts) have amino acid sequence homology in the RefSeq and UniProt databases according to BLASTX search (identity, >30%; coverage, >30%) (Supplemental Table S6). Two unannotated Pi stress-responsive transcripts showed homology; NP_CUFF.161528.1 to an unknown protein and NP_CUFF.2572.1 to a leucine-rich repeat family protein/protein kinase family protein.
Validation experiments by qRT-PCR analyses confirmed the expression of these unannotated transcripts under −P or ++P stress (Fig. 5). NP_CUFF.26799.1 was upregulated in both root and shoot at late −P stress and immediately downregulated after recovery from stress. NP_CUFF.141685.1 was upregulated in root at late ++P stress and immediately downregulated after recovery from stress. Some of these transcripts may be regulated through the P1BS in their flanking regions, although they did not appear to have high homology to one another.

Validation of responsive unannotated transcripts. The expression of OsIPS1 and several unannotated transcripts in response to –P and ++P was validated by qRT-PCR analysis. OsIPS1 is a –P upregulation index gene with statistical significance in the roots (upper) and shoots (lower) as determined by G test. Transcript expression levels were normalized using an internal control (Ubiqutin1) and plotted relative to the expression on day 10 during –P/++P and at 1 day after recovery from stress (Re1). The bars represent the mean ± SE level of transcripts from three experiments. The asterisk indicates the responsive point based on G test (FDR, <0.01). The primers used in qRT-PCR analysis for unannotated transcripts are described in Supplementary Table S5.
Discussion
Identification of responsive non-protein coding transcripts under Pi stress
We used the mRNA-seq approach to characterize the transcriptome of rice under Pi stress at high resolution. This is the first report of a whole transcriptome analysis involving stress due to Pi starvation and over-abundant Pi supply. In addition to the confirmation of almost 77% of RAP transcripts, the results also provide evidence for expression of transcripts not represented in the microarray including transcripts without FL-cDNA support, transcripts based on gene prediction programs, or those identified by homology to other plants (Fig. 3). Furthermore, the Pi stress-responsive transcripts can be functionally characterized based on the expression pattern in root and shoot at early, middle, or late stage of −P or ++P stress. A total of 10,388 transcripts identified using the Cufflinks program have not yet been previously annotated. Some of these unannotated transcripts may have been induced by alternative promoters as they have been identified in the promoter region of RAP transcripts, within the 5′ and 3′ exons supported by ESTs or FL-cDNAs, or within the extended regions of the exons. Although unannotated transcripts may also be found in the introns, we could not differentiate intron-derived transcripts from newly formed exon transcript and the mRNA-seq approach could not define whether the RNA is sense or antisense. In order to characterize intron-derived transcripts, it maybe necessary to obtain longer reads and pair-end sequence data.
We have identified a total of 10,388 unique unannotated transcripts from the root and shoot with 3,006 transcripts showing homology to various proteins (Supplementary Table S6). Among them, 28 transcripts have been found to be rice-specific and 303 transcripts showed homology to other known plant genes. The remaining unannotated transcripts (7,382) without homology to any protein may include non-protein coding transcripts, novel protein transcripts, rare transcripts that are expressed at low copies, transcripts with very low expression levels, or even transcripts which may have lethal functions in Escherichia coli. We particularly focused on the 28 unannotated transcripts, which were differentially expressed under various Pi stress treatments (Supplementary Table S4). These transcripts comprised less than 1% of the responsive transcripts and had much lower expression levels than RAP-representative transcripts for all conditions. For example, the average RPKM values (±SD) for unannotated and RAP-representative transcripts in the root control sample were 0.8 (±2.6) and 23.8 (±94.5), respectively. As these transcripts also showed small differential expression between conditions that could not be detected by statistical tests, it maybe difficult to characterize all these transcripts. Two unannotated transcripts showed protein homology to known proteins, one to leucine family gene and another to an unknown protein. Furthermore, the presence of P1BS cis-acting elements was confirmed in the promoter regions of ten unannotated transcripts. These findings establish the utility of mRNA-Seq in identifying uncharacterized transcripts that are expressed in response to Pi stress.
The expression of approximately 70% of the responsive unannotated transcripts was confirmed by qRT-PCR analysis. Several transcripts (NP_CUFF.26799.1 and NP_CUFF.141685.1) showed a sharp and reversible response to −P or ++P stress (Fig. 5). Most of these transcripts do not contain any ORFs and may function as non-coding transcripts similar to OsIPS1. Some responsive unannotated transcripts could not be validated because of very low expression levels that could not be detected by qRT-PCR. Overall, these validation experiments further suggest that mRNA-Seq can provide strong evidence of transcript expression. It has also been efficiently used in confirming the function of miRNAs such as miR399 in response to Pi stress. In Arabidopsis, the interaction between the Pi- and nitrate-limited signaling pathways affecting E3 ligase gene NLA and anthocyanin synthesis was found to be mediated by miR827, which targets the transcripts of specific proteins that contain the SPX domain (Pant et al. 2009). The rice homolog OsmiR827 exhibits different preferences in tissue expression and regulates different classes of genes sharing a common SPX domain with AtmiR827 (Lin et al. 2010). The existence of miRNA-dependent pathway in distinct aspects of Pi homeostasis in these two species may suggest a species-specific pathway where many unannotated transcripts may play significant roles. It has been reported that miR2111 may target the E3 ligase gene and a calcineurin-like phosphoesterase gene under −P stress (Hsieh et al. 2009). Additionally, miRNAs such as miR169, miR395, and miR398, which respond to various nutritional stresses, were also downregulated in response to Pi stress (Hsieh et al. 2009). Therefore, some of the unannotated transcripts detected here including non-protein coding transcripts and small RNAs may have important functions in the signaling of plant root responses to changing Pi concentrations and the coordination of homeostatic pathways of Pi and other nutrients.
Responsive genes related to morphological and physiological changes
Here, we found that –P stress in rice is associated with several morphological changes such as brown coloration, roughening of the roots and a decreased ratio of shoot-to-root weight (Fig. 1). Under ++P stress, we also observed weak roots and shoots, and an increased ratio of shoot-to-root weight with increased Pi uptake. Interestingly, although the ratio of shoot-to-root weight clearly reversed between 10 and 15 days in the –P and ++P stress-treated plants, the weights of the plants decreased concordantly as reflected in differences in gene expression between the root and shoot (Supplementary Fig. S7). These morphological and physiological changes are associated with the changes in gene expression. The differences in gene expression between −P and ++P stress in root and shoot (Supplementary Figure S7a, b) as well as the differences in gene expression between root and shoot under −P and ++P stress (Supplementary Figure S7c, d) suggest that Pi stress treatments have a significant effect on the overall expression profile of many genes involved in plant response to stress. We have confirmed the response of many previously reported phosphate-related genes. Although some of the genes that showed significant changes in expression may also be involved in the developmental stage, there is no doubt that most of the responsive genes were expressed due to Pi stress as shown by the statistical confirmation of the differences in gene expression over time during −P and ++P treatments. After recovery from stress, we also found that many genes upregulated or downregulated genes during Pi stress, including OsIPS1, reverted to the same level of expression before stress treatments.
Among the –P responsive transcripts, 316 transcripts were commonly expressed in both root and shoot. These transcripts likely function in basic –P signaling cascade that impacts Pi transport and Pi remobilization in plants, and included upregulated transcripts such as OsIPS1, OsIPS2, SQD1, OsRNS3, and PAP10. On the other hand, PHO2/UBC24, which regulates Pi allocation between roots and shoots, was downregulated in the shoots. A total of 1,336 –P responsive transcripts in the roots were not responsive to in the shoots (Supplementary Fig. S7c). These transcripts likely function in the cell-rescue system that impacts Pi uptake, carbon-assimilation reduction and root-system morphology by enhancing root growth, and included upregulated transcripts that encode many phosphate transporters (Pht1;2, Pht1;3; Pht1;6, Pht1;8, Pht1;9, and Pht1;10), an enzyme in the TCA cycle (PEPC/Os08t0366000) and an auxin response factor (Os04t0519700). It has been reported that Pht1;8 and Pht1;9 were located downstream of the PHO2/UBC24-dependent signaling pathway (Bari et al. 2006).
A comparison of the ++P responsive transcripts in root and shoot showed that 358 transcripts were commonly expressed. However, many transcripts with different functions responded in root and shoot under ++P stress. In particular, PHO2/UBC24 was upregulated in root at early ++P stress. This expression may be sufficient to repress the –P upregulated transcripts such as Pht1;8, which is downstream of the PHO2/UBC24-dependent signaling pathway. Pht1;1 and Pht1;4 were also upregulated in root during ++P, which as suggested by Bari et al. (2006) may not be a part of the PHO2/UBC24-dependent signaling pathway. There were 2,248 ++P responsive transcripts in shoot that were not responsive in root. These included upregulated genes such as Pht1;8 and Pht2;1 which may contribute to the establishment of high Pi in shoot. Pht1;8 may also be upregulated by other factors without downregulating PHO2/UBC24 transcription. Protein synthesis (Os01t0678600) and fatty-acid metabolism (Os02t0205500) transcripts were downregulated. A large number of responsive transcripts detected in shoot at early ++P stress may have diverse functions in Pi metabolism, photosynthesis and ATP biosynthesis, and may be transiently activated for plant growth. At the same time, high Pi concentrations may inhibit plant growth because the genes encoding many nuclear proteins, such as poly(A) polymerase (e.g., Os06t0558700), many protein synthesis-related proteins, such as translation initiation factor 2 (e.g., Os03t0296400) and aminoacyl-tRNA synthetases (e.g., Os06t0645400) were downregulated in shoot. The plant may then acclimate and use less energy because the number of responsive transcripts decreases from the middle to the late stage of stress in shoot. Interestingly, Pht1;4 was upregulated in root and shoot under both –P and ++P stress. This finding suggests that Pht1;4 can easily adjust to the cellular Pi condition. In Arabidopsis, it has been reported that AtPHT1;4 was induced by sucrose and is thought to be upstream of the hexokinase sugar-sensing pathways (Lejay et al. 2008). Thus, morphological changes for adaptation in rice due to changes in Pi concentration may be controlled by local and systemic gene expression.
The differences in total phosphorus content of rice plants grown under normal Pi concentration (+P), Pi starvation (−P), and over-abundant Pi supply (++P) are direct manifestations of Pi transporter activities. We identified several responsive transporter genes such as Pht1;4, Pht1;8, and Pht2;1. Genetic modification of Pi transport proteins are maybe the key in developing tolerance to both –P and ++P stress. Britto and Kronzucker (2008) reported that the high-affinity transport system is strongly downregulated under K+-replete conditions and is conversely strongly upregulated under K+-starvation conditions and that the LATS (low-affinity transport system)-range K+ influx may be increased generally by increased K+ (Britto and Kronzucker. 2008). We also observed upregulation of several low- and high-affinity transporter genes such as Pht1;1, Pht1;8, and Pht2;1 under ++P stress. Therefore, a portion of the Pi influx may be regulated by a transport system similar to that of K+ influx.
Promoter analysis and alternative splicing isoforms of responsive genes
The flanking regions of most Pi stress-responsive transcripts contain a P1BS cis-acting element which were significantly over represented in the 1-kb upstream regions of 170 transcripts (33.8%) from the roots and 74 transcripts (34.3%) from the shoots that were upregulated at the late stage of −P stress. In Arabidopsis, the direct targets of PHR1 are greatly enriched in P1BS-containing Pi starvation-induced genes (Bustos et al. 2010). Therefore, PHR1 is maybe upregulated in late stages of −P stress via a P1BS cis-acting element. Since the rest of the genes upregulated in late stage of −P stress do not have a P1BS cis-acting element in the promoter regions, other unknown cis-acting elements may also be involved in upregulation in late stage of −P stress as well as in other expressions patterns in response to Pi stress. Some transcripts may be regulated through the release from repression of PHO2/UBC24 or regulated by other factors such as PHL1. Both PHR1 and PHL1 were found to be partially redundant and have a central role in the control of physiological and molecular responses to –P stress (Bustos et al. 2010).
We searched gene loci with two or more alternative splicing isoforms supported by FL-cDNAs to determine whether alternative splicing mechanisms affect gene expression during Pi stress. Among 34,780 representative loci on the rice genome (IRGSP Build 5 Pseudomolecule), alternative splicing isoforms were confirmed in 5,625 (16.2%) representative loci. To examine changes in the usage of alternative splicing isoforms against Pi stress, the isoforms were compared in terms of Pi stress-responsive expression patterns. As a result, the alternative splicing isoforms of 194 loci (0.6%) showed different expression patterns. This suggests that the Pi stress-responsive expression is also regulated by alternative splicing mechanisms with respect to the timing of the response or tissue specificity. Further analysis of the upstream regions of responsive transcripts and accompanying changes in alternative splicing may reveal expression networks underlying Pi homeostasis in plants.
A genome-wide transcriptional analysis of Pi stress
This study has shown that RNA-Seq can be used to determine transcript expression levels more accurately and capture the transcriptome dynamics associated with Pi stress across different time points and tissues. Interestingly, SIZ1, PHR1, and PHR2, which are located upstream of OsIPS1, were slightly downregulated after 10 days under −P stress in roots and shoots and slightly upregulated at 1 d after recovery in roots, an expression pattern which was in contrast with that of OsIPS1. These genes were previously reported to be upregulated slightly under −P stress based on qRT-PCR analysis and northern analysis (Rubio et al. 2001; Miura et al. 2005). This apparent discrepancy indicates that a more detailed analysis of gene expression during different stages of stress via mRNA-Seq could be useful in detecting subtle changes in gene expression.
Using mRNA-Seq, we were able to identify many novel transcripts which could not be revealed by microarray-based technology. We were also able to characterize even subtle changes in their expression at different stages of stress under –P and ++P treatments. These results will be useful in a detailed functional analysis of an accurate gene set provided in the high-quality map-based genome sequence of the model rice cultivar Nipponbare. Towards this goal, we also constructed transcriptome viewer in GBrowse format to facilitate visualization of all uniquely mapped mRNA-Seq reads in the rice genome under Pi stress (Fig. 6; http://rnaseq-pi.dna.affrc.go.jp/cgi-bin/gb2/gbrowse/RNAseq_p/). A comprehensive rice transcriptome under Pi stress could be an indispensable tool in co-expression analysis of related genes, deciphering gene networks involved in Pi stress, and identifying genes that maybe used to enhance P efficiency and improving P acquisition, which are all critical to crop productivity.

Browser of the rice transcriptome under –P and ++P stress. The mRNA-Seq data mapped on the IRGSP Build 5 pseudomolecules can be accessed in a GBrowse format. The position of the transcript and corresponding microarray probe (if available) is indicated. For each transcript, the expression profiles of mapped reads including RAP transcripts (e.g., OsIPS1) and unannotated transcript under normal Pi concentration (control), –P and ++P stress, and recovery condition are shown as graphs.
Materials and methods
Plant materials and stress treatment
Rice (Oryza sativa ssp. japonica cv. Nipponbare) seeds were germinated and grown by hydroponic culture in nutrient media (Yoshida et al. 1976) in a growth chamber. After 14 days, the seedlings were subjected to Pi stress treatment by transferring in a similar media with the following Pi concentrations: 0 mg Pi/L (18.7 mg NaCl/L; alternatively NaH2PO4) for –P stress, and 100 mg Pi/L (439 mg KH2PO4/L; alternatively NaH2PO4) for ++P stress. The plants were maintained under Pi stress conditions for 30 days at 28°C and 70–80% humidity in a 16-h light/8-h dark cycle with the light period from 6:00 am to 10:00 pm. The plants were moved to different positions in the growth chamber several times during the light period so that all plants in each treatment were exposed to the same amount of light. Root and shoot samples were collected at approximately 9:00 am, frozen in liquid nitrogen, and stored at −80°C until subsequent analyses.
Measurement of dry weight and P uptake
Shoots and roots of seedlings under +P (control), −P, and ++P conditions were separately sampled and oven-dried at 70°C for 2 days. The dry weight of each sample was determined and the phosphorus content was analyzed by the phosphomolybdenum blue reaction using the U-0080D spectrophotometer (Hitachi, Tokyo, Japan) and Spectroquant® phosphate test kit (Merck, Darmstadt, Germany).
Time course analysis of OsIPS1 expression
The expression of OsIPS1 was analyzed by RT-PCR. Total RNA was extracted from root and shoot samples of 1–10-day-old seedlings under –P condition using Trizol® reagent (Life Technologies, Rockville, MD, USA). The RNA was treated with DNase I (Takara, Shiga, Japan), and the first-strand cDNA was synthesized using the SuperScript® III first-strand synthesis system (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The cDNAs produced by reverse transcription were amplified using a pair of gene-specific primers for each gene.
Sequencing and mapping of short reads onto the rice genome
Extraction of total RNA, construction of cDNA, and sequencing with the Illumina Genome Analyzer IIx were performed as described previously (Mizuno et al. 2010). Trimming of Illumina adaptor sequences and low-quality bases (Q < 20) at the 5′ and 3′ ends of each read was performed by the fastx_clipper program in the FASTX-Toolkit and an in-house C language program. Using the pre-processed Illumina reads, the transcript structures were reconstructed by a series of programs, namely, the Bowtie version 0.12.4 for short-read mapping (Langmead et al. 2009), TopHat ver version 1.0.13 for defining exon–intron junctions (Trapnell et al. 2009), and Cufflinks version 0.8.2 for gene structure predictions (Trapnell et al. 2010). Unannotated transcribed regions with no overlap to any known loci was analyzed by comparing known transcript structures annotated in RAP-DB (http://rapdb.dna.affrc.go.jp/) and the MSU Rice Genome Annotation Project database (http://rice.plantbiology.msu.edu/) with the Cufflinks-predicted structures. Those regions that overlapped with rRNA (1 transcript), tRNA (12 transcripts), and repetitive regions with similarity of ≥90% (3,888 transcripts) in RAP-DB were discarded, and the remaining unannotated transcripts were used for further analysis. To estimate the expression levels of each transcript, all pre-processed reads were mapped into the IRGSP Build 5 pseudomolecules (http://rgp.dna.affrc.go.jp/E/IRGSP/Build5/build5.html) by BWA (version 0.5.7) with default parameters (Li and Durbin. 2009). The expression level for each known and unannotated transcript was calculated as RPKM (Reads Per Kilobase exon Model per Million mapped reads) based on the number of uniquely mapped reads that completely overlap with the exonic regions. The resulting mRNA-Seq data were deposited in the DDBJ Sequence Read Archive (accession no. DRA000314).
Rice 44 K microarray analysis
Root RNA samples from plants under under + P (control), −P, and ++P conditions were used for gene expression profiling using the rice 44 K oligomicroarray (Agilent Technologies). Labeling, hybridization and scanning were performed according to the manufacturer’s protocol as described previously (Mizuno et al. 2010). Three biological replicates for each treatment were used for analysis. All the analysis data from microarray experiments are deposited in the NCBI Gene Expression Omnibus (accession no. GSE25647).
Validation of gene expression by qRT-PCR
Expression of unannotated transcripts was analyzed by quantitative RT-PCR using the LightCycler® 480 system (Roche, Basel, Switzerland). Total RNA from root and shoot was used for reverse transcription using the Transcriptor First-Strand synthesis kit. The reaction mixture and reaction condition was described previously (Mizuno et al. 2010). The detection threshold cycle for each reaction was normalized using Ubiquitin1 with 5′-CCAGGACAAGATGATCTGCC-3′ and 5′-AAGAAGCTGAAGCATCCAGC-3′ as primers. Three technical replicates for each treatment were used for analysis.
Analysis of responsive transcripts under Pi stress
To detect stress response transcripts from all RAP transcripts and unannotated transcripts, statistical analysis by G test was conducted between two stages of stress treatments for each transcript. The numbers of mapped reads on a given transcript and those on other regions for two stages were used as variables in 2 × 2 contingency tables for each test. All p values were corrected for a FDR (Benjamini and Hochberg. 1995) of 1% using the R package (ver 2.7.1) and in-house Perl scripts (Benjamini and Hochberg. 1995). The GO terms assigned to each transcript were obtained from RAP-DB for each GO category for biological process, molecular function, and cellular component. We also searched for the P1BS cis-acting element in the 1-kb upstream regions of Pi stress-responsive RAP-representative transcripts using the PLACE database (http://www.dna.affrc.go.jp/PLACE/) and signal scan search program (Prestridge 1991).
Construction of transcriptome viewer
We constructed a browser to show the distribution of mapped mRNA-Seq reads for each condition along with the reference genome sequence and RAP annotation in a GBrowse format
References
Bari R, Datt Pant B, Stitt M, Scheible WR. PHO2, microRNA399, and PHR1 define a phosphate-signaling pathway in plants. Plant Physiol. 2006;141:988–99.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. R Statist Soc B. 1995;57:289–300.
Britto DT, Kronzucker HJ. Cellular mechanisms of potassium transport in plants. Physiol Plantarum. 2008;133:637–50.
Bustos R, Castrillo G, Linhares F, Puga MI, Rubio V, Perez-Perez J, et al. A Central regulatory system largely controls transcriptional activation and repression responses to phosphate starvation in Arabidopsis. Plos Genet. 2010;6(9):e1001102.
Chen YF, Li LQ, Xu Q, Kong YH, Wang H, Wu WH. The WRKY6 transcription factor modulates PHOSPHATE1 expression in response to low Pi stress in Arabidopsis. Plant Cell. 2009;21:3554–66.
Delhaize E, Ryan PR, Hocking PJ, Richardson AE. Effects of altered citrate synthase and isocitrate dehydrogenase expression on internal citrate concentrations and citrate efflux from tobacco (Nicotiana tabacum L.) roots. Plant Soil. 2003;248:137–44.
Devaiah BN, Karthikeyan AS, Raghothama KG. WRKY75 transcription factor is a modulator of phosphate acquisition and root development in Arabidopsis. Plant Physiol. 2007;143:1789–801.
Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, et al. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet. 2007;39:1033–37.
Fujii H, Chiou TJ, Lin SI, Aung K, Zhu JK. A miRNA involved in phosphatestarvation response in Arabidopsis. Curr Biol. 2005;15:2038–43.
Gojon A, Nacry P, Davidian JC. Root uptake regulation: a central process for NPS homeostasis in plants. Curr Opin Plant Biol. 2009;12:328–38.
Hammond JP, White PJ. Sucrose transport in the phloem: integrating root responses to phosphorus starvation. J Exp Bot. 2008;59:93–109.
He GM, Zhu XP, Elling AA, Chen LB, Wang XF, Guo L, et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell. 2010;22:17–33.
Hsieh LC, Lin SI, Shih ACC, Chen JW, Lin WY, Tseng CY, et al. Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Physiol. 2009;151:2120–32.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25.
Lejay L, Wirth J, Pervent M, Cross JMF, Tillard P, Gojon A. Oxidative pentose phosphate pathway-dependent sugar sensing as a mechanism for regulation of root ion transporters by photosynthesis. Plant Physiol. 2008;146:2036–53.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li LH, Qiu XH, Li XH, Wang SP, Lian XM. The expression profile of genes in rice roots under low phosphorus stress. Sci China Ser C. 2009;52:1055–64.
Lin SI, Chiou TJ, Santi C, Jobet E, Lacut E, El Kholti N, et al. Complex regulation of two target genes encoding SPX-MFS proteins by rice miR827 in response to phosphate starvation. Plant Cell Physiol. 2010;51:2119–31.
Lu TT, Lu GJ, Fan DL, Zhu CR, Li W, Zhao QA, et al. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res. 2010;20:1238–49.
MacIntosh GC, Hillwig MS, Meyer A, Flagel L. RNase T2 genes from rice and the evolution of secretory ribonucleases in plants. Mol Genet Genomics. 2010;283:381–96.
Miura K, Rus A, Sharkhuu A, Yokoi S, Karthikeyan AS, Raghothama KG, et al. The Arabidopsis SUMO E3 ligase SIZ1 controls phosphate deficiency responses. Proc Natl Acad Sci USA. 2005;102:7760–5.
Mizuno H, Kawahara Y, Sakai H, Kanamori H, Wakimoto H, Yamagata H, et al. Massive parallel sequencing of mRNA in identification of unannotated salinity stress-inducible transcripts in rice (Oryza sativa L.). BMC Genomics. 2010;11:683.
Morcuende R, Bari R, Gibon Y, Zheng W, Pant BD, Blasing O, et al. Genome-wide reprogramming of metabolism and regulatory networks of Arabidopsis in response to phosphorus. Plant Cell Environ. 2007;30:85–112.
Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8.
Nacry P, Canivenc G, Muller B, Azmi A, Van Onckelen H, Rossignol M, et al. A role for auxin redistribution in the responses of the root system architecture to phosphate starvation in Arabidopsis. Plant Physiol. 2005;138:2061–74.
Pant BD, Musialak-Lange M, Nuc P, May P, Buhtz A, Kehr J, et al. Identification of nutrient-responsive Arabidopsis and rapeseed microRNAs by comprehensive real-time polymerase chain reaction profiling and small RNA sequencing. Plant Physiol. 2009;150:1541–55.
Paszkowski U, Kroken S, Roux C, Briggs SP. Rice phosphate transporters include an evolutionarily divergent gene specifically activated in arbuscular mycorrhizal symbiosis. Proc Natl Acad Sci USA. 2002;99:13324–9.
Prestridge DS. SIGNAL SCAN: a computer program that scans DNA sequences for eukaryotic transcriptional elements. Comput Appl Biosci. 1991;7:203–6.
Rubio V, Linhares F, Solano R, Martin AC, Iglesias J, Leyva A, et al. A conserved MYB transcription factor involved in phosphate starvation signaling both in vascular plants and in unicellular algae. Gene Dev. 2001;15:2122–33.
Smith AP, Jain A, Deal RB, Nagarajan VK, Poling MD, Raghothama KG, et al. Histone H2A.Z regulates the expression of several classes of phosphate starvation response genes but not as a transcriptional activator. Plant Physiol. 2010;152:217–25.
Tesfaye M, Temple SJ, Allan DL, Vance CP, Samac DA. Overexpression of malate dehydrogenase in transgenic alfalfa enhances organic acid synthesis and confers tolerance to aluminum. Plant Physiol. 2001;127:1836–44.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–U174.
Vance CP, Uhde-Stone C, Allan DL. Phosphorus acquisition and use: critical adaptations by plants for securing a nonrenewable resource. New Phytol. 2003;157:423–47.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009a;10:57–63.
Wang C, Ying S, Huang H, Li K, Wu P, Shou H. Involvement of OsSPX1 in phosphate homeostasis in rice. Plant J. 2009b;57:895–904.
Wissuwa M, Gamat G, Ismail AM. Is root growth under phosphorus deficiency affected by source or sink limitations? J Exp Bot. 2005;56:1943–50.
Xie Q, Frugis G, Colgan D, Chua NH. Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Gene Dev. 2000;14:3024–36.
Yi K, Wu Z, Zhou J, Du L, Guo L, Wu Y, et al. OsPTF1, a novel transcription factor involved in tolerance to phosphate starvation in rice. Plant Physiol. 2005;138:2087–96.
Yoshida S, Forno AD, Cock HJ, Gomez AK. Laboratory manual for physiological studies of rice. 3rd ed. Manila: International Rice Research Institute; 1976.
Acknowledgments
We thank Ms. F. Aota, Ms. K. Ohtsu, and Ms. K. Yamada for technical assistance, and Dr. Y. Nagamura and Ms. R. Motoyama for assistance with the microarray experiments. This work was supported by the Ministry of Agriculture, Forestry and Fisheries of Japan (Genomics for Agricultural Innovation, RTR-0001).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.

Supplementary Fig. S1
{kind=link}
Overview of Pi stress treatments of rice seedlings for transcriptome analysis by mRNA-seq. Seeds were germinated and grown in Yoshida’ nutrient medium. After 2 weeks, the seedlings were subjected to Pi starvation (−P) and over-abundant Pi supply (++P) stress treatments. Root and shoot samples from 8 plants were collected at 1, 5, and 10 days after stress treatments, and 1 day recovery after 10 days of stress and used for construction of mRNA libraries for mRNA-Seq analysis. Microarray-based transcriptome analysis was performed using root samples in + P (control), −P (1, 5, and 10 days), and ++P (1, 5, 10 days) treatments in three biological replicates. (JPEG 329 kb)

Supplementary Fig. S2
{kind=link}
Quantification of gene expression by mRNA-Seq and microarray analysis in roots under −P and ++P stresses after 1, 5, and 10 days. Each scatter plot shows RPKM in mRNA-Seq and the signal intensity (average of three biological replicates) for the microarray of the roots. The abscissa shows the RPKM (log2) of each treatment; the ordinate shows the signal intensity (log2) of each treatment. In all treatments A, high-correlation coefficient (>0.8) was obtained in all treatments. (JPEG 506 kb)

Supplementary Fig. S3
{kind=link}
Expression patterns of representative transcripts in each of the 24 expression groups. The expression patterns were grouped based on significant differences in expression at different periods of stress as determined by G test (FDR, <0.01). The responsive point is indicated by an asterisk. (JPEG 1050 kb)

Supplementary Fig. S4
{kind=link}
Distribution of GO categories (biological processes) for Pi-responsive genes as categorized into 24 expression patterns. (JPEG 1076 kb)

Supplementary Fig. S5
{kind=link}
Expression pattern of –P responsive transcripts identified in root and shoot at different stages of stress. Upregulated and downregulated genes are indicated in red and blue, respectively. (JPEG 1777 kb)

Supplementary Fig. S6
{kind=link}
Expression pattern of ++P responsive transcripts identified in root and shoot at different stages of stress. Upregulated and downregulated genes are indicated in red and blue, respectively. (JPEG 2036 kb)

Supplementary Fig. S7
{kind=link}
Venn diagrams showing the expression specificity of Pi stress-responsive genes based on common expression in (a) −P and ++P stress in root, (b) −P and ++P stress in shoot, (c) root and shoot under –P stress, and (d) root and shoot under ++P stress. Representative transcripts are shown in the graph. Upregulated and downregulated genes are indicated in red and blue, respectively. (JPEG 851 kb)
ESM 1
(XLS 654 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Oono, Y., Kawahara, Y., Kanamori, H. et al. mRNA-Seq Reveals a Comprehensive Transcriptome Profile of Rice under Phosphate Stress. Rice 4, 50–65 (2011). https://doi.org/10.1007/s12284-011-9064-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12284-011-9064-0