
Abstract
Introduction
Monoamine-based antidepressants inhibit neurotransmitter reuptake within short time. However, it commonly takes several weeks until clinical symptoms start to resolve—indicating the involvement of effects distant from reuptake inhibition.
Objective
To unravel other mechanisms involved in drug action, a “reverse” pharmacological approach was applied to determine antidepressant-induced alterations of hippocampal gene expression.
Materials and methods
The behavioral response to long-term paroxetine administration of male DBA/2Ola mice was assessed by the forced swim test (FST), the modified hole board (mHB), and the dark/light box. Hippocampi of test-naive mice were dissected, and changes in gene expression by paroxetine treatment were investigated by means of microarray technology.
Results and discussion
Robust effects of paroxetine on passive stress-coping behavior in the FST were observed. Furthermore, anxiolytic properties of long-term antidepressant treatment could be identified in DBA mice in both, the mHB and dark/light box. Analysis of microarray results revealed a list of 60 genes differentially regulated by chronic paroxetine treatment. Preproenkephalin 1 and inhibin beta-A showed the highest level of transcriptional change. Furthermore, a number of candidates involved in neuroplasticity/neurogenesis emerged (e.g., Bdnf, Gfap, Vim, Sox11, Egr1, Stat3). Seven selected candidates were confirmed by in situ hybridization. Additional immunofluorescence colocalization studies of GFAP and vimentin showed more positive cells to be detected in long-term paroxetine-treated DBA mice.
Conclusion
Candidate genes identified in the current study using a mouse strain validated for its responsiveness to long-term paroxetine treatment add, in our opinion, to unraveling the mechanism of action of paroxetine as a representative for SSRIs.
Similar content being viewed by others


Introduction
Monoamine-based antidepressants enhance neurotransmission by various mechanisms within a short time, while resolution of depressive symptomatology takes several weeks, sometimes months. This discrepancy in time course suggests that the well-characterized initial mechanisms trigger a number of adaptive responses that finally allow for incremental improvements of psychopathology. For example, under naturalistic clinical conditions, only about 35% of depressed patients achieve full clinical remission within 8−12 weeks of treatment with citalopram, a selective serotonin reuptake inhibitor (Trivedi et al. 2006). Even after sequential treatments with alternatives, including augmentation strategies, a considerable percentage of the patients fail to remit (Fava and Davidson 1996; Carvalho et al. 2007). Apparently, the various antidepressants induce cellular mechanisms beyond monoamine receptors, and successive time-consuming changes in neuronal and glial circuitries determine the clinical effects of an antidepressant medication (Nestler et al. 2002). Over the last years, the search for novel antidepressants that act more focused followed two complementary strategies: (1) accumulated neuroscience findings were translated into focused targets, e.g., the well founded stress-hormone (glucocorticoids, corticotrophin-releasing hormone CRH) excess among depressive patients was translated into drugs that block glucocorticoid and CRH receptors (Holsboer and Ising 2008); and (2) unbiased approaches were employed where human genetic studies contrasting depressed patients with healthy controls identified genetic variations that, when associated with gain or loss of gene function, may serve as novel targets (Craddock and Forty 2006).
The latter approach emerged from decoding the human genome and was long considered to be the gateway to personalized medicine where patients are treated according to their genetic makeup. Some interesting novel drug targets came up by these strategies (e.g. Lucae et al. 2006; McQuilin et al. 2008), but overall, the success in using genotype information for identification of valid drug targets is limited so far. The main reason for this disenchantment is that the genotype provides little information about changes in gene activity following a cellular challenge. Environmental factors ranging from early childhood trauma to previous diseases, drug exposure, acute life events, or nutrition, may all impact upon signal-induced changes in gene activity. Such epigenetic modulations can be reversible, enduring, or even transgenerational (e.g., Tsankova et al. 2007; Mill and Petronis 2007). The introduction of microarrays, a technique that profiles levels of many thousand mRNA transcripts, was embraced because mRNA expression levels are influenced by both, genetic polymorphisms and environmental factors. A major drawback for studies of human depression is the inaccessibility of diseased tissue and the limitation imposed on extrapolations from expression profiles in peripheral cells (e.g., lymphocytes) to neurons or glial cells in defined brain areas (Sullivan et al. 2006). Therefore, gene expression profiles relied on post mortem studies where defined brain areas of affected individuals were contrasted with those of non-affected controls (e.g., Mirnics et al. 2001; Sequeira et al. 2007). Studies interrogating the effects of drugs on gene activity in selected brain areas cannot use such a strategy because the effects of a given drug and the underlying disease cannot be differentiated with certainty. Therefore, studies aiming at discovery of genes that are regulated by antidepressants mainly rely on rodent brains (e.g., Conti et al. 2007; Nakatani et al. 2004; Wong and Licinio 2004; Yamada et al. 2005), as genetically homogenous animals can be used, and most confounding factors can be controlled.
Validation of genes emerging from microarray analysis is a major challenge and might benefit from the generation or the use of already existing knockout models. Despite this fact, pharmacogenomic studies rarely have been conducted in mice. In the current study, we used a DBA/2Ola (DBA/2) mouse line as we hypothesized that antidepressant-induced changes in gene expression are more relevant if (1) the animal model exhibits already a phenotype resembling some signs and symptoms of the human disease, and (2) these features are responsive to the kind of drugs under study. The chosen DBA/2 strain of mice shows a high level of anxiety-like behavior (Yilmazer-Hanke et al. 2003; Ohl et al. 2003). Anxiolytic properties of chronic antidepressant treatment have been reported in mice (Mirza et al. 2007) and human patients (Pillay and Stein 2007). Furthermore, the DBA/2Ola mice display a reduced inhibitory HPA axis feedback compared to another inbred mouse strain (Thoeringer et al. 2007). This neuroendocrine impairment is a characteristic feature among patients with depression (for review see Holsboer 2001).
The present microarray analysis focused on the hippocampus, a brain area implicated in the neuropathology and psychopathology of affective disorders (Campbell and Macqueen 2004; Engin and Treit 2007). Moreover, the hippocampus displays morphological alterations in depressed patients that can be influenced by antidepressant treatment (Duman and Monteggia 2006). We treated DBA/2 mice with paroxetine or vehicle by oral application and examined paroxetine-induced behavioral changes in parallel with changes of hippocampal gene expression. Seven candidate genes that emerged from the microarray experiment were chosen and confirmed by in situ hybridization. Finally, we used immunofluorescence colocalization studies to support that some of the paroxetine-induced changes in gene activity affect genes that are involved in neurogenesis and supposedly also in neuronal plasticity.
Materials and methods
Subjects
Male DBA/2OlaHsd mice (Harlan Winkelmann, Germany) aged 8−9 weeks at arrival were kept under standard laboratory conditions (21 ± 2°C, 45−55% humidity, 12:12 h light cycle, with lights on at 7:00 a.m.) with food and water available ad libitum. The animals were housed singly in standard cages (30 × 20 × 14 cm), and treatment started after a habituation period of 2 weeks after arrival. Animal experiments were performed in accordance with the NIH Guide for the Care and Use of Mammals in Neuroscience and Behavioural Research and the Guide for the Care and Use of Laboratory Animals of the Government of Bavaria, Germany.
Treatment
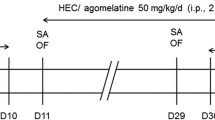
Animals were randomly distributed to the treatment group vehicle or paroxetine (10 mg/kg). Paroxetine was prepared freshly before treatment by diluting it in tap water (1 mg/ml), and before each application, the preparation was thoroughly mixed. Drug or vehicle was given orally by gavaging twice per day, i.e., in the morning between 9:00 and 10:00 and in the afternoon between 6:00 and 7:00 p.m., over a treatment period of 28 days. On day 29, the animals were treated in the morning, and depending on their assignment to the various experimental groups, behavioral tests were performed. Subsequent to testing, animals were euthanized by an overdose of isofluran, and the brains were taken. The brains of test-naïve mice were processed for microarray experiments or in situ hybridization /immunohistochemistry.
Behavioral testing
Analysis of behavioral effects of the chronic paroxetine application was done in different experimental groups (1−3, see below) of test-naïve animals. The forced swim test (FST) was applied, as it is a commonly used test to assess antidepressant-like properties of compounds (Cryan and Holmes 2005; Porsolt et al. 1977). We used the modified hole board test (mHB) in order to obtain a comprehensive overview on behavioral changes under mild stressful conditions. The dark/light box was used to specifically address drug-induced changes in anxiety-like behavior. The FST and testing in the dark/light box were performed 4 h after last application in order to confirm the behavioral effects of the treatment at the same time point when the brains of test-naïve-treated mice were taken for microarray analysis. The mHB was performed 30 min after last application as many published results on behavioral effects of antidepressants were assessed at this time point (e.g., Lucki et al. 2001; David et al. 2003; Crowley et al. 2005).
During the 5-min test periods, behavior of the animals was recorded by means of a video camera, and a trained observer directly scored the respective parameters. Data were statistically analysed by Mann–Whitney U tests.
Experimental group 1 (FST)
FST1 was performed on day 29 of treatment (12:30−2:00 p.m.), 4 h after the oral treatment with either vehicle (n = 7) or paroxetine (n = 7). Each animal was placed into a beaker (diameter 12 cm, height 24 cm) filled with water (temperature 25−26°C) to a height of 12 cm for a test period of 5 min. The parameters struggling (vigorous attempts to escape), swimming, and floating (immobile position with only small movements to keep balance) were scored by a trained observer blind to the treatment. As we were interested in the effects of a first FST exposure on the stress-coping strategy, all animals were retested the next day (24 h after the FST1). Animals received either vehicle or paroxetine at the evening of day 29 and in the morning of day 30, 4 h before they were exposed to the second FST.
Experimental group 2 (mHB)
The mHB (for details, see Ohl et al. 2001) was performed on treatment day 29 (9:30–11:20 a.m.), 30 min after oral treatment with vehicle (n = 7) or paroxetine (n = 5, as two animals of the paroxetine group were stopped to be treated after they showed a bodyweight loss of around 20% at weeks 3–4). The day before the experiment, each animal received three oak flakes for habituation as an oak flake was used as familiar food in the test; a piece of almond represented the novel food and was placed together with the oak flake in one corner of the test apparatus. The animals were placed into the outer area of the test apparatus, facing the board. During the 5-min test period, the following behaviors were scored: line crossings, stretched attends, rearing, self-grooming, board visits, hole exploration, exploration and intake of familiar and novel food, and defecation.
Experimental group 3 (dark/light box)
Testing in the dark/light box was performed on treatment day 29 (12:30−2:00 p.m.), 4 h after oral application of vehicle (n = 7) or paroxetine (n = 7). Each animal was placed in the dark compartment (15 × 20 × 25 cm) of the test apparatus, facing the tunnel (4 × 7 × 10 cm) connecting the dark to the brightly lit (680–700 lx, 30 × 20 × 25 cm) compartment (for details see Timpl et al. 1998). During the 5-min test period, the time spent in each compartment (dark, tunnel, and lit), the latency until the first full entry (with four paws), and the number of full entries into the lit compartment were assessed.
Brain area dissection and RNA isolation
Experimental group 4
Animals were treated with either vehicle (n = 10) or paroxetine (n = 10) in parallel to the animals of experimental group 1 (FST), and 4 h after the last oral treatment, the animals were euthanized by an overdose of isofluran. The brains were removed, and five brains of each treatment were immediately frozen and stored at −80°C until sectioning for validation purpose (in situ hybridization or immunohistochemistry, see below). The hippocampi of the remaining five brains of each treatment were dissected, weighed, and immediately processed.
Total RNA was extracted with 300 µl of TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The tissue was homogenized by using a pipette, 1 µl linear Acrylamid (Ambion, Huntington, UK), and 60 µl chloroform (Merck, Darmstadt, Germany) was added to the homogenate which subsequently was centrifuged for 5 min at 13,000 rpm. The aqueous phase was decanted, and an equal volume of isopropanol (∼180 µl) was added to this aliquot. The mixture was allowed to precipitate for 30 min at −80°C. The precipitate was collected by centrifugation at 13,000 rpm for 30 min. The pellet was washed with ethanol (70%), briefly centrifuged, air dried, and afterwards resuspended in 11 µl Ampuwa water (Fresenius, Bad Homburg, Germany).
Gene expression profiling
Total RNA obtained from hippocampi of individuals of the treatment groups vehicle (n = 5) and paroxetine (n = 5) was apportioned and used on three different microarray platforms (Affymetrix, Codelink, MPIP) to analyze treatment-induced changes in gene expression in hippocampi of DBA/2 mice.
Affymetrix mouse MOE430A GeneChips (oligonucleotide-based) and Codelink Mouse Uniset I BioArrays (oligonucleotide-based) were purchased from the manufacturer. In-house mouse cDNA-based microarrays were printed with the Chipwriter Pro (Biorad) on Corning GAPS II glass slides using BMAP (brain mouse anatomy project) and MMSV (Mus musculus sequence verified) libraries (Research Genetics) as well as 6,000 cDNA clones obtained from the RZPD resource center (http://www.rzpd.de). Additional control and reference cDNAs were included resulting in a total of 19,048 cDNAs representing 12,847 unique unigene clusters (based on unigene build no. 150). Using Unigene annotation, we found a total of 4,862 genes represented on all three platforms.
For the MPIP microarrays, all bacterial clones were amplified by polymerase chain reaction (PCR), purified, and verified by gel electrophoresis. The spotting buffer contained 3× standard saline citrate (SSC) and 1.5 M Betaine. After spotting, the arrays where heated to 80°C for 10 s and crosslinked using a UV Stratalinker 2400 at 60 mJ (Stratagene). Before blocking, the slides were dried at 65°C for 30 min. Immediately after this, the slides were incubated in blocking solution (50% Formamide, 5× SSC, 0.1% sodium dodecyl sulfate (SDS), 0.1 mg/ml bovine serum albumin) at 42°C for 1 h. The blocking was followed by shortly shaking the slides in H2O, then in isopropanol. The arrays were dried by centrifugation and stored at room temperature.
Probe construction, microarray hyridization, and data acquisition
For the Affymetrix GeneChips, 10 µg of total RNA of each individual were amplified and labeled using the Megascript T7 kit (Ambion). For each animal of the vehicle (n = 5) or paroxetine (n = 5) group, 5 µg of the biotin-labeled fragmented aRNA were hybridized to a single MOE430A GeneChip. Subsequent steps were performed according to the manufacturer’s protocol.
Correspondingly, ten BioArrays from Codelink were hybridized with non-pooled biotin-labeled aRNA obtained by amplification of 5 µg total RNA per mouse using the manufacturer’s protocols and reagents.
The cDNA microarray (MPIP) experiment was performed on group-wise pooled material, using dual color (Cy3/Cy5) design with direct sample comparison of indirectly labeled aRNA, including dye swapping. For this, 2 µg of total RNA of each individual were amplified, labeled and cleaned-up using Ambion’s MessageAmp Aminoallyl mRNA amplification and labeling kit and Amersham’s Cy3 and Cy5 monoreactive dyes. Ten cDNA microarrays were hybridized with pools of aRNA from experimental and control animals, respectively. Hybridization was carried out for 16 h at 50°C under a coverslip using 5 µg of labeled aRNA per dye and a hybe buffer containing 50% Formamide, 5× SSC, 0.1% SDS, 0.1 mg/ml mouse COT1-DNA, and 5 µg Poly(dA) per array. Slides were washed for 5 min at 42°C with 2× SSC/0,1% SDS, followed by a wash at RT with 0.1× SSC/0.1% SDS for 10 min and a third washing step at RT with 0.1× SSC for 1 min. The arrays were rinsed in 0.01× SSC for up to 10 s and dried by centrifugation. Fluorescent array images were acquired using a ScanArray 4000 scanner (Perkin Elmer; ScanArray Version 3.1) with 87% Laser power (Cy3), 70% Laser power (Cy5), and PMT gain 80% (Cy3, Cy5). Data were quantified using Quantarray Software (Perkin Elmer; Version 2.1.0.0) with fixed circle mode and transferred into a PostgreSQL database for further processing. Following identification of significantly regulated genes, the respective spots on the arrays were visually inspected (see Supplementary Table S1).
Data analysis
Affymetrix data were analyzed using the DNA-Chip Analyzer Software (dCHIP, Version: Sep 15, 2005) which is suitable for analysis of multiple GeneChips (Li and Wong 2001; http://www.dCHIP.org). Using outlier images, one chip (vehicle treated) was identified as problematic and was therefore removed from further analysis. The following criteria for detection of differentially expressed genes were chosen: perfect match only model, no log transformation, Experiment/Baseline > 1.35 or Baseline/Experiment > 1.35; Present call percent of Baseline ≥ 75; Present call percent of Experiment ≥ 75; p value ≤ 0.02; not using the lower 90% confidence bound of fold-change criterium. The p value was chosen in a way that the false discovery rate (FDR), calculated using 1,000 permutations, constituted 12.5%.
Codelink data were normalized by median centring using Amersham’s analysis software. Further analysis was performed using Significance Analysis of Microarrays (SAM, Version 2.10; Tusher et al. 2001). For this, present/absent calls were generated analogous to the Affymetrix system. A feature was assigned “present”, if quality was “good” and above_thr was “true”; all other features were assigned “absent”. The following settings were chosen for the analysis: response type: 2 class, unpaired; unlogged data; using T statistic; median centring; 1,000 permutations; automatic s0 factor estimation; delta: 1.0; false discovery rate: 12.13; Present call percent of Baseline ≥ 75; Present call percent of Experiment ≥ 75; with no fold change hurdle (see Larsson et al. 2005). Using an additional absolute fold change threshold of 1.35, 16 genes could be detected as differentially expressed.
Raw data from MPIP cDNA microarrays were normalized according to the procedure outlined in Yang et al. (2002) and subjected to a t test for detection of differential expression. The obtained p values were corrected for multiple testing using Benjamini–Hochberg’s FDR procedure (Benjamini and Hochberg 1995). Genes were considered as differentially expressed when the FDR was <0.1, and the absolute fold change was >1.35.
In situ hybridization
Radioactive in situ hybridization experiments were performed according to standard protocols (Droste et al. 2003). Probes were generated using templates created by PCR from sequence verified clones of the microarray cDNA library (Penk1: M13227; Bdnf: AI854196; Gfap: AI836096; Sox11: AI836553; Vim: AI845820; Calb1: AI452326; Tex261: AI843892). Tissue slices of corresponding brain areas from vehicle and antidepressant-treated animals were positioned on the same slide to ensure identical processing conditions.
Immunohistochemistry
Colocalization studies of GFAP and vimentin were performed on 4% paraformaldehyde-fixed tissue sections prepared from frozen mouse brains using the primary antibodies rabbit-anti-GFAP (1:250, #Z0334, DakoCytomation) and mouse-anti-VIM (1:50, #V5255, Sigma). Secondary antibodies used were Alexa Fluor 488 goat-anti-rabbit (1:500, Molecular Probes) and biotinylated goat-anti-mouse IgG (1:300, VectorLabs #BA-9200) in combination with Texas Red Avidin D (1:60) from Vector Fluorescent Avidin Kit (VectorLabs #A-1100). Sections were counterstained with bis-Benzimide (Sigma, #B2883).
Results
Forced swim test
Mice treated chronically with paroxetine compared to vehicle-treated mice showed a significant reduction in time floating (FST1: p = 0.001; FST2: p = 0.004). Time Struggling was increased by paroxetine but did not reach the significance level (FST1: p = 0.2; FST2: p = 0.16), and time swimming was slightly enhanced in FST1 (p = 0.2) and significantly enhanced in FST2 (p = 0.001; Fig. 1).

Behavioral effects of chronic paroxetine treatment in DBA/2Ola mice assessed by the forced swim test (FST1, a) on day 29 of treatment, 4 h after last application. The second FST (FST2, b) was performed 24 h after FST1 and 4 h after application of paroxetine. (grey bars: vehicle, n = 7; black bars: paroxetine 10 mg/kg, twice per day, n = 7), *p < 0.05
A second exposure to the FST induced an increase in passive floating behavior in both treatment groups, but a significant change was only observed in the animals treated with vehicle (comparison time floating FST1 to FST2 within vehicle group: p = 0.01).
Modified hole board
In the mHB test, parameters indicating general activity/exploration were not affected by the chronic paroxetine treatment (line crossings: p = 0.4, number of rearings: p = 0.6). The paroxetine group showed a significant reduction in risk assessment behavior, i.e., the number of stretched attends was lower (p = 0.04). The animals showed a trend to spend more time in the exposed area (percent time on board: p = 0.1); the latency until first entry on board was only slightly reduced (p = 0.2). The latencies for the intake of familiar or novel food were decreased but did not reach significance level (p = 0.1; p = 0.06). Furthermore, the latency until the animals showed grooming behavior and the number of boli were nonsignificantly reduced in the paroxetine group (Fig. 2).

Behavioral effects of chronic paroxetine treatment in DBA/2Ola mice assessed by the modified hole board on day 29 of treatment, 30 min after last application (grey bars: vehicle, n = 7; black bars: paroxetine 10 mg/kg, twice per day, n = 5), *p < 0.05
Dark/light box
Changes in anxiety-like behavior observed in the mHB test could be confirmed by use of a more specific anxiety test, the dark/light box. Animals chronically treated with paroxetine made significantly more entries into the lit compartment of the apparatus (percent entries: p = 0.012), spent more time in it (percent time: p = 0.014), and the latency until the first entry was lower (p = 0.01). Furthermore, it was observed that paroxetine-treated animals spent more time in the connecting tunnel between the dark and lit compartment (p = 0.004), and they displayed an overall increase in the number of entries into either the dark or the lit compartment (p = 0.005), the latter is interpreted as a result of behavioral disinhibition (Fig. 3).

Behavioral effects of chronic oral paroxetine treatment in DBA/2Ola mice assessed by the dark/light box on day 29 of treatment, 4 h min after last application (grey bars: vehicle, n = 7; black bars: paroxetine 10 mg/kg, twice per day, n = 7), *p < 0.05
Microarray results
According to the criteria selected for the analysis of the data and the detection of significant gene regulation (see “Materials and methods” section) by the chronic treatment, a total of 60 genes was detected (Table 1).
By using Affymetrix Genechips and the appropriate analysis, 28 genes were discovered as differentially expressed in mice chronically treated with paroxetine (18 up-, ten down-regulated). For the Codelink Genechips, 16 genes could be detected as differentially expressed (six up-, ten down-regulated) and by means of the MPIP platform, 27 genes were found to be differentially expressed (21 up-, six down-regulated). In 28 cases, the significant regulation on one platform was supported by a comparable fold change on at least one more platform. Of these, 11 genes also matched the chosen significance criteria on a second platform. Fourteen significantly regulated genes were only represented on one of the platforms, and in 18 cases, probes for a gene were present on at least two platforms but did not show a comparable fold-change. This finding could be explained by methodological or analytical differences or by a platform-dependent probe bias.
Significantly regulated candidates and the absolute fold change detected are listed in Table 1; additional annotational data and information on respective p values and standard errors are given in Supplementary Table S1.
Based on Gene Ontology information, the various gene candidates were assigned to different functional groups, whereby it has to be mentioned that for the product of several genes, a variety of functions were attributed by Gene Ontology.
Validation of selected genes by in situ hybridization and immunohistochemistry
Seven candidates were selected due to different levels of up- or down-regulation in the different Microarray platforms (Affymetrix, Codelink, MPIP): Penk1 (preproenkephalin 1, threefold up-regulated, Affymetrix), Bdnf (brain-derived neurotrophic factor, 2.1-fold up-regulated, MPIP), Gfap (glial fibrillary acidic protein, 2.2-fold up-regulated, Affymetrix), Sox11 (SRY-box containing gene 11, 1.9-fold up-regulated, MPIP), Vim (vimentin, twofold up-regulated, Affymetrix and 2.3-fold up-regulated, MPIP), Calb1 (calbindin-28 K, 1.5-fold down-regulated, Affymetrix and Codelink), and Tex261 (testis expressed gene 261, 1.4-fold down-regulated, MPIP). Verification of the expression level of each candidate in hippocampus slices of four control and four paroxetine treated animals by in situ hybridization confirmed the microarray results, and representative pictures for each comparison are shown in Fig. 4.

Validation of selected microarray results by in situ hybridization in vehicle- and paroxetine-treated DBA/2Ola mice. The mRNA signals of Penk1 (preproenkephalin 1), Bdnf (brain-derived neurotrophic factor), Gfap (glial fibrillary acidic protein), Vim (vimentin), Sox11 (SRY-box containing gene 11), Calb1 (calbindin -28 K), and Tex261 (testis expressed gene 261) in the hippocampus are shown. The genes were selected due to different levels of up- or down-regulation on the three microarray platforms (Affymetrix, Codelink, MPIP; see Table 1)
The colocalization study on the protein products of Gfap and Vim (Fig. 5) in the hippocampus (eight slices per individual) of vehicle and paroxetine-treated mice (n = 5 per group) confirmed that the transcriptional changes detected were also reflected on the protein level. Furthermore, analysis of the cells positive for both immunofluorescence signals (Fig. 5a,b) revealed for each of the paroxetine-treated mouse a higher number of cells (Fig. 5c) co-expressing GFAP and vimentin in the dentate gyrus.

Colocalization of GFAP (glial fibrillary acidic protein) and vimentin in the hippocampus of vehicle- (a) and paroxetine- (b) treated DBA/2Ola mice. Sections were double stained by immunofluorescence for GFAP (green signal) and vimentin (red), bis-benzimide was used as counterstain (blue signal). Cells positive for both signals, GFAP and Vimentin, are marked by the circles. Individual analyses of five animals per treatment (c) revealed for each of the paroxetine-treated mice (black bars; grey bars: vehicle-treated) a higher number of cells co-expressing GFAP and Vimentin in the subgranular layer of the hippocampus
Discussion
Using three different behavioral tests, we corroborated that chronic paroxetine treatment of DBA/2 mice, that show an anxiety-like phenotype at baseline, induces behavioral changes consistent with an antidepressant-like profile. In the FST, the animals showed a paroxetine-induced decrease in passive behavior, which is in agreement with previous reports demonstrating that monoamine-based antidepressants induce such behavioral changes (for review see Jacobson and Cryan 2007). In addition, anxiolytic effects have been shown in two independent behavioral tests, the mHB and the dark/light box. In the only mildly aversive mHB test, risk assessment behavior as well as the overall pattern of avoidance behavior was reduced after application of paroxetine—indicating an anxiolytic effect of this treatment (Blanchard et al. 1993; Ohl et al. 2001). In the dark/light box, a commonly used specific anxiety test (Bourin and Hascoet 2003), the reduction of anxiety-like behavior in DBA/2 mice by chronic paroxetine treatment was even more pronounced: paroxetine-treated animals showed more entries into the brightly lit compartment, and they spent more time in this aversive compartment. This finding is in line with previous behavioral studies using this antidepressant drug (Mirza et al. 2007; Hascoet et al. 2000). Ambiguous results concerning the sensitivity of DBA/2 mice towards the effects of antidepressants were reported. Some studies found the DBA/2 strain to be antidepressant-sensitive (Lucki et al. 2001; Crowley et al. 2005), whereas others did not (David et al. 2003; Cervo et al. 2005). The underlying reasons for this discrepancy still remain to be determined. One difference between the present study and those reporting that the DBA/2 strain did not respond to acute antidepressant treatment is related to the duration of drug treatment. A 28-day treatment period was chosen here due to the objective to identify paroxetine-induced effects on gene expression within a time frame that might be meaningful for the human situation.
In the microarray analysis, we identified a total of 60 genes that were significantly regulated by the paroxetine treatment, with the highest change of transcript level for preproenkephalin (threefold up-regulated) and inhibin beta-A (2.7-fold up-regulated). Although the same tissue samples were probed in three different microarray platforms, none of the candidate genes reached significance on all three of them. However, 18% of the candidates were significantly regulated on two platforms, and a total of 47% of the candidate genes showed comparable fold-regulations (above the fold-change cut-off) on at least one more platform, revealing a considerable true miss rate. Less stringent data analysis would substantially amend the overlap—but on the expense of a higher false discovery rate. Of the candidate genes, 23% were present on one platform only, and 30% were present on at least two platforms but not comparably regulated. The latter could be explained by platform-specific probe biases or technical variances. Noteworthy, many of the genes differentially regulated in our analysis were also described to be significantly altered in a very recently published study by Miller et al. (2008). They used a similar treatment procedure (orally via drinking water over 21 days), the closely related DBA/2 J mouse strain, and a SSRI (fluoxetine). Comparing the lists of antidepressant-regulated genes, a total of 20 candidates was found in both the study of Miller et al (2008) and our study including candidates like Penk1, Inhba, or neuronal pentraxin 2 (Nptx2).
For validation purposes, we have chosen seven genes (Penk1, Bdnf, Gfap, Sox11, Vim, Calb 1, Tex261) that were regulated between 1.4- and threefold and represent the different platforms used. By in situ hybridization, we confirmed that Penk1, Bdnf, Gfap, Vim, and Sox11 were up-regulated and that expression of Calb1 and Tex261 was decreased by paroxetine.
In Table 1, we have listed, by functional classification, those transcripts that were significantly changed following paroxetine treatment. When functionally classifying transcripts, we observed that differentially regulated genes could be separated into 12 main categories: calcium binding, channel activity, enzymatic activity, growth factor activity, hormone activity, immune system, opioid peptide activity, receptor activity, signal transduction, structural molecules, transcription factor activity, and transporter activity. Genes that were not attributable to one of these categories where included in the category entitled “others”. As it is obvious from Table 1, antidepressant-induced effects are consonant with known interactions including signal transduction, ion channels, receptors, neuropeptides and transporters as well as hormonal and immune systems. The exact mechanisms by which the listed transactions are regulated by paroxetine and which biochemical effects these changes in gene activity may have, remains to be elucidated. In the following, we will confine ourselves to a number of observed candidates that are better understood in the context of previous reports.
Paroxetine enhances preproenkephalin 1 (Penk1) expression
We found the transcript of Penk1, the precursor of Met- and Leu-enkephalin, to be threefold up-regulated following paroxetine treatment. Enkephalins belong to the family of endogenous opioids and are involved in, e.g., reward/motivation (Barbano and Cador 2007) and the regulation of the stress response (Drolet et al. 2001). An involvement of the endogenous opioidergic system in the mediation of several symptoms of depression has long been discussed, and it has been shown that enkephalin catabolism inhibitors (RB101) induce antidepressant-like effects (Roques 2000). A recent study showed that deletion of Penk1 in mice resulted in elevated levels of anxiety (Bilkei-Gorzo et al. 2004) and supports the idea that the anxiolytic effect of long-term paroxetine treatment found in the present study might be partly related to a marked increase of Penk1 expression. In seeming contrast to the present study, repeated application of fluoxetine did not alter Penk1 expression in male Wistar rats as measured by in situ hybridization in different brain regions (Dziedzicka-Wasylewska et al. 2002). The conflicting results might be explained by several methodological differences, of which the shorter treatment period might be the most critical factor (Boehm et al. 2006; Shishinka et al. 2007). A recent study reports the effects of enkephalins on behavior and BDNF expression in the hippocampus (Zhang et al. 2006). In our study, we also determined a robust increase in Bdnf expression in the hippocampus of paroxetine-treated DBA/2 mice.
Paroxetine-induced effects on neurotrophic factors
Current hypotheses on causality of depression and antidepressant mechanisms of action include the monoamine hypothesis built upon antidepressants pharmacology (Hirschfeld 2000), the corticosteroid receptor hypothesis explaining behavioral changes as sequel to defunct adaptation to stressors (Holsboer 2000; de Kloet et al. 2005) and the neurotrophic hypothesis (for review see Nestler et al. 2002). All three hypotheses are complementary to each other. The neurotrophic hypothesis of depression and antidepressant mechanism of action was formulated as stressed rats showed decreased levels of hippocampal BDNF, and antidepressants were shown to produce opposite effects, thus compensating the stress-related BDNF suppression (e.g. Nibuya et al. 1995). The finding of opposite effects of antidepressants on stress-induced BDNF suppression is supported by a recent study that indicated an antidepressant-reversible up-regulation of apoptotic processes by chronic mild stress in rats (Bergstrom et al. 2007). Because hippocampi of depressive patients were found to be smaller than those of controls, it was suggested that the stress-induced decrease of BDNF accounts for structural damage and reduced neurogenesis among these patients (Manji and Duman 2001). Vice versa, it is proposed that antidepressants reinstate hippocampal size and function through elevation of BDNF and other neurotrophic factors (see Duman and Monteggia 2006). Interestingly, an interaction of antidepressant treatment effects and elevated expression of neurotrophic factors is not limited to pharmacological manipulation. Voluntary exercise was suggested to have antidepressant properties in animal models (Duman et al. 2008) and mood-elevating actions in humans (Babyak et al. 2000). A recent microarray study showed that also voluntary exercise up-regulated several neurotrophic factors (Hunsberger et al. 2007). Although more research is needed to validate the neurotrophic hypothesis, we were impressed by how many genes implicated in neurotrophic actions were found to be regulated by paroxetine.
Among others, the transcript of BDNF was up-regulated, while expression of neurotrophin-3 (Ntf3, NT-3) was down-regulated. Interestingly, stress, often precipitating depressive episodes, has been shown to display opposite effects on BDNF and NT-3 expression in the hippocampus of rodents: BDNF was down-regulated, while expression of NT-3 was up-regulated (Smith 1996). The altered expression of neurotrophins might be related to the observation that chronic stress leads to atrophy in hippocampal CA3 neurons (McEwen 2005). These stress effects have been suggested to be related to the development of depression and might be antagonized by application of various antidepressants. As mentioned above, it has been repeatedly shown that several antidepressants increase BDNF in rodents and humans (see Duman and Monteggia 2006; but also see Coppell et al. 2003). In addition, intrahippocampal application of BDNF, but also NT-3, has been shown to have antidepressant-like effects on rats in the FST and learned helplessness paradigm (Shirayama et al. 2002). In our experiment, we found a twofold decrease of NT-3 mRNA levels in the hippocampus in animals treated chronically with paroxetine. NT-3 levels have been reported to be increased in the hippocampus and in the locus coeruleus by repeated severe stress and decreased by electroconvulsive seizures (ECS) or by long-term treatment with desipramine or imipramine, but not with fluoxetine or trazodone (Smith et al. 1995; Smith 1996). Karege et al. (2005) reported a significant decrease of BDNF and NT-3 protein levels in the hippocampus of drug-free suicide victims compared with non-suicide controls. In drug-treated suicide victims, neurotrophin levels were not significantly different from non-suicide controls. This observation further indicates that both neurotrophins might be involved in antidepressant drug activity. On the other hand, it was found that NT-3 inhibits both FGF2-induced neurosphere growth and bromodeoxyuridine (BrdU) incorporation in a dose-dependent manner (Jin et al. 2005). The latter finding would fit with our results and would suggest that the decrease of NT-3 might be supportive for neurogenesis.
The survival and function of neurons in the central nervous system are dependent on an ever-growing list of factors, and besides the candidates BDNF and NT-3, we found some additional interesting factors involved in neurogenesis/neuronal plasticity to be regulated by antidepressant treatment.
Paroxetine-induced effects on glial cells—implicationsfor neurogenesis
In our study, the transcript level of glial fibrillary acidic protein (GFAP), of which the protein product is almost exclusively expressed in astrocytes, was significantly up-regulated by chronic paroxetine application. Alterations of GFAP expression are observed in response to a variety of events, and an increase might be associated to gliosis, neurodegeneration or neuroregeneration, and also neurogenesis. Analysis of post mortem tissue from patients with major depression has revealed significant reductions of GFAP protein or in GFAP/glial cell counts in various brain regions, suggesting a decrease of glial function in this disorder (Fatemi et al. 2004). In a study investigating male tree shrews that were submitted to 5 weeks of psychosocial stress, a significant decrease of both the number and somal volume of astroglia was detected. Additionally, it was found that 28-days treatment with fluoxetine prevented the stress-induced numerical decrease of astrocytes. The antidepressant treatment had no effects on the shrinkage of somal volume. These changes of astroglial structural plasticity in response to stress and antidepressant treatment support the notion that glial changes may contribute to the pathophysiology of affective disorders as well as the cellular activity of antidepressants (Czeh et al. 2006; Manev et al. 2003).
Astrocytes provide structural as well as trophic support to neurons. They also have an important role in the immune response of the brain, in synaptic function, clearance of cellular ions and transmitters, in neuronal metabolism, and in neuronal migration (Allen and Barres 2005; Araque 2006; Mennerick and Zorumski 1994; Pellerin and Magistretti 2004; Verkhratsky et al. 1998; Vernadakis 1996). More recently, astrocytes have been recognized to also play a role as neural progenitor cells themselves in both the developing and mature CNS (Goldman 2003; Song et al. 2002), and to promote neurogenesis. For example, Song et al. (2002) cultured neural progenitors from the adult rat hippocampus with either primary cultures of hippocampal neurons or astrocytes. They found that neural progenitors cultured with hippocampal astrocytes were induced to differentiate into neurons. Our observation of a prominent up-regulation of Gfap in the paroxetine-treated animals by microarray analysis and in situ hybridization raises the question whether this might indicate neurogenesis, especially as vimentin (another intermediate filament protein, dynamically regulated during development) turned out to be up-regulated as well. Our double-labeling study in brain slices, to detect GFAP and vimentin positive cells within the dentate gyrus, showed that some of the cells positive for GFAP also express vimentin and are located in the subgranular zone. Moreover, the number of those cells was twice as high in the paroxetine-treated mice. According to the study of Garcia et al. (2004), this indicates that the double-labeled cells of the subgranular zone are GFAP-expressing neural progenitors and supports the notion that increased neurogenesis occurs in hippocampi of chronically paroxetine-treated mice.
Paroxetine-induced effects on miscellaneous genes potentially involved in neuroplasticity
Other candidates of our list seemingly involved in neuroplasticity processes include the SYR-box containing gene 11 (Sox11) and the signal transducer and activator of transcription 3 (Stat3), both of which we found to be up-regulated by the paroxetine treatment. The transcription factor Sox11 is known to be expressed during later stages of neural development in areas of the brain in which neurons undergo differentiation (Kuhlbrodt et al. 1998). In vitro studies suggest that Sox11 is centrally involved in regulating events that promote neurite growth and neuron survival (Jankowski et al. 2006). In the adult brain, Sox11 expression is weak under basal conditions but has been found to be up-regulated after experimentally induced seizures or nerve injury and has been linked to injury-induced neuritogenesis (Elliott et al. 2003; Tanabe et al. 2003). So far, Sox11 has rarely been associated to antidepressant-induced alterations in gene expression, but a more recent study reported an up-regulation of Sox11 within several hours following electroconvulsive shock (ECS) in the dentate gyrus, the piriform cortex, and in the amygdala (Sun et al. 2005). The effect of Sox11 up-regulation in the latter two brain regions is not known. Its activation in the subgranular layer of the dentate gyrus by ECS (Sun et al. 2005) as well as our finding supports the notion that Sox11 is contributing to antidepressant-induced neuronal plasticity.
STAT3 is known to regulate gene transcription in response to cytokines and growth factors. It has been shown to be activated in reactive astrocytes (Xia et al. 2002) and to be involved in 5-HT1A-receptor-mediated neurite outgrowth (Fricker et al. 2005). Ng et al. (2006) identified STAT3 as an essential component of neurotrophin signaling and functions, i.e., inhibition of STAT3 expression decreased BDNF-promoted neurite outgrowth in primary hippocampal neurons. Furthermore, it was suggested that neurotrophin-induced increase in STAT3 activation underlies several downstream functions of neurotrophin signaling. Sleep deprivation, which has short-term antidepressant effects in human patients, affects a number of genes in the cortex of rats, among which mRNA levels of STAT3 as well as BDNF and TrkB were found to be up-regulated (Cirelli and Tononi 2000). Additionally, they also report an enhanced transcript level of nerve growth factor-inducible protein A (NGFI-A).
NGFI-A (alias Egr1, zif268, krox24, ZENK) has been shown to be up-regulated following chronic treatment with a variety of different antidepressants (Bjartmar et al. 2000) and is another candidate on our list of genes regulated by chronic paroxetine application. Alterations in NGFI-A expression have been observed in different brain regions and seem to be responsive to a variety of stimuli (e.g., Thompson and Rosen 2006). This factor has been associated to serotonin-induced DNA demethylation, and it is hypothesized that NGFI-A contributes to epigenetic programming of glucocorticoid receptor expression (Weaver et al. 2007).
Conclusion
This study explores paroxetine-induced hippocampal gene expression in mice that exhibit antidepressant-like behavioral changes according to three independent tests. We used three different microarray platforms and confirmed drug-induced changes in gene activity by in situ hybridization. Within the limitations of the available technology, we conclude that paroxetine exerts a number of effects upon transcripts implicated in causality and treatment of depression. Many of the paroxetine-regulated genes are currently linked to neuronal plasticity, and the effects seem to occur via different mechanisms. Intriguingly, paroxetine seems to interfere not only with neurons but also with glial cells indicating an increase in progenitor cells and other factors necessary for neuronal differentiation. The many changes in neurotrophic factors, receptor constituents, and transcription factors may also promote neuroplasticity in and between existing neurons.
We are aware that on this level of investigation, the given interpretations are speculative. Nevertheless, studies as the current one will help us to gain insight into the mechanisms of action of current antidepressants, disentangle the effects which contribute to the resolution of depressive symptomatology, and ultimately might lead us to those genes that are causally involved in depression.
References
Allen NJ, Barres BA (2005) Signaling between glia and neurons: focus on synaptic plasticity. Curr Opin Neurobiol 15:542–548
Araque A (2006) Astrocyte-neuron signaling in the brain-implications for disease. Curr Opin Investig Drugs 7:619–624
Babyak M, Blumenthal JA, Herman S, Khatri P, Doraiswamy M, Moore K, Craighead WE, Baldewicz TT, Krishnan KR (2000) Exercise treatment for major depression: maintenance of therapeutic benefit at 10 months. Psychosom Med 62:633–638
Barbano MF, Cador M (2007) Opioids for hedonic experience and dopamine to get ready for it. Psychopharmacology 191:497–506
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B 57:289–300
Bergstrom A, Jayatissa MN, Thykjaer T, Wiborg O (2007) Molecular pathways associated with stress resilience and drug resistance in the chronic mild stress rat model of depression: a gene expression study. J Mol Neurosci 33:201–215
Bilkei-Gorzo A, Racz I, Michel K, Zimmer A, Klingmuller D, Zimmer A (2004) Behavioral phenotype of pre-proenkephalin-deficient mice on diverse congenic backgrounds. Psychopharmacology 176:343–352
Bjartmar L, Johansson IM, Marcusson J, Ross SB, Seckl JR, Olsson T (2000) Selective effects on NGFI-A, MR, GR and NGFI-B hippocampal mRNA expression after chronic treatment with different subclasses of antidepressants in the rat. Psychopharmacology 151:7–12
Blanchard RJ, Yudko EB, Rodgers RJ, Blanchard DC (1993) Defense system psychopharmacology: an ethological approach to the pharmacology of fear and anxiety. Behav Brain Res 58:155–165
Boehm C, Newrzella D, Herberger S, Schramm N, Eisenhardt G, Schenk V, Sonntag-Buck V, Sorgenfrei O (2006) Effects of antidepressant treatment on gene expression profile in mouse brain: cell type- specific transcription profiling using laser microdissection and microarray analysis. J Neurochem 97(Suppl 1):44–49
Bourin M, Hascoet M (2003) The mouse light/dark box test. Eur J Pharmacol 463:55–65
Campbell S, Macqueen G (2004) The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci 29:417–426
Carvalho AF, Cavalcante JL, Castelo MS, Lima MC (2007) Augmentation strategies for treatment-resistant depression: a literature review. J Clin Pharm Ther 32:415–428
Cervo L, Canetta A, Calcagno E, Burbassi S, Sacchetti G, Caccia S, Fracasso C, Albani D, Forloni G, Invernizzi RW (2005) Genotype-dependent activity of tryptophan hydroxylase-2 determines the response to citalopram in a mouse model of depression. J Neurosci 25:8165–8172
Cirelli C, Tononi G (2000) Gene expression in the brain across the sleep-waking cycle. Brain Res 885:303–321
Conti B, Maier R, Barr AM, Morale MC, Lu X, Sanna PP, Bilbe G, Hoyer D, Bartfai T (2007) Region-specific transcriptional changes following the three antidepressant treatments electro convulsive therapy, sleep deprivation and fluoxetine. Mol Psychiatry 12:176–189
Coppell AL, Pei Q, Zetterstrom TS (2003) Bi-phasic change in BDNF gene expression following antidepressant drug treatment. Neuropharmacology 44:903–910
Craddock N, Forty L (2006) Genetics of affective (mood) disorders. Eur J Hum Genet 14:660–668
Crowley JJ, Blendy JA, Lucki I (2005) Strain-dependent antidepressant-like effects of citalopram in the mouse tail suspension test. Psychopharmacology 183:257–264
Cryan JF, Holmes A (2005) The ascent of mouse: advantages in modelling human depression and anxiety. Nat Rev Drug Discov 4:775–790
Czeh B, Simon M, Schmelting B, Hiemke C, Fuchs E (2006) Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology 31:1616–1626
David DJ, Renard CE, Jolliet P, Hascoet M, Bourin M (2003) Antidepressant-like effects in various mouse strains in the forced swimming test. Psychopharmacology 166:373–382
de Kloet ER, Joëls M, Holsboer F (2005) Stress and the brain: from adaptation to disease. Nat Rev Neurosci 6:463–475
Drolet G, Dumont EC, Gosselin I, Kinkead R, Laforest S, Trottier JF (2001) Role of endogenous opioid system in the regulation of the stress response. Prog Neuro-psychopharmacol Biol Psychiatry 25:729–741
Droste SK, Gesing A, Ulbricht S, Müller MB, Linthorst AC, Reul JM (2003) Effects of long-term voluntary exercise on the mouse hypothalamic-pituitary-adrenocortical axis. Endocrinology 144:3012–3023
Duman RS, Monteggia LM (2006) A neurotrophic model for stress-related mood disorders. Biol Psychiatry 59:1116–1127
Duman CH, Schlesinger L, Russell DS, Duman RS (2008) Voluntary exercise produces antidepressant and anxiolytic behavioral effects in mice. Brain Res 1199:148–158
Dziedzicka-Wasylewska M, Dlaboga D, Pierzchala-Koziec K, Rogoz Z (2002) Effect of tianeptine and fluoxetine on the levels of Met-enkephalin and mRNA encoding proenkephalin in the rat. J Physiol Pharmacol 53:117–125
Elliott RC, Miles MF, Lowenstein DH (2003) Overlapping microarray profiles of dentate gyrus gene expression during development- and epilepsy-associated neurogenesis and axon outgrowth. J Neurosci 23:2218–2227
Engin E, Treit D (2007) The role of hippocampus in anxiety: intracerebral infusion studies. Behav Pharmacol 18:365–374
Fatemi SH, Laurence JA, Raghi-Niknam M, Stary JM, Schulz SC, Lee S, Gottesman II (2004) Glial fibrillary acidic protein is reduced in cerebellum of subjects with major depression, but not schizophrenia. Schizophr Res 69:317–323
Fava M, Davidson KG (1996) Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 19:179–200
Fricker AD, Rios C, Devi LA, Gomes I (2005) Serotonin receptor activation leads to neurite outgrowth and neuronal survival. Brain Res Mol Brain Res 138:228–235
Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV (2004) GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci 7:1233–1241
Goldman S (2003) Glia as neural progenitor cells. Trends Neurosci 26:590–596
Hascoet M, Bourin M, Nic Dhonnchadha BA (2000) The influence of buspirone, and its metabolite 1-PP, on the activity of paroxetine in the mouse light/dark paradigm and four plates test. Pharmacol Biochem Behav 67:45–53
Hirschfeld RM (2000) History and evolution of the monoamine hypothesis of depression. J Clin Psychiatry 61(Suppl 6):4–6
Holsboer F (2000) The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23:477–501
Holsboer F (2001) Stress, hypercortisolism and corticosteroid receptors in depression: implications for therapy. J Affect Disord 62:77–91
Holsboer F, Ising M (2008) Central CRH system in depression and anxiety – Evidence from clinical studies with CRH(1) receptor antagonists. Eur J Pharmacol 583:350–357
Hunsberger JG, Newton SS, Bennett AH, Duman CH, Russell DS, Salton SR, Duman RS (2007) Antidepressant actions of the exercise-regulated gene VGF. Nat Med 13:1476–1482
Jacobson LH, Cryan JF (2007) Feeling strained? Influence of genetic background on depression-related behavior in mice: a review. Behav Genet 37:171–213
Jankowski MP, Cornuet PK, McIlwrath S, Koerber HR, Albers KM (2006) SRY-box containing gene 11 (Sox11) transcription factor is required for neuron survival and neurite growth. Neuroscience 143:501–514
Jin L, Hu X, Feng L (2005) NT3 inhibits FGF2-induced neural progenitor cell proliferation via the PI3 K/GSK3 pathway. J Neurochem 93:1251–1261
Karege F, Vaudan G, Schwald M, Perroud N, La Harpe R (2005) Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res 136:29–37
Kuhlbrodt K, Herbarth B, Sock E, Enderich J, Hermans-Borgmeyer I, Wegner M (1998) Cooperative function of POU proteins and SOX proteins in glial cells. J Biol Chem 273:16050–16057
Larsson O, Wahlestedt C, Timmons JA (2005) Considerations when using the significance analysis of microarrays (SAM) algorithm. BMC Bioinformatics 6:129
Li C, Wong WH (2001) Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci 98:31–36
Lucae S, Salyakina D, Barden N, Harvey M, Gagné B, Labbé M, Binder EB, Uhr M, Paez-Pereda M, Sillaber I, Ising M, Brückl T, Lieb R, Holsboer F, Müller-Myhsok B (2006) P2RX7, a gene coding for a purinergic ligand-gated ion channel, is associated with major depressive disorder. Hum Mol Genet 15:2438–2445
Lucki I, Dalvi A, Mayorga AJ (2001) Sensitivity to the effects of pharmacologically selective antidepressants in different strains of mice. Psychopharmacology 155:315–322
Manev H, Uz T, Manev R (2003) Glia as putative target for antidepressant treatment. J Affect Disord 75:59–64
Manji HK, Duman RS (2001) Impairments of neuroplasticity and cellular resilience in severe mood disorders: implications for the development of novel therapeutics. Psychopharmacol Bull 35:5–49
McEwen BS (2005) Glucocorticoids, depression, and mood disorders: structural remodelling in the brain. Metabolism 54:20–23
McQuilin A, Bass NJ, Chouhury K, Puri V, Kosmin M, Lawrence J, Curtis D, Gurling HM (2008) Case-control studies show that a non-conservative amino-acid change from glutamine to arginine in the P2RX7 purinergic receptor protein is associated with both bipolar- and unipolar-affective disorders. Mol Psychiatry doi:10.1038/mp.2008.6
Mennerick S, Zorumski CF (1994) Glial contributions to excitatory neurotransmission in cultured hippocampal cells. Nature 368:59–62
Mill J, Petronis A (2007) Molecular studies of major depressive disorder: the epigenetic perspective. Mol Psychiatry 12:799–814
Miller BH, Schultz LE, Gulati A, Cameron MD, Pletcher MT (2008) Genetic regulation of behavioral and neuronal responses to fluoxetine. Neuropsychopharmacology 33:1312–1322
Mirnics K, Middleton FA, Lewis DA, Levitt P (2001) Analysis of complex brain disorders with gene expression microarrays: schizophrenia as a disease of the synapse. Trends Neurosci 24:479–486
Mirza NR, Nielsen EO, Troelsen KB (2007) Serotonin transporter density and anxiolytic-like effects of antidepressants in mice. Prog Neuro-psychopharmacol Biol Psychiatry 31:858–866
Nakatani N, Aburatani H, Nishimura K, Semba J, Yoshikawa T (2004) Comprehensive expression analysis of a rat depression model. Pharmacogenomics 4:114–126
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002) Neurobiology of depression. Neuron 34:13–25
Ng YP, Cheung ZH, Ip NY (2006) STAT3 as a downstream mediator of Trk signaling and functions. J Biol Chem 281:15636–15644
Nibuya M, Morinobu S, Duman RS (1995) Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 15:7539–7547
Ohl F, Sillaber I, Binder E, Keck ME, Holsboer F (2001) Differential analysis of behavior and diazepam-induced alterations in C57BL/6N and BALB/c mice using the modified hole board test. J Psychiatr Res 35:147–154
Ohl F, Roedel A, Binder E, Holsboer F (2003) Impact of high and low anxiety on cognitive performance in a modified hole board test in C57BL/6 and DBA/2 mice. Eur J Neurosci 17:128–136
Pellerin L, Magistretti PJ (2004) Neuroenergetics: calling upon astrocytes to satisfy hungry neurons. Neuroscientist 10:53–62
Pillay NS, Stein DJ (2007) Emerging anxiolytics. Expert Opin Emerg Drugs 12:541–554
Porsolt RD, Bertin A, Jalfre M (1977) Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther 229:327–336
Roques BP (2000) Novel approaches to targeting neuropeptide systems. Trends Pharmacol Sci 21:475–483
Sequeira A, Klempan T, Canetti L, ffrench-Mullen J, Benkelfat C, Rouleau GA, Turecki G (2007) Patterns of gene expression in the limbic system of suicides with and without major depression. Mol Psychiatry 12:640–655
Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS (2002) Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci 22:3251–3261
Shishinka GT, Kalinina TS, Dygalo NN (2007) Up-regulation of tryptophan hydroxylase-2 mRNA in the rat brain by chronic fluoxetine treatment correlates with its antidepressant effect. Neuroscience 150:404–412
Smith MA (1996) Hippocampal vulnerability to stress and aging: possible role of neurotrophic factors. Behav Brain Res 78:25–36
Smith MA, Makino S, Altemus M, Michelson D, Hong SK, Kvetnansky R, Post RM (1995) Stress and antidepressants differentially regulate neurotrophin 3 mRNA expression in the locus coeruleus. Proc Natl Acad Sci 92:8788–8792
Song H, Stevens CF, Gage FH (2002) Astroglia induce neurogenesis from adult neural stem cells. Nature 417:39–44
Sullivan PF, Fan C, Perou CM (2006) Evaluating the comparability of gene expression in blood and brain. Am J Med Genet B Neuropsychiatr Genet 141:261–268
Sun W, Park KW, Choe J, Rhyu IJ, Kim IH, Park SK, Choi B, Choi SH, Park SH, Kim H (2005) Identification of novel electroconvulsive shock-induced and activity-dependent genes in the rat brain. Biochem Biophys Res Commun 327:848–856
Tanabe K, Bonilla I, Winkles JA, Strittmatter SM (2003) Fibroblast growth factor-inducible-14 is induced in axotomized neurons and promotes neurite outgrowth. J Neurosci 23:9675–9686
Thoeringer CK, Sillaber I, Roedel A, Erhardt A, Mueller MB, Ohl F, Holsboer F, Keck ME (2007) The temporal dynamics of intrahippocampal corticosterone in response to stress-related stimuli with different emotional and physical load: an in vivo microdialysis study in C57BL/6 and DBA/2 inbred mice. Psychoneuroendocrinology 32:746–757
Thompson BL, Rosen JB (2006) Immediate–early gene expression in the central nucleus of the amygdala is not specific for anxiolytic or anxiogenic drugs. Neuropharmacology 50:57–68
Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JMHM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W (1998) Impaired stress response and reduced anxiety in lacking a functional corticotrophin-releasing hormone receptor 1. Nat Genet 19:162–166
Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz B, McGrath PJ, Shores-Wilson K, Biggs MM, Balasubramani GK, Fava M, STAR*D Study Team (2006) Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 163:28–40
Tsankova N, Renthal W, Kumar A, Nestler EJ (2007) Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 8:355–367
Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci 98:5116–5121
Verkhratsky A, Orkand RK, Kettenmann H (1998) Glial calcium: homeostasis and signaling function. Physiol Rev 78:99–141
Vernadakis A (1996) Glia-neuron intercommunications and synaptic plasticity. Prog Neurobiol 49:185–214
Weaver ICG, Alessio ACD, Brown SE, Hellstrom IC, Dymov S, Sharma S, Szyf M, Meaney MJ (2007) The transcription factor nerve growth factor-inducible protein A mediates epigenetic programming: altering epigenetic marks by immediate early genes. J Neurosci 27:1756–1768
Wong ML, Licino J (2004) From monoamines to genomic targets: a paradigm shift for drug discovery in depression. Nat Rev Drug Discov 3:136–151
Xia XG, Hofmann HD, Deller T, Kirsch M (2002) Induction of STAT3 signaling in activated astrocytes and sprouting septal neurons following entorhinal cortex lesion in adult rats. Mol Cell Neurosci 21:379–392
Yamada M, Yamada M, Higuchi T (2005) Antidepressant-elicited changes in gene expression: remodelling of neuronal circuits as a new hypothesis for drug efficacy. Prog Neuro-psychopharmacol Biol Psychiatry 29:999–1009
Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30:e15
Yilmazer-Hanke DM, Roskoden T, Zilles K, Schwegler H (2003) Anxiety-related behaviour and densities of glutamate, GABAA, acetylcholine and serotonin receptors in the amygdala of seven inbred mouse strains. Behav Brain Res 145:145–159
Zhang H, Torregrossa MM, Jutkiewicz EM, Shi Y, Rice KC, Woods JH, Watson SJ, Ko MC (2006) Endogenous opioids upregulate brain-derived neurotrophic factor mRNA through δ- and μ-opioid receptors independent of antidepressant-like effects. Eur J Neurosci 23:984–994
Acknowledgements
We would like to thank Amersham for kindly providing equipment and support for this experiment and Sabine Ulbricht, Katja Mayer and Yvonne Grübler for excellent technical assistance. Furthermore, we would like to thank Dr. Marianne Müller and Dr. Susanne Lucae for stimulating discussions. The experiments comply with the current laws of Germany.
This work has partly been funded by the Federal Ministry of Education and Research (BMBF) in the framework of the National Genome Research Network (NGFN), Förderkennzeichen 01GS0481.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors are responsible for the contents of this publication.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
Provides additional annotation information (platform specific probe identifier and Entrez Gene ID), statistical data and, for the inhouse cDNA microarray platform, composite spot images. These images served for quality control purposes and were generated in the following way: First, the brightness of all array scan images was normalized. Second, the coordinates of each probe on each array as given by the scanner software were used to extract images of each spot of a given probe. The resulting spot images were colored and assembled in four rows, the first one showing the Cy3 and the second the Cy5 signal. Row three is an overlay of the Cy3 and Cy5 images, and row four depicts the scan area used to collect the intensity data with the fixed circle method. Columns 1 to 5 represent one labeling combination (vehicle-treated–Cy3 / paroxetine-treated–Cy5), columns 6 to 10 the dye flip. Where necessary, the brightness of the composite images was increased to facilitate visual inspection. (↑: up-regulated; ↓: down-regulated; n.p. Probe not present or below quality threshold, asterisks significantly regulated by Paroxetine, parentheses regulation level below fold change cutoff; m log2 ratio of group means, Sd standard deviation, 1 non-FDR corrected p value, 2 FDR-corrected p value, see data analysis section) (XLS 1.489 MB).
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sillaber, I., Panhuysen, M., Henniger, M.S.H. et al. Profiling of behavioral changes and hippocampal gene expression in mice chronically treated with the SSRI paroxetine. Psychopharmacology 200, 557–572 (2008). https://doi.org/10.1007/s00213-008-1232-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-008-1232-6