
Abstract
Purpose
A real-time immunocapture PCR (RT-IPCR) has been fabricated for the detection of Staphylococcus aureus enterotoxin B (SEB) from food and environmental samples.
Methods
Considering the fact, anti-SEB immunoglobulin G (IgG) has affinity towards protein A, produced by nearly all S. aureus, and generates false-positive read out in all immuno-based assay. We have employed avian anti-SEB antibody (SEB-IgY) as capture probe, since IgY interact less efficiently to protein A and biotinylated SEB-specific monoclonal antibody (SEB -MAb) conjugated with reporter DNA as revealing probe for real-time PCR amplification and signal generation. Sensitivity and selectivity of the assay were evaluated employing closely related enterotoxins and other toxins.
Results
The RT-IPCR is highly specific and sensitive (100 fg/mL). The practical applicability of the assay was tested using spiked food sample as well as naturally contaminated food samples. The sensitivity and specificity of RT-IPCR were not compromised by the foods tested and was able to detect SEB conveniently. Further, the assay was validated comparing with the in-house developed PCR, and plausible result was obtained.
Conclusion
The developed assay can be utilized as a low-cost detection system of SEB in routine food testing laboratories.
Similar content being viewed by others


Introduction
Superantigenic staphylococcal enterotoxins (SEs) (SEA, SEB, SEC, and remaining until SEV) and toxic shock syndrome toxin (TSST) are considered as the major cause of food-borne illness worldwide (Argudín et al. 2010; Ortega et al. 2010, and Fratamico et al. 2005). Among them, SEB is a potent etiological agent for food poisoning and considered as a category B biological warfare agent due to its aerosol incapacitating characteristics, toxicity, and easy to produce in bulk (Wu et al. 2016; Greenfield et al. 2002). Moreover, consumption and exposure to SEB in very low concentration result in clinical manifestation (emesis, abdominal cramps, and diarrhea) within a few hours (Xu and Mccormick 2012). The lethal manifestations caused by SEB warrants their early detection for proper management of cases. Therefore, detection and quantification of SEB at extremely low concentrations in a variety of food samples are needs of the hour. To date, several methods like conventional culture-based methods and various immunological and PCR-based platforms have been developed and validated for the detection of SEB from food and biological samples (Klotz et al. 2003; Schmitz et al. 1998). However, these traditional methods lack the accuracy and sensitivity required for low-level detection. The PCR-based assays allow specific identification of toxin encoding genes but the presence of polymerase inhibitory compounds in complex matrices hinders the sensitivity of the assays (Mondal et al. 2018). Almost all S. aureus strains produce a number of enterotoxins, but this does not conclude their toxin production capability in food as their toxin-producing ability varies based on the food environment. Hence, direct detection of toxin in food can provide higher diagnostic relevance evidence for food poisoning (Ramlal et al. 2018). Considering this fact, to date, various immunoassays targeting various toxins from diverse matrices have been developed and some of which are also available commercially (Alefantis et al. 2004; Rong-Hwa et al. 2010). Although these assays detect the actual toxin component but lack the sensitivity of PCRs, involve extensive sample preparation procedures generate false-negative results due to the use of mammalian origin antibodies (monoclonal and polyclonal). Immunoglobulin (IgG) has tremendous specificity and binding affinity towards antigens (Jin et al. 2013). However, the Fc domain of IgG has an affinity to staphylococcal protein A (SpA) and produce false-positive results in all immunoassays. SpA also has affinity towards Fab region in 15–20% of IgG, IgA, and IgM antibodies of humans (Ljungberg et al. 1993; Nagaraj et al. 2016). Immunoglobulin Y (IgY) found in the egg yolk and serum of bird has equivalent antigen-binding affinities like IgG. Hence, this IgY exhibits low affinity towards SpA in immunoassays (Carlander et al. 1999). However, only IgY-based immunoassay for the detection of SEB does not exist till date. The immuno-polymerase chain reaction (IPCR) is a sensitive technique which combines the PCR exponential signal amplification power with the versatility and flexibility of immunoassays like ELISA. Moreover, it also replaces the enzyme conjugated to reporter antibody in ELISA with an oligomer/reporter DNA (Zhang et al. 2008). The technique has been adapted for sensitive detection of several bacterial (Mehta et al. 2014), viral pathogens (Nolasco et al. 1993), bacterial toxins (Fischer et al. 2007), mycotoxins (Babu and Muriana 2011), and disease-specific antibodies (Niemeyer et al. 2005). Due to the inclusion of several washing steps before the addition of DNA marker, IPCRs are less prone to non-specific amplification when compared to quantitative PCRs. On the other hand, real-time PCR (RT-PCR) can enhance the sensitivity of PCR and is faster for qualitative and quantitative detection (Fitzmaurice et al. 2004). Considering all these facts, in the present work, a real-time immunocapture PCR (RT-IPCR)-based platform was devised employing IgY as capturing probe and monoclonal anti-SEB antibody conjugated with DNA as revealing probe for detection and quantification of enterotoxin B (SEB). The amplification of revealing DNA probe and detection signal was monitored by RT-PCR. The sensitivity and specificity of the assay were evaluated and compared with in-house developed PCR kit. The practicability and robustness of the developed system were evaluated with pure toxins, cultures supernatants, and naturally contaminated foods samples. Altogether, the developed method can be a suitable tool for qualitative and quantitative detection of SEB.
Material and method
Bacterial cultures and media
All the bacterial cultures used in the study were obtained from the repository of DFRL as listed in Table 2. The Staphylococcus aureus strains were grown in Tryptic Soy Broth (TSB, Hi-media, India) for 24–36 h at 37 °C. All other bacterial cultures were maintained in Brain Heart Infusion (BHI) agar/broth unless mentioned otherwise. SEB-specific monoclonal antibody was obtained from DFRL, Mysore.
Immunization schedule and extraction of IgY antibodies
Specific pathogen-free white leghorn chicken was immunized via intramuscular injection with denatured r-SEB proteins with an equal volume of Freund’s complete adjuvant (FCA). Subsequently, the same concentration of toxin emulsified with Freund’s incomplete adjuvant (FIA) was administrated as booster dose on days 14, 28, and 42. On completion of immunization, eggs were collected and stored at 4 °C. IgY was extracted from the eggs by PEG precipitation method (Pauly et al. 2011) and was purified by affinity chromatography as per manufacturer’s instructions (AminoLink Plus Immobilization kit, Thermo Scientific, India), and antibody titer was checked by indirect ELISA.
Labeling and confirmation of biotin for SEB-specific monoclonal antibodies
SEB-specific monoclonal antibody was biotinylated using antibody biotinylation kit (Thermo, India) as per manufacturer’s protocol. Biotinylation of the antibody was confirmed by dot blot. Briefly, biotinylated antibodies (10 μL) were coated on nitrocellulose membrane, air-dried and blocked with 3% skimmed milk, and probed with streptavidin-HRP conjugate (Invitrogen, CA, USA) for development of chromogenic dot. Nitrocellulose membranes coated with monoclonal antibodies probed with streptavidin-HRP conjugate were used as controls.
Biotinylation of reporter DNA
Biotinylated DNA oligomer was prepared by PCR using the primers mentioned in Table 1. Briefly, 80 bp DNA was synthesized and was amplified with biotin forward and normal reverse primer. The PCR amplification was carried out in 20 μL reaction volume containing 1 μL of template DNA, 1X PCR buffer, 1.5 mM MgCl2, 200 μM dNTPs, 10 μM each of primer, and 1.0 U Taq polymerase in Eppendrof pro thermocycler (Eppendrof, USA). The PCR parameter was standardized with the initial denaturation of 94 °C for 4 min and then 94 °C for a 30-s denaturation, 56 °C for a 30-s annealing, and 72 °C for a 30-s extension for 30 cycles and last with a final extension of 72 °C for 5 min. The PCR amplicons were purified by a gel extraction kit (Qiagen, India), quantified, and stored at − 20 °C till further use.
Preparation of antibody-DNA conjugates
Conjugation of biotin-modified oligomers with antibody is the most crucial step for the success of immunocapture PCR. Strongest non-covalent interaction of biotin-streptavidin molecules was used for the generation of antibody-DNA conjugates. Streptavidin provides four binding sites for biotin, hence act as a bridge for the interaction of biotinylated revealing antibody (Btn-MAb-SEB) and biotin-modified oligomers. The bridge complex was prepared using 1:1000 dilution of biotinylated monoclonal antibody (2 mg/mL), 1:1000 dilution of streptavidin (1 mg/mL), and 1:1000 dilutions of biotinylated DNA complex (50 ng/μL). In all RT-IPCR, 20 μL bridge complexes (Btn-MAb-SEB-DNA) was used for signal generation.
Standardization of Immunocapture real-time PCR for detection of SEB
The assay format of RT-IPCR was illustrated in Fig. 1. Briefly, RT-IPCR for the detection of SEB was standardized using SEB-IgY antibodies for antigen capture, followed by Btn-MAb-SEB antibodies as revealing antibodies conjugated with 80 bp oligo for signal generation. Briefly, SEB-IgY antibodies were diluted to 1:1000 in carbonate-bicarbonate buffer (100 μL) and coated in well of Microtiter PCR plates (Nunc/Eppendrof, India). After incubation, the plate was blocked with 200 μL of 1% BSA at 4 °C overnight. The plate was washed thrice with PBST (tween 20-0.05%), air dried, sealed, and stored at − 20 °C until further use. For testing, the wells were incubated with 100 μL of SEB and incubated at 37 °C for 1 h followed by washing thrice with PBST. Then, bridge complex (Btn-MAb–SEB DNA) was added to the well and incubated at 37 °C for 1 h. Post-incubation, the plates were washed thrice with PBST and finally twice with double distilled water, and RT-PCR was performed. Evagreen master mix (Bio-Rad, India) was used for real-time-based monitoring of template amplification. Amplification was carried out: for each 10 μL of reaction contains 5 μL of 2× Evagreen master mix, 1 μL of each primer (0.5 μM), 1 μL of template DNA, and 3 μL of water. Amplification condition as follows: 30 sec at 95 °C, followed by 40 PCR cycles of denaturation at 95 °C for 10 sec, and 60 °C for 10 s for the annealing and extension phases. One no-template control contained the real-time PCR master mix without reporter DNA. Real-time amplification parameter was analyzed using the CFX software (Bio-Rad, India).

Illustration of RT-IPCR assay format
Specificity testing of RT-IPCR
The specificity of RT-IPCR was carried out by testing other related enterotoxins of S. aureus (r-SEA, r-SEG, and r-SEC) and culture supernatants extract of various staphylococcal and non-staphylococcal cultures listed in Table 2. Briefly, the cultures were inoculated in Brain Heart Infusion (BHI) broth and incubated in shaker incubator at 37 °C. The toxin was extracted by TCA precipitation from the 48-h old cultures grown in BHI broth (Nguyen 2014). The immunocapture sandwich complex with reporter DNA was carried out in PCR ELISA plate employing toxin and culture extracted toxin as described earlier. Real-time PCR based amplification was performed with the standardized condition, and the results were monitored.
Sensitivity testing of RT-IPCR
The sensitivity of the real-time immunocapture was determined using various concentrations of r-SEB (10 fg to 1 ng). Briefly, SEB was added to pre-coated microtiter plate wells with anti-SEB IgY, assay was performed as mentioned earlier, and the result was observed.
Evaluation of RT-IPCR on spiked food sample and natural samples and comparison with available kit
Working efficiency of developed RT-IPCR was evaluated in spiked milk samples as well as naturally contaminated samples. Various concentrations of toxin (10 fg to 1 ng) spiked in 1 mL of milk. The toxin was extracted from spiked milk samples and RT-IPCR as mentioned earlier. For evaluation from natural samples, 45 natural contaminated samples were enriched in TSB broth at 37 °C for 30–48 h, the toxin was extracted by the TCA precipitation method, and RT-PCR assay was performed. Moreover, a total of 91 numbers of laboratory repository biochemically confirmed S. aureus was also tested by RT-IPCR, and results were compared with in-house developed PCR kit.
Statistical analysis
All the experiments were done in triplicate; results were tested for significance using the parametric of one-way analysis of variance (ANOVA), and those with p values less than 0.05 were considered significant.
Result
Generation of anti-SEB IgY and determination of affinity
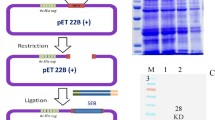
For the generation of anti-SEB-IgY, purified r-SEB protein was immunized into chicks along with Freund’s adjuvant. IgY was purified employing PEG method (Fig. 2a). Significant antibody titre was observed even at 1:32000 dilutions after 42 days of immunization. The affinity of IgY to native SEB toxins was confirmed by Western blot analysis using purified r-SEB which showed a clear band of SEB at 28.3 kDa (Fig. 2b).

a Extraction and purification of IgY antibody. Extracted IgY (L1), purified IgY (L2–L5), and L6 marker. b Reactivity of IgY by Western blot. L1: native SEB, L2: denatured SEB, and L3: marker. c Biotinylation of anti SEB monoclonal antibody (P1–P5) various elution of monoclonal antibody, P6: non-biotinylated antibody, (P7, P8): control
Generation of biotinylated anti-SEB monoclonal antibody
Biotinylation of anti-SEB antibodies was confirmed by dot blot assay. The biotinylated antibodies and non-biotinylated antibodies were coated on nitrocellulose membrane followed by probing with streptavidin-HRP conjugate and developed with TMB/H2O2. The position where biotinylated antibodies were spotted, there was a development of brown dots in the presence of TMB/H2O2, whereas no chromogenic reaction was observed with the non-biotinylated antibodies (Fig. 2c).
Standardization of reporter DNA amplification and sensitivity of RT-IPCR
The real-time PCR amplification was carried out in standardized conditions. The real-time amplification of reporter DNA started at a Cq greater than 12 which was confirmed by agarose gel electrophoresis (Fig. 3a). The sensitivity of RT-IPCR was evaluated employing various concentrations of r-SEB. A linear increased Cq range approximately 12 to 26 with the increased toxin concentration was observed which allowed quantitative detection of SEB (Fig. 3b). The assay could detect 100 fg/mL of toxins without any hindrance (Fig. 3c). Higher amplification was observed at higher concentration of toxin and reduced with the decrease in the antigen concentration.

Sensitivity of RT-IPCR. a Monitoring of amplification in agarose gel (L1-L6)1 ng to 10 fg of toxin, L7: control. b Real-time-based monitoring for amplification of various concentration of toxin. c Linear correlation for various concentration of toxin
Specificity of RT-IPCR
The specificity of the RT-IPCR was confirmed by testing with other related enterotoxins of S. aureus (r-SEA, r-SEC, and r-SEG), and TCA precipitated culture supernatants of related and other pathogen listed in Table 2. The real-time analysis confirmed that the assay was able to detect enterotoxin B producing S. aureus strain without any difficulty which was further confirmed by PCR-based kits. No amplification was observed with the other related toxins or culture supernatant of various bacterial cultures.
Sensitivity of RT-IPCR in spiked milk sample
In an infection scenario, milk products are regarded as S. aureus major source of infection and also in the milk, and they are present along with other non-specific bacterial proteins or milk-related proteins which might inhibit detection thereby resulting in false-negative results. Hence, applicability of the assay was proven from the spiked milk sample. The developed RT-IPCR assay can detect 1 pg/mL of toxin that proves the efficiency of the newly described assay (Fig. 4a–c).

Sensitivity of RT-IPCR in spiked sample. a Monitoring of amplification in agarose gel (L1–L4)1 ng to 1 pg of toxin, L5: control. b Real-time-based monitoring for amplification of various concentration of spiked toxin. c Linear correlation for various concentration of spiked toxin
Evaluation of RT-IPCR on natural samples
To validate the robustness and practical application of the assay, samples were collected from various sources (N = 45) over different periods and processed separately for the presence of SEB. The assay was able to detect confirmed SEB from a naturally contaminated source which is further validated with in-house developed PCR kit, and an exact similar result was obtained (Table 3). Moreover, this RT-PCR assay could detect all the (N-91) SEB positive strains of repository isolates without exhibiting any non-specificity (Table 4).
Discussion
Enterotoxigenic S. aureus is considered the most threatening pathogen due to its efficiency to produce a large number of toxins and possesses a threat of foodborne infection as well as clinical symptoms. Among the enterotoxins, SEB is the most considered as a biowarfare agent due to its potency, toxicity, superantigenicity, easy to aerosolize, and can be produced in bulk. Hence, detection and quantification of lower concentration SEB are the highest priority for food safety and national security application (Argudín et al. 2010; Ortega et al. 2010; Fratamico et al. 2005). Therefore, in this study, we have attempted for the development of RT-IPCR-based platform for detection and quantification of SEB. The assay combines immune-based capture of target and exponential signal amplification capability of real-time PCR using revealing antibody probed with DNA. Capturing IgY probe was generated by immunizing r-SEB in chicks.
S. aureus is a commensal that often produces diverse toxins in a similar environment, food matrices, and infection scenarios. The inherent affinity of the mammalian IgG Fc region with S. aureus protein A(SpA) leads to false-positive results. The avian (IgY) antibodies have no inherent affinity towards protein A and are more humane, convenient, inexpensive, and are therefore a preferred alternative in the development of specific detection assays (Nagaraj et al. 2016; Jin et al. 2013; Carlander et al. 1999, and Ljungberg et al. 1993). Therefore, the RT-IPCR was designed and developed using antigen-capture IgY antibodies to avoid the background signal or cross reactivity. Moreover, the PCR plates were blocked with 0.5% bovine serum albumin to avoid non-specific binding of analytes to sites. The exponential signal amplification power of RT-PCR was enhanced by multiple washing steps to minimize non-specific binding of the monoclonal revealing antibody. For revealing probe, we have employed biotinylated anti-SEB monoclonal antibodies conjugated with distinguishable size 80 bp biotinylated oligomers via a streptavidin bridge for signal amplification. The oligomers were randomly synthesized and free from restriction which also minimized the chance of nonspecific amplification and false-positive results encountering in bacterial specimens/samples. The immunocapture and PCR steps were performed in 96 well PCR plates to overcome the necessity of thermal or chemical disruption of antibody-DNA complex for RT-IPCR purposes which would contrarily be necessary in case of immune-capture in microtiter wells. The newly described and developed RT-IPCR is highly specific to SEB without exhibiting cross reactivity with other related (viz., SEC, SEG, and SEA) and TCA precipitate culture supernatants of non-target bacterial species. Moreover, this RT-IPCR is more sensitive than normal PCR with the sensitivity of 100 fg/mL.
To date, the major challenges in identifying toxins from environmental matrices are due to the low concentration of toxin required with inhibitory substances in complex environmental and clinical matrices. The developed RT-IPCR could directly detect the SEB from spiked/artificially contaminated samples accurately avoiding the need for pre-enrichment or extensive toxin extraction procedures. Moreover, laboratory repository (Mondal et al. 2018) isolates testing and comparison with seb specific in-house PCR-based detection confirmed the assay feasibility and practicability. The major enhancement of LOD by the RT-IPCR over the traditional detection method was probably is one of the important crucial merits of the system. Firstly, the capture antibody facilitated the concentration of the analytes and removal of other contaminants by washing thereby ensuring high-affinity oriented binding of SEB from complex environmental samples, or food matrices by. Secondly, multiple washing steps removed the components from the sample matrix that might interfere with the detection. Thirdly, incorporation of an optimized streptavidin-reporter DNA combination in the assay design was instrumental in improving assay sensitivity by real-time PCR. Therefore, this highly sensitive RT-IPCR demonstrates its applicability to serve as an efficient and rapid detection platform for SEB from food and environmental samples for routine use in microbiological laboratories especially for the identification of potentially low concentration of target which cannot be monitored by ELISA. Furthermore, sensitivity, specificity, and cost, the assay is comparable with the available detection system. Hence, the developed assay platform can be utilized for detection of other pathogens and toxins for qualitative and quantitative detection in routine microbiological laboratory.
References
Alefantis T, Grewal P, Ashton J, Khan AS, Valdes JJ, Del Vecchio VG (2004) A rapid and sensitive magnetic bead-based immunoassay for the detection of staphylococcal enterotoxin B for high-through put screening. Mol Cell Probes 18(6):379–382
Argudín MÁ, Mendoza MC, Rodicio MR (2010) Food poisoning and S. aureus enterotoxins. Toxins 2(7):1751–1773
Babu D, Muriana PM (2011) Immunomagnetic bead-based recovery and real time quantitative PCR (RT iq-PCR) for sensitive quantification of aflatoxin B1. J Microbiol Methods 86(2):188–194
Carlander D, Stålberg J, Larsson A (1999) Chicken antibodies: a clinical chemistry perspective. Ups J Med Sci 104(3):179–189
Fischer A, von Eiff C, Kuczius T, Omoe K, Peters G, Becker K (2007) A quantitative real-time immuno-PCR approach for detection of staphylococcal enterotoxins. J Mol Med 85(5):461–469
Fitzmaurice J, Glennon M, Duffy G, Sheridan JJ, Carroll C, Maher M (2004) Application of real-time PCR and RT-PCR assays for the detection and quantitation of VT 1 and VT 2 toxin genes in E. coli O157: H7. Mol Cell Probes 18(2):123–132
Fratamico PM, Bhunia AK, Smith JL (eds) (2005) Foodborne pathogens: microbiology and molecular biology. Horizon Scientific Press
Greenfield RA, Slater LN, Bronze MS, Brown BR, Jackson R, Iandolo JJ, Hutchins JB (2002) Microbiological, biological, and chemical weapons of warfare and terrorism. Am J Med Sci 323(6):326–340
Jin W, Yamada K, Ikami M, Kaji N, Tokeshi M, Atsumi Y, Baba Y (2013) Application of IgY to sandwich enzyme-linked immunosorbent assays, lateral flow devices, and immunopillar chips for detecting staphylococcal enterotoxins in milk and dairy products. J Microbiol Methods 92(3):323–331
Klotz M, Opper S, Heeg K, Zimmermann S (2003) Detection of S. aureus enterotoxins A to D by real-time fluorescence PCR assay. J Clin Microbiol 41(10):4683–4687
Ljungberg UK, Jansson B, Niss U, Nilsson R, Sandberg BE, Nilsson B (1993) The interaction between different domains of staphylococcal protein A and human polyclonal IgG, IgA, IgM and F (ab') 2: separation of affinity from specificity. Mol Immunol 30(14):1279–1285
Mehta PK, Raj A, Singh NP, Khuller GK (2014) Detection of potential microbial antigens by immuno-PCR (PCR-amplified immunoassay). J Med Microbiol 63(5):627–641
Mondal B, Ramlal S, Kingston J (2018) Colorimetric DNAzyme biosensor for convenience detection of enterotoxin B harboring S. aureus from food samples. J Agric Food Chem 66(6):1516–1522
Nagaraj S, Ramlal S, Kingston J, Batra HV (2016) Development of IgY based sandwich ELISA for the detection of staphylococcal enterotoxin G (SEG), an egc toxin. Int J Food Microbiol 237:136–141
Nguyen PK (2014) Investigating the rate of the S. aureus nasal carriage in Vietnamese community. Doctoral dissertation, International University, Vietnam
Niemeyer CM, Adler M, Wacker R (2005) Immuno-PCR: high sensitivity detection of proteins by nucleic acid amplification. Trends Biotechnol 23(4):208–216
Nolasco G, De Blas C, Torres V, Ponz F (1993) A method combining immunocapture and PCR amplification in a microtiter plate for the detection of plant viruses and subviral pathogens. J Virol Methods 45(2):201–218
Ortega E, Abriouel H, Lucas R, Gálvez A (2010) Multiple roles of S. aureus enterotoxins: pathogenicity, superantigenic activity, and correlation to antibiotic resistance. Toxins 2(8):2117–2131
Pauly D, Chacana PA, Calzado EG, Brembs B, Schade R (2011) IgY technology: extraction of chicken antibodies from egg yolk by polyethylene glycol (PEG) precipitation. J Visual Exp 51
Ramlal S, Mondal B, Lavu PS, Bhavanashri N, Kingston J (2018) Capture and detection of S. aureus with dual labeled aptamers to cell surface components. Int J Food Microbiol 265:74–83
Rong-Hwa S, Shiao-Shek T, Der-Jiang C, Yao-Wen H (2010) Gold nanoparticle-based lateral flow assay for detection of staphylococcal enterotoxin B. Food Chem 118(2):462–466
Schmitz FJ, Steiert M, Hofmann B, Verhoef J, Hadding U, Heinz HP, Köhrer K (1998) Development of a multiplex-PCR for direct detection of the genes for enterotoxin B and C, and toxic shock syndrome toxin-1 in S. aureus isolates. J Med Microbiol 47(4):335–340
Wu S, Duan N, Gu H, Hao L, Ye H, Gong W, Wang Z (2016) A review of the methods for detection of S. aureus enterotoxins. Toxins 8(7):176
Xu SX, Mccormick J (2012) Staphylococcal superantigens in colonization and disease. Front Cell Infect Microbiol 2:52
Zhang W, Bielaszewska M, Pulz M, Becker K, Friedrich AW, Karch H, Kuczius T (2008) New immuno-PCR assay for detection of low concentrations of Shiga toxin 2 and its variants. J Clin Microbiol 46(4):1292–1297
Acknowledgements
The first author is indebted to the Department of Science and Technology (DST, New Delhi, India) for financial assistance through INSPIRE program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Research involving human participants and/or animals
NA
Informed consent
NA
Funding
All the authors are indebted to DFRL, DRDO for providing laboratory and other facility to carry out the research work.
Author information
Authors and Affiliations
Contributions
BM carried out the design of the study, performing experiments, result analysis, and drafted the manuscript. SR carried out the immunoassays, results analysis, and drafted the manuscript. KS participated in the design of the experiments and performed the analysis. AS was involved in real-time PCR data analysis and performing the experiments. SA carried out the experiment and drafted the manuscript. MP carried out data analysis and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The animal experimental performed in the present research was in accordance with the animal usage protocols approved by the Institutional Animal Ethical Committee, Defence Food Research Laboratory (DFRL/28/IAEC/CPCSEA), completely accredited by Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), India.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial and non-financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mondal, B., Ramlal, S., Setlem, K. et al. A real-time immunocapture PCR (RT-IPCR) without interference of protein A for convenient detection of staphylococcal enterotoxin B from food and environmental samples. Ann Microbiol 70, 25 (2020). https://doi.org/10.1186/s13213-020-01567-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13213-020-01567-8