
Abstract
Background
The association of scaling and root planing (SRP) with systemic metronidazole (MTZ) plus amoxicillin (AMX) has shown to be an effective treatment protocol, particularly for periodontitis stages III and IV, generalized. More recently, probiotics have also been suggested as a promising adjunctive treatment for periodontal diseases due to their antimicrobial and anti-inflammatory properties. Therefore, the aim of this randomized clinical trial (RCT) is to evaluate the clinical, microbiological, and immunological effects of probiotics as adjuncts to SRP alone or with MTZ+AMX in the treatment of periodontitis.
Methods
Subjects with periodontitis are being randomly assigned to receive (i) SRP alone, or with (ii) two probiotic lozenges/day for 90 days (Prob), (iii) MTZ (400 mg) and AMX (500 mg) thrice a day (TID) for 14 days (MTZ+AMX), or (iv) Prob and MTZ+AMX. Subjects are being monitored for up to 12 months post-treatment. Nine subgingival plaque samples per patient are being collected at baseline and at 3, 6, and 12 months post-therapy and analyzed by checkerboard DNA–DNA hybridization for 40 bacterial species. Peripheral blood and gingival crevicular fluid (GCF) of four randomly selected periodontal sites will be analyzed by means of a multiplex fluorescent bead-based immunoassay for 17 cyto/chemokines.
Statistical analyses
The significance of differences in each group (over the course of the study) will be sought using repeated measures ANOVA or Friedman tests and among groups (at each time point) using either ANOVA/ANCOVA or Kruskal-Wallis tests, depending on normality of the data. The chi-square test will be used to compare differences in the frequency of subjects achieving the clinical endpoint for treatment (≤ 4 sites with PD ≥ 5 mm) at 1 year and of self-perceived adverse effects. A stepwise forward logistic regression analysis will be performed in order to investigate the impact of different predictor variables on the percentage of patients achieving the clinical endpoint for treatment. The Number Needed to Treat (NNT) with different treatment protocols will be also calculated. Statistical significance will be set at 5%.
Trial registration
ClinicalTrials.gov NCT03733379. Registered on November 7, 2018.
Similar content being viewed by others


Background
Scaling and root planing (SRP), the standard of care for treatment of periodontitis, leads to an overall improvement in clinical parameters, including reduction in probing depth (PD), percentage of sites with bleeding on probing (BOP), and gain in clinical attachment [1,2,3]. However, SRP does not lead to major and sustainable clinical improvements in all subjects and periodontal sites, especially in cases of severe periodontitis [1, 4,5,6]. This is probably because SRP alone is unable to produce a sufficiently deep change in the oral microbial ecology (rebiosis) in order to produce a new stable climax biofilm community compatible with periodontal health [1, 2, 6,7,8,9]. Therefore, adjunctive therapies, such as systemic antimicrobials [6, 10,11,12,13,14,15,16,17,18,19] and probiotics [20,21,22,23,24], have been proposed with the purpose of potentiating the effects of SRP.
Among all the systemic antibiotics tested to date in the treatment of periodontitis, the combination of metronidazole (MTZ) and amoxicillin (AMX) has been shown to offer the greatest clinical and microbiological benefits. There is consistent scientific evidence showing that the adjunctive use of MTZ+AMX improves the outcomes of SRP, including a greater reduction in the number of deep pockets after treatment and, consequently, in the need for surgical interventions [19]. A number of ecological benefits have been associated with the use of MTZ+AMX during the active phase of treatment. This adjunctive therapy is effective in reducing key periodontal pathogens, such as Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans [25,26,27,28,29,30] in periodontal pockets and on other oral surfaces, and to foster an increase in the proportions of beneficial microorganisms [6].
More recently, probiotics have also been suggested as promising adjunctive treatments for periodontitis [20,21,22,23,24, 31,32,33,34,35]. The effects of probiotics in periodontal treatment have been investigated in vitro and in vivo [36,37,38]. Subgingival recolonization by periodontopathogens [36] and the level of local inflammation [37] are delayed or prevented with the use of probiotics. Probiotics also had a favorable impact on bone density within periodontal pockets [39]. However, few randomized clinical trials (RCTs) have evaluated the effectiveness of systemic probiotics as adjuncts to SRP in periodontal treatment [40,41,42,43,44,45,46,47,48]. The results of these RCTs have indicated an overall improvement in clinical parameters with the adjunctive use of systemic probiotics in the treatment of periodontitis. However, to date, no robust RCT have evaluated the effects of systemic probiotics in periodontal treatment or have compared the outcomes of this treatment with those obtained with the adjunctive use of systemic antibiotics.
Methods/design
Aim, design, and setting of the study
The aim of this study is to evaluate the clinical, microbiological, and immunological effects of systemic probiotics as an adjunct to SRP alone or in combination with MTZ+AMX in the treatment of periodontitis. In order to address this aim, a double-blinded, four-armed, placebo-controlled, and bicentric RCT was designed. This study is being conducted at Guarulhos University (UNG; Guarulhos, SP, Brazil) and Federal University of Parana (UFPR; Curitiba, PR, Brazil). The study protocol was developed according to the Standard Protocol Items: Recommendations for Interventional Studies (SPIRIT) guidelines and using the SPIRIT checklist (Additional file 1) [49] and was registered in the ClinicalTrials.gov (NCT03733379) database.
Ethical considerations
This trial is being conducted according to the principles of the Declaration of Helsinki for studies involving human subjects. The protocol was approved by the Institutional Review Board of UNG (Research Ethics Committee, CAAE: 74049717.7.1001.5506) and UFPR (Research Ethics Committee, CAAE: 74049717.7.3001.0102). All eligible volunteers are being informed about the nature, potential risks, and benefits of their participation in this study, and they sign a term of informed consent (Additional file 2).
Subject population and inclusion/exclusion criteria
Systemically healthy volunteers with untreated generalized severe periodontitis (periodontitis stages III or IV, generalized [50];) are being selected from the Center for Clinical Trials of UNG and Periodontal Clinic of UFPR. Subjects are being selected according to the following inclusion criteria: presence of at least 15 teeth (excluding third molars and teeth indicated for extraction), at least 30% of the teeth with PD and CAL ≥ 4 mm, and a minimum of 6 teeth with at least one interproximal site each with PD and clinical attachment level (CAL) ≥ 5 mm. Exclusion criteria are as follows: pregnancy, breastfeeding, current smokers and former smokers—for at least 5 years [51], systemic diseases that could affect the progression of periodontitis (e.g., diabetes, immunological disorders, osteoporosis), SRP in the previous 12 months, antibiotic therapy in the previous 6 months, long-term intake of anti-inflammatory drugs, need for antibiotic pre-medication for dental treatment, use of orthodontic appliances, extensive prosthetic rehabilitation, and allergy to MTZ and/or AMX.
Interventions
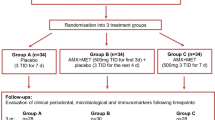
At baseline, all volunteers fulfill a structured questionnaire to collect information about demographic, oral, and general health data. Subsequently, a complete periodontal clinical assessment and subgingival biofilm sampling are performed. One week later, gingival crevicular fluid (GCF) and peripheral blood samples are collected. Patients receive oral hygiene instruction (OHI), including tooth brushing and interproximal dental hygiene (using dental floss and interdental brushes). Other elements may be indicated according to individual needs, such as subjects with removable prostheses. In addition, supragingival scaling using ultrasonic scaler (Cavitron® Select™ Ultrasonic Scaler, Denstply, New York, USA) and curettes (Millenium, GOLGRAN, São Caetano do Sul, SP, Brazil) are performed. The plaque index is repeated 1 week after OHI and supragingival plaque removal. Subsequently, each volunteer is randomly allocated to one of the following therapeutic groups: (i) control: SRP +two antibiotic placebo pills three times a day (TID) for 14 days + two probiotic placebo lozenges a day for 90 days; (ii) probiotic (test 1): SRP + two antibiotic placebo pills TID for 14 days + two probiotic lozenges a day for 90 days; (iii) antibiotic (test 2): SRP + MTZ 400 mg and AMX 500 mg TID for 14 days + two probiotic placebo lozenges a day for 90 days; and (iv) antibiotic + probiotic (test 3): SRP + MTZ 400 mg and AMX 500 mg TID for 14 days + two probiotic lozenges a day for 90 days (Figs. 1 and 2).

Experimental design of the study. AMX, amoxicillin (500 mg, 3 times × day); CLIN, clinical assessment; OHI, oral hygiene instruction; IMUNO, immunological assessment; MICRO, microbiological assessment; MTZ, metronidazole (400 mg, 3 times × day); Prob, probiotic; PMT, periodontal maintenance therapy; SRP, scaling and root planing

Schedule of volunteers’ enrolment, interventions, and assessments according to the SPIRIT statement. AMX, amoxicillin; Mo, months; MTZ, metronidazole
The administration of all medications (probiotic and antibiotics) and placebos starts immediately after the first SRP session. Antibiotics (or placebos) are given TID for 14 days and include two tablets, one of MTZ and one of AMX. Antibiotics and placebos are being specially prepared for this study by the same pharmacy (Gallen Pharmacy and Manipulation Ltda, Maringá, PR, Brazil). All tablets have the same color and size and are stored in properly coded opaque plastic bottles, containing 22 capsules/lozenges. Volunteers, therapists, and examiners are blind to the interventions applied.
Probiotic (or placebo) are administered two times per day for 90 days and include one lozenge every 12 h after brushing and flossing. The second lozenge is administered just before going to sleep. The lozenges are slowly dissolved in the volunteer’s oral cavity. Probiotic and placebo are being prepared for this study by BioGaia AB (Stockholm, Sweden). Probiotic lozenges contain 2 different strains of Lactobacillus reuteri: L. reuteri DSM 17938 and L. reuteri ATCC (PTA 5289), each at a minimum amount of 1 × 108 CFU per lozenge. Placebo probiotic lozenges are identical to the active lozenges in taste, texture, and shape, but they do not contain L. reuteri. All lozenges are kept refrigerated at a temperature below 25 °C until they are handed to the patient.
SRP is performed using Gracey curettes (conventional and mini-fives) 5/6, 7/8, 11/12, and 13/14 under local anesthesia, and the treatment is completed in four to six sessions of approximately 1 h, distributed over a period of 14 days. At the end of each session, the clinical coordinator of each center evaluates the effectiveness of SRP using the outcome “smoothness of the scaled roots.” All volunteers will receive clinical monitoring at baseline and at 3, 6, and 12 months post-therapies. In addition, all subjects will receive periodontal maintenance every 3 months post-treatment until the end of the study. These sessions include OHI and supragingival/subgingival biofilm/calculus removal, as necessary. If any participant shows periodontal attachment loss ≥ 2 mm in 3 or more sites during the maintenance phase, they will be excluded from the study and referred for periodontal re-treatment.
Monitoring of compliance and adverse events
A study assistant in each clinical center is monitoring the compliance with intake of probiotic/antibiotics/placebos by calling the patients 2 times a week during the first 3 weeks of medication intake. After that, participants are monitored once a week for the next 9 weeks. Volunteers should take 21 antibiotics tablets in the first week and 21 in the second week. They are instructed to return the bottles at beginning of the second week of treatment and receive new bottles containing the same amount of medication to take during the second week. Volunteers are not informed about the total number of tablets in each bottle; therefore, at the end of each week, it is possible to confirm if any residual capsule (22nd) is left in the bottle, to enable compliance to be monitored. Subjects are also asked to return the bottles at 45 days and 90 days post-SRP, and the bottles are checked. On the 14th and at 90 days of the medication intake, subjects answer a questionnaire about any self-perceived side effects.
Clinical examination (baseline, 3, 6, and 12 months)
Two calibrated examiners, one from each center (UNG and UFPR), are performing the clinical evaluations and collection of biofilm, GCF, and blood samples. The following periodontal parameters are being evaluated: visible plaque [52], gingival bleeding (0/1), BOP (0/1), SUP (0/1), PD (mm), and CAL (mm) at six sites per tooth (mesiobuccal, buccal, distobuccal, distolingual/palatal, lingual/palatal and mesiolingual/palatal) in all teeth, excluding third molars. PD and CAL measurements are rounded to the nearest millimeter using a North Carolina periodontal probe (Hu-Friedy, Chicago, IL, USA).
Investigator’ calibration
The two examiners (B.R.V. and G.M.S.S.) were trained and calibrated prior to the trial, in order to achieve maximum reproducibility in the measurements. The calibration exercise for both centers was conducted at UNG before the beginning of the study, as previously described [53]. This exercise will be repeated once a year. Briefly, the standard error of measurement for continuous periodontal clinical parameters (PD and CAL) is evaluated. For the other clinical variables, the average level of agreement between the examiners is determined and considered satisfactory when the value is higher than 90% (Kappa test).
Microbiological monitoring (baseline, 3, 6, and 12 months)
Sample collection
Nine subgingival samples are being collected per volunteer, three in each of the following categories: shallow (PD ≤ 3 mm), moderate (PD = 4–6 mm), and deep (PD ≥ 7 mm). The selected sites are located on non-contiguous interproximal teeth, preferably distributed across the four quadrants and subset according to baseline PD. Teeth with open margins and overhangs, furcation lesions, extensive caries, and/or endo-periodontal lesions are not selected for sampling. After recording the clinical parameters, supragingival plaque is removed and the subgingival samples are collected with individual sterile mini-Gracey curettes #11–12, positioned at the most apical portion of the subgingival sulcus/pocket and with a single apical-coronal movement. The samples are immediately placed in separate Eppendorf tubes containing 150 ul of TE buffer solution (10 mM Tris-HCl, 1 mM EDTA, pH 7.6), and then 100 ul of 0.5 M NaOH is added to each tube and the samples are dispersed using a vortex mixer. The samples are being stored at − 20 °C. The tubes containing the samples are previously identified with the volunteer’s code, date, and site collected.
Processing of microbiological samples
Counts and proportion of 40 bacterial species will be determined in each sample, using the Checkerboard DNA–DNA hybridization technique [54, 55]. The microbiological analysis will be entirely performed at the Laboratory of Microbiology of UNG. Briefly, the suspensions containing the bacterial biofilm samples will be boiled for 10 min and neutralized using 0.8 ml of 5 M ammonium acetate, and the DNA is released in the solution. The released DNA will be then placed into the extended slots of a Minislot 30 apparatus (Immunetics, Cambridge, MA, USA) concentrated on a 15 × 15 cm positively charged nylon membrane (Boehringer Mannheim, Indianapolis, IN, USA) and fixed to the membrane by baking it at 120 °C for 20 min. Subsequently, the membrane will be placed in a Miniblotter 45 (Immunetics, Cambridge, MA, USA) with the lanes of DNA at 900 to the lanes of the device. Digoxigenin-labeled whole genomic DNA probes for 40 bacterial species are hybridized in individual lanes of the Miniblotter 45. After hybridization, the membranes are washed in highly astringent solution and the DNA probes are detected using the antibody to digoxigenin conjugated with alkaline phosphatase and chemiluminescence detection. The last two lanes in each run contain standards at concentrations of 105 and 106 cells of each species. Signals will be evaluated visually by comparison with the standards for the test species on the same membrane by a calibrated examiner (k test = 93%). The sensitivity of this assay will be adjusted to allow detection of 104 cells of a given species by adjusting the concentration of each DNA probe [54,55,56].
Immunological monitoring (baseline and 12 months)
Serum and GCF sampling
Peripheral blood samples and GCF are being collected 1 week after clinical examination. Two non-contiguous diseased sites (i.e., PD and CAL > 5 mm, BoP and no furcation involvement) and two non-contiguous healthy sites (i.e., PD and CAL < 4 mm without BOP and/or gingival bleeding) are being randomly chosen per patient for GCF sampling (among the same sites selected for microbiological sampling). After removal of the supragingival biofilm, the selected sites are isolated with cotton rolls and gently dried with an air syringe to eliminate saliva. GCF is collected by inserting standard paper strips (Periopaper, Oraflow Inc., Smithtown, NY, USA) up to approximately 2 mm into the sulcus/pocket for 30 s. Strips visually contaminated with blood are discarded. The GCF volume is measured in a calibrated device (Periotron 8000, Proflow Inc., Amityville, NY, USA), and the readings are converted into an actual volume (microliter, μl) by reference to a standard curve. Strips from the four selected sites are immediately placed into separate microcentrifuge tubes and stored at − 80 °C for subsequent assays. Peripheral blood samples are into an appropriate tube (Serum BD Vacutainer Plus Plastic Serum Tubes, BD, Franklin Lakes, NJ). Fasted samples are obtained in the morning for all patients. Immediately after blood collection, the serum is separated from blood by centrifugation (10 min at 1300 rpm) and stored in aliquots at − 80 °C.
Processing of serum and GCF samples
The tubes containing the samples will be vortexed for 15 s and centrifuged for 5 min at 1500×g in order to elute. Samples will be analyzed using a Multiplex Bead Immunoassay (MAGPIX® System, Merck Millipore, Billerica, MA, USA) by means of a multiplex fluorescent bead-based immunoassay for 17 cyto/chemokines (eotaxin, GCSF, GM-CSF, interferon (IFN)-γ, IL-1β, IL-2, IL-6, IL-7, IL-8, IL-10, IL-12, protein chemoattractant monocyte [MCP]-1, MIP-1α, and tumor necrosis factor [TNF]-α) using commercially available kits (Milliplex Human Cytokine/Chemokine, EMD Millipore, Billerica, MA, USA) and a plate reader (Luminex. 100TM, EMD Millipore, Billerica, MA, USA), in accordance with the manufacturer’s recommendations. The amount of protein in each sample will extrapolate from standards using an appropriate software (Beadview EMD Millipore, Billerica, MA, USA). The minimum detectable concentrations for eotaxin, G-CSF, GM-CSF, IFN-γ, IL-1β, IL-2, IL-6, IL-7, IL-8, IL-10, IL-12, MCP-1, MIP-1α, and TNF-α are as follows: 1.2 pg/ml, 0.5 pg/ml, 9.5 pg/ml, 0.1 pg/ml, 0.4 pg/ml, 0.3 pg/ml, 0.3 pg/ml, 1.8 pg/ml, 0.2 pg/ml, 0.3 pg/ml, 10.5 pg/ml, 0.9 pg/ml, 3.5 pg/ml, and 0.1 pg/ml, respectively. The results will be reported as total amount (pg/site/30 s) and concentrations of cyto/chemokines per volume of GCF (pg/μl) and serum (pg/ml).
Primary and secondary outcome variables
The primary outcome variable is the difference among groups for the percentage of volunteers achieving the clinical endpoint for therapy (≤ 4 sites with PD ≥ 5 mm) at 1 year post-treatment [57].
Secondary outcome variables are as follows: difference between baseline and 12 months post-therapy for mean CAL gain and PD reduction (in the full mouth and in different PD categories), mean number and percentage of volunteers with 0, 1–2, or ≥ 3 sites with different thresholds of residual pockets (e.g., PD ≥ 6 and ≥ 7 mm) at 12 months post-therapy; mean number (and mean reductions) of different thresholds of residual pockets at all post-treatment time points; difference among groups for the percentage of volunteers achieving different thresholds of BOP (e.g., ≤ 10%, 20% and 30% of bleeding sites in the mouth) at 1 year post-treatment; differences in the occurrence of adverse events among therapeutic groups; and differences in the counts and proportions of 40 bacterial species in the biofilm samples at all post-treatment time points and in the levels of 17 chemokines in the GCF and peripheral blood samples at 1 year post-therapy.
Sample size calculation
The ideal sample size to assure adequate power for this study was based on the percentage of volunteers achieving “≤ 4 sites with PD ≥ 5 mm” at 1 year post-treatment [57]. Considering a difference of 44 percentage points between groups for volunteers achieving this clinical endpoint at 1 year post-treatment [5] and a significance level of 5%, 17 subjects per group would be necessary to provide a power of 80%. Considering an attrition of 25%, it was established that at least 22 subjects should be included in each treatment group. Considering a difference of 30 percentage points between groups for volunteers achieving this clinical endpoint at 1-year post-treatment and a significance level of 5% 37 subjects per group would be necessary to provide a power of 80%. Considering an attrition rate of 15%, it was established that 44 volunteers should be included in each treatment group (total 176 subjects).
Randomization
The study coordinator (M.Fe.), who is not involved in the inclusion and treatment of patients, assigned the study participants to one of the four treatment groups by means of a computer-generated random sequence (Random Allocation Software, http://random-allocation-software.soft). Randomization was stratified by center using permuted blocks of 4, 8, and 12. The study coordinator also organizes the bottles with the antibiotics tablets in opaque plastic bags labeled with the volunteer’s number. In addition, one clinical investigator and the study coordinator sent the randomization list to the BioGaia AB Company (Stockholm, Sweden). The company made and packed the probiotic (and placebo) lozenges in boxes labeled with each volunteer’s number. The bags are handed directly to the volunteer by the study coordinator. This sequence of procedures assures allocation concealment.
Data collection and management
Clinical data are entered directly onto electronic spreadsheets (Microsoft® Excel 2011®, version 14.4.8, Copyright© 1990, Microsoft, Santa Rosa, California, USA), at the time of clinical examination, by a single investigator in each center. Data quality will be validated by checking missing data, out-of-range values, and invalid responses. The analyses will be conducted using a statistical program developed by Sigmund Socransky (Forsyth, USA), as well as SAS and GraphPad Prism (version 7.0) software.
Statistical analysis
Clinical monitoring
The chi-square test will be used to compare the differences in the frequency of gender and to compare the differences in the frequency of subjects achieving the clinical endpoint for treatment (≤ 4 sites with PD ≥ 5 mm) at 1-year (primary outcome variable) and of self-perceived adverse effects. Each individual continue/categorical variable will be computed per subject and then across subjects in each group. The significance of clinical differences over the course of the study will be sought using repeated measures ANOVA or Friedman multiple comparison tests (and their respective post-hoc tests, when necessary), and at each time point (among groups), using either ANOVA or Kruskal-Wallis multiple comparison tests (and their respective post-hoc tests when necessary) or ANCOVA with adjustments for the baseline values. Parametric or non-parametric tests will be chosen based on the normality of the data. A stepwise forward logistic regression analysis will be performed to investigate the impact of predictor variables on the clinical endpoint for treatment, i.e., “presence of ≤ 4 sites with PD ≥ 5 mm at 1-year post-therapy (yes/no).” The Number Needed to Treat (NNT) with each treatment protocol in order to obtain treatment success (≤ 4 sites with PD ≥ 5 mm) will be calculated using the following formula: NNT = 1/ARR, where ARR = |CER − EER| and CER = control group event rate and EER = experimental group event rate (http://ktclearinghouse.ca/cebm/glossary/nnt). The level of significance will be set at 5%. The data will be evaluated using intention-to-treat analysis with last observation carried forward.
Microbiological and immunological monitoring
Microbiological data will be expressed in counts (levels) and proportion counts of DNA probes. Mean counts will be expressed as counts × 105. Microbial data will be calculated at each site for each volunteer and then across volunteers within each group, at each time point. The sum of the mean proportions of individual bacterial species will be computed for each microbial complex described by Socransky et al. [58]. The concentration and total amount of each cytokine/chemokine in the GCF will be assessed per volunteer and then per volunteer within each group at each study time point. Immunological data will be calculated at each site for each volunteer and then across volunteers within each group, at each time point. The significance of differences in microbiological and immunological parameters over the course of the study will be sought using repeated measures ANOVA or Friedman multiple comparison tests (and their respective post-hoc tests, when necessary) and at each time point (among groups) using either ANOVA or Kruskal-Wallis multiple comparison tests (and their respective post-hoc tests when necessary) or ANCOVA with adjustments for the baseline values. Parametric or non-parametric tests will be chosen based on the normality of the data.
Trial status
The study protocol was approved by Guarulhos University (CAAE: 74049717.7.1001.5506) and Federal University of Parana (CAAE: 74049717.7.3001.0102) Ethics Committees on October 3 (2017) and February 28 (2018) respectively.
The recruitment of participants started on November 5 (2018), and the estimated study completion date is December 30 (2021).
Availability of data and materials
The authors of this manuscript declare that the study protocol was registered at ClinicalTrials.gov with register number NCT03733379 on November 7, 2018. The data during the current study will be available in the following clinical trial website: https://clinicaltrials.gov/ct2/show/NCT03733379?term=Retamal-Valdes&draw=2&rank=1
Abbreviations
- SRP:
-
Scaling and root planing
- BOP:
-
Bleeding on probing
- SUP:
-
Suppuration
- PD:
-
Probing depth
- MTZ:
-
Metronidazole
- AMX:
-
Amoxicillin
- RCT:
-
Randomized clinical trial
- UNG:
-
Guarulhos University
- UFPR:
-
Federal University of Parana
- SPIRIT:
-
Standard Protocol Items: Recommendations for Interventional Studies
- CAL:
-
Clinical attachment level
- GCF:
-
Gingival crevicular fluid
- OHI:
-
Oral hygiene instruction
- TID:
-
Thrice a day
- ATCC:
-
American Type Culture Collection
- CFU:
-
Colony forming unit
- M:
-
Molar concentration
- DNA:
-
Deoxyribonucleic acid
- GEE:
-
Generalized estimating equation
References
Haffajee AD, Cugini MA, Dibart S, Smith C, Kent RL Jr, Socransky SS. The effect of SRP on the clinical and microbiological parameters of periodontal diseases. J Clin Periodontol. 1997;24(5):324–34. https://doi.org/10.1111/j.1600-051X.1997.tb00765.x.
Cugini MA, Haffajee AD, Smith C, Kent RL Jr, Socransky SS. The effect of scaling and root planing on the clinical and microbiological parameters of periodontal diseases: 12-month results. J Clin Periodontol. 2000;27(1):30–6. https://doi.org/10.1034/j.1600-051x.2000.027001030.x.
Cobb CM. Clinical significance of non-surgical periodontal therapy: an evidence-based perspective of scaling and root planing. J Clin Periodontol. 2002;29(Suppl 2):6–16.
Goodson JM, Haffajee AD, Socransky SS, Kent R, Teles R, Hasturk H, et al. Control of periodontal infections: a randomized controlled trial I. The primary outcome attachment gain and pocket depth reduction at treated sites. J Clin Periodontol. 2012;39(6):526–36. https://doi.org/10.1111/j.1600-051X.2012.01870.x.
Feres M, Soares GM, Mendes JA, Silva MP, Faveri M, Teles R, et al. Metronidazole alone or with amoxicillin as adjuncts to non-surgical treatment of chronic periodontitis: a 1-year double-blinded, placebo-controlled, randomized clinical trial. J Clin Periodontol. 2012;39(12):1149–58. https://doi.org/10.1111/jcpe.12004.
Feres M, Figueiredo LC, Soares GM, Faveri M. Systemic antibiotics in the treatment of periodontitis. Periodontol 2000. 2015;67(1):131–86.
Soares GM, Mendes JA, Silva MP, Faveri M, Teles R, Socransky SS, et al. Metronidazole alone or with amoxicillin as adjuncts to non-surgical treatment of chronic periodontitis: a secondary analysis of microbiological results from a randomized clinical trial. J Clin Periodontol. 2014;41(4):366–76. https://doi.org/10.1111/jcpe.12217.
Silva MP, Feres M, Sirotto TA, Soares GM, Mendes JA, Faveri M, et al. Clinical and microbiological benefits of metronidazole alone or with amoxicillin as adjuncts in the treatment of chronic periodontitis: a randomized placebo-controlled clinical trial. J Clin Periodontol. 2011;38(9):828–37. https://doi.org/10.1111/j.1600-051X.2011.01763.x.
Teughels W, Feres M, Oud V, Martín C, Matesanz P, Herrera D. Adjunctive effect of systemic antimicrobials in periodontitis therapy: A systematic review and meta-analysis. J Clin Periodontol. 2020;47(Suppl 22):257–81. https://doi.org/10.1111/jcpe.13264.
Haffajee AD, Socransky SS, Gunsolley JC. Systemic anti-infective periodontal therapy. A systematic review. Ann Periodontol. 2003;8(1):115–81. https://doi.org/10.1902/annals.2003.8.1.115.
Herrera D, Sanz M, Jepsen S, Needleman I, Roldan S. A systematic review on the effect of systemic antimicrobials as an adjunct to scaling and root planing in periodontitis patients. J Clin Periodontol. 2002;29(Suppl 3):136–59; discussion 60-2. https://doi.org/10.1034/j.1600-051X.29.s3.8.x.
Sgolastra F, Gatto R, Petrucci A, Monaco A. Effectiveness of systemic amoxicillin/metronidazole as adjunctive therapy to scaling and root planing in the treatment of chronic periodontitis: a systematic review and meta-analysis. J Periodontol. 2012;83(10):1257–69. https://doi.org/10.1902/jop.2012.110625.
Sgolastra F, Petrucci A, Gatto R, Monaco A. Effectiveness of systemic amoxicillin/metronidazole as an adjunctive therapy to full-mouth scaling and root planing in the treatment of aggressive periodontitis: a systematic review and meta-analysis. J Periodontol. 2012;83(6):731–43. https://doi.org/10.1902/jop.2011.110432.
Zandbergen D, Slot DE, Cobb CM, Van der Weijden FA. The clinical effect of scaling and root planing and the concomitant administration of systemic amoxicillin and metronidazole: a systematic review. J Periodontol. 2013;84(3):332–51. https://doi.org/10.1902/jop.2012.120040.
Keestra JA, Grosjean I, Coucke W, Quirynen M, Teughels W. Non-surgical periodontal therapy with systemic antibiotics in patients with untreated aggressive periodontitis: a systematic review and meta-analysis. J Periodontal Res. 2015;50(6):689–706. https://doi.org/10.1111/jre.12252.
Keestra JA, Grosjean I, Coucke W, Quirynen M, Teughels W. Non-surgical periodontal therapy with systemic antibiotics in patients with untreated chronic periodontitis: a systematic review and meta-analysis. J Periodontal Res. 2015;50(3):294–314. https://doi.org/10.1111/jre.12221.
Rabelo CC, Feres M, Goncalves C, Figueiredo LC, Faveri M, Tu YK, et al. Systemic antibiotics in the treatment of aggressive periodontitis. A systematic review and a Bayesian network meta-analysis. J Clin Periodontol. 2015;42(7):647–57. https://doi.org/10.1111/jcpe.12427.
Zandbergen D, Slot DE, Niederman R, Van der Weijden FA. The concomitant administration of systemic amoxicillin and metronidazole compared to scaling and root planing alone in treating periodontitis: =a systematic review=. BMC Oral Health. 2016;16(1):27. https://doi.org/10.1186/s12903-015-0123-6.
Teughels W, Feres M, Oud V, Martin C, Matesanz P, Herrera D. Adjunctive effect of systemic antimicrobials in periodontitis therapy. A systematic review and meta-analysis. J Clin Periodontol. 2020.
Teughels W, Van Essche M, Sliepen I, Quirynen M. Probiotics and oral healthcare. Periodontol 2000. 2008;48:111–47.
Teughels W, Loozen G, Quirynen M. Do probiotics offer opportunities to manipulate the periodontal oral microbiota? J Clin Periodontol. 2011;38(Suppl 11):159–77. https://doi.org/10.1111/j.1600-051X.2010.01665.x.
Martin-Cabezas R, Davideau JL, Tenenbaum H, Huck O. Clinical efficacy of probiotics as an adjunctive therapy to non-surgical periodontal treatment of chronic periodontitis: a systematic review and meta-analysis. J Clin Periodontol. 2016;43(6):520–30. https://doi.org/10.1111/jcpe.12545.
Ikram S, Hassan N, Raffat MA, Mirza S, Akram Z. Systematic review and meta-analysis of double-blind, placebo-controlled, randomized clinical trials using probiotics in chronic periodontitis. J Investig Clin Dent. 2018;9(3):e12338. https://doi.org/10.1111/jicd.12338.
Vives-Soler A, Chimenos-Kustner E. Effect of probiotics as a complement to non-surgical periodontal therapy in chronic periodontitis: a systematic review. Med Oral Patol Oral Cir Bucal. 2020;25(2):e161–e7.
van Winkelhoff AJ, Rodenburg JP, Goene RJ, Abbas F, Winkel EG, de Graaff J. Metronidazole plus amoxycillin in the treatment of Actinobacillus actinomycetemcomitans associated periodontitis. J Clin Periodontol. 1989;16(2):128–31. https://doi.org/10.1111/j.1600-051X.1989.tb01626.x.
Goené RJ, Winkel EG, Abbas F, Rodenburg JP, van Winkelhoff AJ, de Graaff J. Microbiology in diagnosis and treatment of severe periodontitis. A report of four cases. J Periodontol. 1990;61(1):61–4. https://doi.org/10.1902/jop.1990.61.1.61.
van Winkelhoff AJ, Tijhof CJ, de Graaff J. Microbiological and clinical results of metronidazole plus amoxicillin therapy in Actinobacillus actinomycetemcomitans-associated periodontitis. J Periodontol. 1992;63(1):52–7. https://doi.org/10.1902/jop.1992.63.1.52.
Berglundh T, Krok L, Liljenberg B, Westfelt E, Serino G, Lindhe J. The use of metronidazole and amoxicillin in the treatment of advanced periodontal disease. A prospective, controlled clinical trial. J Clin Periodontol. 1998;25(5):354–62. https://doi.org/10.1111/j.1600-051X.1998.tb02455.x.
Winkel EG, van Winkelhoff AJ, van der Velden U. Additional clinical and microbiological effects of amoxicillin and metronidazole after initial periodontal therapy. J Clin Periodontol. 1998;25(11 Pt 1):857–64. https://doi.org/10.1111/j.1600-051X.1998.tb02382.x.
Flemmig TF, Milián E, Karch H, Klaiber B. Differential clinical treatment outcome after systemic metronidazole and amoxicillin in patients harboring Actinobacillus actinomycetemcomitans and/or Porphyromonas gingivalis. J Clin Periodontol. 1998;25(5):380–7. https://doi.org/10.1111/j.1600-051X.1998.tb02459.x.
Gruner D, Paris S, Schwendicke F. Probiotics for managing caries and periodontitis: systematic review and meta-analysis. J Dent. 2016;48:16–25. https://doi.org/10.1016/j.jdent.2016.03.002.
Matsubara VH, Bandara HM, Ishikawa KH, Mayer MP, Samaranayake LP. The role of probiotic bacteria in managing periodontal disease: a systematic review. Expert Rev Anti-Infect Ther. 2016;14(7):643–55. https://doi.org/10.1080/14787210.2016.1194198.
Ouwehand AC, Invernici MM, Furlaneto FAC, Messora MR. Effectiveness of multistrain versus single-strain probiotics: current status and recommendations for the future. J Clin Gastroenterol. 2018;52 Suppl 1, Proceedings from the 9th Probiotics, Prebiotics and New Foods, Nutraceuticals and Botanicals for Nutrition & Human and Microbiota Health Meeting, held in Rome, Italy from September 10 to 12, 2017:S35-s40.
Ho SN, Acharya A, Sidharthan S, Li KY, Leung WK, McGrath C, et al. A systematic review and meta-analysis of clinical, immunological, and microbiological shift in periodontitis after nonsurgical periodontal therapy with adjunctive use of probiotics. J Evid Based Dent Pract. 2020;20(1):101397. https://doi.org/10.1016/j.jebdp.2020.101397.
Nguyen T, Brody H, Lin GH, Rangé H, Kuraji R, Ye C, et al. Probiotics, including nisin-based probiotics, improve clinical and microbial outcomes relevant to oral and systemic diseases. Periodontol 2000. 2020;82(1):173–85.
Teughels W, Newman MG, Coucke W, Haffajee AD, Van Der Mei HC, Haake SK, et al. Guiding periodontal pocket recolonization: a proof of concept. J Dent Res. 2007;86(11):1078–82. https://doi.org/10.1177/154405910708601111.
Maekawa T, Hajishengallis G. Topical treatment with probiotic Lactobacillus brevis CD2 inhibits experimental periodontal inflammation and bone loss. J Periodontal Res. 2014;49(6):785–91. https://doi.org/10.1111/jre.12164.
Nissen L, Sgorbati B, Biavati B, Belibasakis GN. Lactobacillus salivarius and L. gasseri down-regulate Aggregatibacter actinomycetemcomitans exotoxins expression. Ann Microbiol. 2014;64(2):611–7. https://doi.org/10.1007/s13213-013-0694-x.
Nackaerts O, Jacobs R, Quirynen M, Rober M, Sun Y, Teughels W. Replacement therapy for periodontitis: pilot radiographic evaluation in a dog model. J Clin Periodontol. 2008;35(12):1048–52. https://doi.org/10.1111/j.1600-051X.2008.01333.x.
Shimauchi H, Mayanagi G, Nakaya S, Minamibuchi M, Ito Y, Yamaki K, et al. Improvement of periodontal condition by probiotics with Lactobacillus salivarius WB21: a randomized, double-blind, placebo-controlled study. J Clin Periodontol. 2008;35(10):897–905. https://doi.org/10.1111/j.1600-051X.2008.01306.x.
Mayanagi G, Kimura M, Nakaya S, Hirata H, Sakamoto M, Benno Y, et al. Probiotic effects of orally administered Lactobacillus salivarius WB21-containing tablets on periodontopathic bacteria: a double-blinded, placebo-controlled, randomized clinical trial. J Clin Periodontol. 2009;36(6):506–13. https://doi.org/10.1111/j.1600-051X.2009.01392.x.
Vivekananda MR, Vandana KL, Bhat KG. Effect of the probiotic Lactobacilli reuteri (Prodentis) in the management of periodontal disease: a preliminary randomized clinical trial. J Oral Microbiol. 2010;2(1). https://doi.org/10.3402/jom.v2i0.5344.
Teughels W, Durukan A, Ozcelik O, Pauwels M, Quirynen M, Haytac MC. Clinical and microbiological effects of Lactobacillus reuteri probiotics in the treatment of chronic periodontitis: a randomized placebo-controlled study. J Clin Periodontol. 2013;40(11):1025–35. https://doi.org/10.1111/jcpe.12155.
Laleman I, Yilmaz E, Ozcelik O, Haytac C, Pauwels M, Herrero ER, et al. The effect of a streptococci containing probiotic in periodontal therapy: a randomized controlled trial. J Clin Periodontol. 2015;42(11):1032–41. https://doi.org/10.1111/jcpe.12464.
Tekce M, Ince G, Gursoy H, Dirikan Ipci S, Cakar G, Kadir T, et al. Clinical and microbiological effects of probiotic lozenges in the treatment of chronic periodontitis: a 1-year follow-up study. J Clin Periodontol. 2015;42(4):363–72. https://doi.org/10.1111/jcpe.12387.
Morales A, Carvajal P, Silva N, Hernandez M, Godoy C, Rodriguez G, et al. Clinical effects of Lactobacillus rhamnosus in non-surgical treatment of chronic periodontitis: a randomized placebo-controlled trial with 1-year follow-up. J Periodontol. 2016;87(8):944–52. https://doi.org/10.1902/jop.2016.150665.
Morales A, Gandolfo A, Bravo J, Carvajal P, Silva N, Godoy C, et al. Microbiological and clinical effects of probiotics and antibiotics on nonsurgical treatment of chronic periodontitis: a randomized placebo- controlled trial with 9-month follow-up. J Appl Oral Sci. 2018;26:e20170075.
Invernici MM, Salvador SL, Silva PHF, Soares MSM, Casarin R, Palioto DB, et al. Effects of Bifidobacterium probiotic on the treatment of chronic periodontitis: a randomized clinical trial. J Clin Periodontol. 2018;45(10):1198–210. https://doi.org/10.1111/jcpe.12995.
Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gotzsche PC, Krleza-Jeric K, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200–7. https://doi.org/10.7326/0003-4819-158-3-201302050-00583.
Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Clin Periodontol. 2018;45(Suppl 20):S162–s70. https://doi.org/10.1111/jcpe.12946.
Lang NP, Tonetti MS. Periodontal risk assessment (PRA) for patients in supportive periodontal therapy (SPT). Oral Health Prev Dent. 2003;1(1):7–16.
Silness J, Loe H. Periodontal disease in pregnancy. II. Correlation between oral hygiene and periodontal condition. Acta Odontol Scand. 1964;22(1):121–35. https://doi.org/10.3109/00016356408993968.
Araujo MW, Hovey KM, Benedek JR, Grossi SG, Dorn J, Wactawski-Wende J, et al. Reproducibility of probing depth measurement using a constant-force electronic probe: analysis of inter- and intraexaminer variability. J Periodontol. 2003;74(12):1736–40. https://doi.org/10.1902/jop.2003.74.12.1736.
Socransky SS, Smith C, Martin L, Paster BJ, Dewhirst FE, Levin AE. “Checkerboard” DNA-DNA hybridization. Biotechniques. 1994;17(4):788–92.
Mestnik MJ, Feres M, Figueiredo LC, Duarte PM, Lira EA, Faveri M. Short-term benefits of the adjunctive use of metronidazole plus amoxicillin in the microbial profile and in the clinical parameters of subjects with generalized aggressive periodontitis. J Clin Periodontol. 2010;37(4):353–65. https://doi.org/10.1111/j.1600-051X.2010.01538.x.
Tamashiro NS, Duarte PM, Miranda TS, Maciel SS, Figueiredo LC, Faveri M, et al. Amoxicillin plus metronidazole therapy for patients with periodontitis and type 2 diabetes: a 2-year randomized controlled trial. J Dent Res. 2016;95(7):829–36. https://doi.org/10.1177/0022034516639274.
Feres M, Retamal-Valdes B, Faveri M, Duarte P, Shibli J, Soares GMS, et al. Proposal of a clinical endpoint for periodontal trials: the treat-to-target approach. J Int Acad Periodontol. 2020;22(2):41–53.
Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL Jr. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25(2):134–44. https://doi.org/10.1111/j.1600-051X.1998.tb02419.x.
Acknowledgements
The authors of this manuscript entitled would like to thank the BioGaia AB Company (Stockholm, Sweden) for supporting this study. The authors also acknowledge the Brazilian Coordination for the Improvement of Higher Education Personnel (CAPES) and the National Council for Scientific and Technological Development (CNPq, Brazil) for supporting some researchers involved in this study.
Funding
The authors of this manuscript declare that this study is being supported by BioGaia AB Company, Stockholm, Sweden. Biogaia contributed to study conception and design and will review the final manuscript.
Author information
Authors and Affiliations
Contributions
MF and BRV drafted the manuscript. MF, WT, and BRV contributed to the study conception and design. MF is the general coordinator. BRV, LMO, RNS, AF, GMSS, and NT are responsible for the recruitment of the participants, clinical assessment, and collection of microbiological and immunological samples. AF, PG, and CPF perform the periodontal treatment. PMD will be responsible for processing the immunological samples. LCF will be responsible for processing the microbiological samples. HD will perform the clinical and immunological data analysis. MF will perform the microbiological data analysis. All authors critically revised the manuscript, gave final approval, and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The authors of this manuscript declare that the Guarulhos University Clinical Research Ethics Committee (CAAE: 74049717.7.1001.5506) and Federal University of Parana Ethics Committee (CAAE: 74049717.7.3001.0102) approved the study protocol. In addition, all participants have signed the informed consent term and approved their participation in the study.
Consent for publication
“Not applicable.”
Competing interests
This study is being supported by BioGaia AB Company, Stockholm, Sweden. The authors certify that have no commercial or associative interest that represents a conflict of interest in connection with the manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
SPIRIT 2013 Checklist: Recommended items to address in a clinical trial protocol and related documents*.
Additional file 2.
Informed consent term.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Retamal-Valdes, B., Teughels, W., Oliveira, L.M. et al. Clinical, microbiological, and immunological effects of systemic probiotics in periodontal treatment: study protocol for a randomized controlled trial. Trials 22, 283 (2021). https://doi.org/10.1186/s13063-021-05246-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-021-05246-0