
Abstract
Friedreich ataxia (FRDA), the most common inherited ataxia, is caused by transcriptional silencing of the nuclear FXN gene, encoding the essential mitochondrial protein frataxin. Currently, there is no approved therapy for this fatal disorder. Gene silencing in FRDA is due to hyperexpansion of the triplet repeat sequence GAA·TTC in the first intron of the FXN gene, which results in chromatin histone modifications consistent with heterochromatin formation. Frataxin is involved in mitochondrial iron homeostasis and the assembly and transfer of iron-sulfur clusters to various mitochondrial enzymes and components of the electron transport chain. Frataxin insufficiency leads to progressive spinocerebellar neurodegeneration, causing symptoms of gait and limb ataxia, slurred speech, muscle weakness, sensory loss, and cardiomyopathy in many patients, resulting in death in early adulthood. Numerous approaches are being taken to find a treatment for FRDA, including excision or correction of the repeats by genome engineering methods, gene activation with small molecules or artificial transcription factors, delivery of frataxin to affected cells by protein replacement therapy, gene therapy, or small molecules to increase frataxin protein levels, and therapies aimed at countering the cellular consequences of reduced frataxin. This review will summarize the mechanisms involved in repeat-mediated gene silencing and recent efforts aimed at development of therapeutics.
Similar content being viewed by others

Introduction
Over 40 neurodegenerative and neuromuscular diseases are caused by expansion of simple repetitive nucleotide sequences in various genes throughout the human genome [1,2,3]. These include trinucleotide repeats (causing, for example, Huntington’s disease, many of the spinocerebellar ataxias, Friedreich ataxia, myotonic dystrophy type 1 (DM1), and fragile X syndrome, among others), tetranucleotide repeats (myotonic dystrophy type 2, DM2), pentanucleotide repeats (spinocerebellar ataxia type 10, SCA10), and even hexanucleotide repeats (C9orf72-related amyotrophic lateral sclerosis and frontotemporal dementia [4]). Recent evidence also suggests that the common eye disease Fuchs endothelial corneal dystrophy (FECD) is largely due to an expanded triplet repeat sequence [5]. Repeat expansions affect many different aspects of gene/protein function, including generation of toxic proteins and/or loss of the normal protein function (as in Huntington’s disease [2]), toxic RNA species that sequester essential splicing factors (as in DM1, DM2 and FECD; [6, 7]), generation of small toxic repeat proteins due to repeat-associated non-ATG or RAN translation (reviewed in [8]) and inhibition of gene transcription as in fragile X syndrome and Friedreich ataxia [9].
In Friedreich ataxia (FRDA, OMIM#229300), a GAA·TTC triplet repeat sequence leads to the loss of the essential mitochondrial protein frataxin in affected individuals. The repeats are located within the first intron of the nuclear FXN gene, and expansion above a certain threshold (above ~ 60 repeats) causes inhibition of gene transcription, leading to lower levels of FXN mRNA and protein. Although ~ 96–98% of FRDA patients are homozygous for the GAA·TTC triplet repeat expansion in the FXN gene [10], a small number of patients have one expanded allele and a second allele harboring either a premature stop codon, various point mutations, frameshift mutations, deletions, or splice site mutations [10,11,12,13,14]. Patient repeat alleles are most often between 600 and 900 repeats but repeats as long as 1700 have been reported (reviewed in [15]). Longer repeats are associated with more severe gene repression [16], lower levels of frataxin protein and earlier onset and increased severity of the disease [10, 17]. Individuals who are heterozygous for the mutation also have somewhat reduced levels of frataxin relative to nonaffected individuals but are asymptomatic.
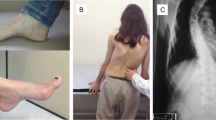
Available evidence supports a role for frataxin in the biogenesis of iron-sulfur (Fe–S) clusters in mitochondria, and in particular frataxin has been reported to be an allosteric activator that binds the Fe–S assembly complex and stimulates the cysteine desulfurase subunit and Fe–S cluster assembly activities [18]. Loss of frataxin in FRDA causes a reduction in Fe–S enzymes, such as components of the electron transport chain and other essential mitochondrial enzymes, resulting in altered cellular iron metabolism with iron accumulation in mitochondria, decreased mitochondrial energy production, and increased oxidative stress ([19,20,21]; for a recent review, see [15]). Frataxin insufficiency leads to progressive spinocerebellar neurodegeneration, causing symptoms of gait and limb ataxia, slurred speech, muscle weakness, and sensory loss, with scoliosis, cardiomyopathy, and diabetes in a number of patients. The prevalence of FRDA is around 1–2/100,000 and onset is usually in adolescence. At present, there is no effective treatment for FRDA, and generally within 10 to 15 years after the first appearance of symptoms, affected individuals are confined to a wheelchair. In later stages, patients become completely incapacitated. Individuals with FRDA generally die in early adulthood from the associated heart disease, the most common cause of death in FRDA [22]. This review will summarize our current state of knowledge of the mechanisms, whereby the repeats cause FXN gene silencing and the development of therapeutics, based on countering gene silencing, the low frataxin levels in patients, and the downstream consequences of loss of the essential mitochondrial protein frataxin.
Mechanistic Studies of FXN Gene Silencing
Shortly after the discovery of the expanded GAA·TTC repeats as the genetic basis for FRDA [10], transcriptional silencing of the FXN gene by the repeats was established [17, 23]. Cells from affected individuals generally have between ~ 10–30% of the levels of FXN mRNA and frataxin protein of healthy individuals, and these levels correlate strongly with GAA·TTC repeat lengths and inversely with age of onset [24]. Early studies also considered whether the repeats have an effect on splicing of the primary FXN transcript. Using RNase protection assays, Bidichandani and colleagues reported no differences between FRDA and unaffected cells, at least for intron 1 splicing [17]. Although one publication using a reporter construct did find an effect of the repeats on RNA splicing in an artificial system, this study did not extend their results to the endogenous FXN gene in patient cells [25]. Additionally, one report has provided evidence for alternatively spliced FXN mRNA isoforms, which were found to be largely similar between patient and unaffected cells but with some minor differences in the heart and cerebellum [26]. However, other data do not support a splicing defect in FRDA cells [17, 27, 28]. Notably, no stable alternatively spliced FXN transcript can be detected in FRDA cells by northern blot analysis [28]. Similarly, there is no evidence that long GAA repeat intron 1 RNA is stable and could lead to an RNA toxicity disease, such as found in DM1 and FECD [5, 6, 27, 29]. Absence of stable intron 1 transcripts also suggests that RAN translation is unlikely to be involved in FRDA [27]. Having established that RNA transcription is impaired by the GAA·TTC repeats, the question remained as to how the repeats interfere with RNA polymerase at the FXN gene (Fig. 1).

Mechanisms of FXN gene silencing. (A) In cells from unaffected individuals, the FXN gene, with short lengths of GAA·TTC repeats, is packaged in open chromatin, allowing RNA polymerase II (pol II) access to the promoter and allowing elongation through the repeats. Nucleosomes bear highly acetylated histone (blue marks on the amino-terminal tails of the histones). (B) In FRDA cells, the FXN gene is packaged in condensed heterochromatin, having particular types of histone methylation marks, such as H3K9me3 (red boxes on the amino-terminal tails of histone H3), which forms the binding site for heterochromatin protein HP1, leading to chromatin condensation. Several models have been proposed to account for heterochromatin formation by long GAA·TTC repeats, including triplexes and/or sticky DNA; R-loops that recruit the heterochromatin machinery; short RNA transcripts that recruit components of the RNA interference machinery; and an antisense transcript called FAST-1, which depletes the chromatin boundary protein CTCF, leading to heterochromatin. All of these mechanisms could lead to promoter silencing and blocking transcription elongation through the repeats
Extensive biochemical studies documented that expanded GAA·TTC repeats adopt unusual DNA structures ([17, 23, 30], and additional references therein) (Fig. 1B). In vitro, plasmids containing long GAA·TTC repeats form triplexes containing two purine (R) GAA strands along with one pyrimidine (Y) TTC strand, flanking a single-stranded pyrimidine region. Additionally, RRY triplexes have been shown to adopt an unusual structure known as “sticky” DNA [30], which inhibits transcription both in vitro and in transfection assays in mammalian cells [31]. Molecules that reverse the sticky DNA conformation adopted by long GAA·TTC repeats induce FXN gene expression in FRDA lymphoblast cell lines [32]. Other investigators have proposed that a DNA-RNA triplex is responsible for blocking transcription elongation [33, 34]. These early biochemical results are consistent with the observed correlation between repeat length, triplex formation, and the age at onset and severity of disease. Further, Gromak and colleagues have provided evidence for the enrichment of R-loops at FRDA FXN alleles compared to unaffected alleles and a role for R-loops in the silencing mechanism [35]. R-loops are DNA-RNA duplexes with the nontemplate DNA as single stranded (Fig. 1B). Gromak and colleagues propose that R-loops initiate gene silencing through heterochromatin ([35] and see below).
Studies from several laboratories have focused on the possibility of additional RNA-mediated silencing mechanisms in neurodegenerative diseases [36]. Bidichandani and colleagues described an antisense transcript of FXN, termed FAST-1 (FXN antisense transcript 1) and evidence points to a role for this RNA in FXN gene silencing in FRDA [37]. A tag corresponding to FAST-1 RNA is found in the human antisense transcriptome (position 70840691 on the “-” strand of chromosome 9, at the FXN exon 1/intron 1 boundary; UCSC Genome Browser [38]), but given the location of the start site for this RNA, it would not include the repeats. FAST-1 RNA overlaps with the binding site for the chromatin insulator protein CCCTC-binding factor CTCF, and patient cells were reported to harbor higher levels of FAST-1 along with depletion of CTCF. The reciprocal relationship between low levels of FXN mRNA in patient cells and CTCF depletion could be reproduced in unaffected cells through knockdown of CTCF [37]. Reduced CTCF occupancy at the FRDA locus has also been extended to the cerebellum of the YG8R FRDA mouse model containing a human FXN transgene containing ~ 200 repeats [39]. These results are consistent with a model whereby FAST-1 transcription and concomitant CTCF depletion leads to heterochromatin formation and subsequent FXN gene silencing (Fig. 1). A recent study has confirmed these findings and shown that over-expression of FAST-1 in trans can lead to CTCF depletion and gene silencing at the FXN locus [40]. Just how the repeats cause increases in FAST-1 RNA levels in patient cells has not been elucidated. In addition to FAST-1, which does not overlap the GAA·TTC repeats, Corey and colleagues have proposed that RNA transcripts of the repeats (sense or antisense, forming R-loops), recruit the RNA silencing complex (RISC) component Argonaut 2 (Ago2) to initiate silencing [41], and suggest a therapeutic approach based on this mechanism (see below). Evidence for the role of short, 24-nucleotide siRNAs in GAA·TTC mediated gene silencing has also been presented in Arabidopsis thaliana, and knocking down components of the RNA interference (RNAi) pathway can reverse gene silencing in this system as well [42].
Although additional effort will be needed to fully elucidate the mechanisms that induce silencing at FXN genes harboring long GAA·TTC repeats, it is widely accepted that silencing involves a transition of the FXN gene from open chromatin to condensed heterochromatin, thereby blocking essential steps in the transcription cycle (Fig. 1B). The first evidence for the role of heterochromatin in FRDA came from a study from Festenstein and colleagues employing artificial transgenes for the lymphoid cell surface marker protein hCD2 linked to expanded GAA·TTC repeats [43]. The repeats were reported to induce repressive heterochromatin, in a manner reminiscent of position effect variegated gene silencing (PEV) [43]. PEV occurs when a gene is located within or near regions of heterochromatin, and silent heterochromatin is characterized by the presence of particular types of histone modifications (H3 lysine 9 di- and trimethylation (H3K9me2/3), H3K27me3, and histone tail hypoacetylation), the presence of histone deacetylases, DNA methyltransferases, chromodomain proteins, such as members of the HP-1 family of repressors, and polycomb group proteins [44]. Festenstein and coworkers [43] also found that the hCD2 transgene harboring GAA·TTC repeats is resistant to DNase I digestion, compared to the normal transgene lacking GAA·TTC repeats. Additionally, GAA·TTC repeat-mediated repression was enhanced by co-expression of the heterochromatin protein HP1 [43]. It is possible that non-B DNA structures, such the triplexes and/or the sticky DNA conformation adopted by GAA·TTC repeats, or the RNA-based mechanisms discussed above, are signals that initiate heterochromatin formation and FXN gene silencing in FRDA (Fig. 1B).
To assess the heterochromatin model for gene silencing by expanded GAA·TTC repeats, our laboratory and other groups examined histone modifications on FXN gene chromatin using chromatin immunoprecipitation (ChIP) methods, with antibodies to various histone modifications and cell lines derived from FRDA patients ([45, 46] and reviewed in [47, 48]) and in mouse models for the disease [49, 50]. The first intron of active FXN alleles in normal cell lines is enriched in acetylated histones H3 and H4, compared to the inactive alleles in FRDA cells. Additionally, H3K9 is highly trimethylated in FRDA cells compared to unaffected cells [28, 45, 46, 51]. Importantly, although early studies were conducted in FRDA lymphoid cells and fibroblasts, the same epigenetic differences between active and inactive FXN alleles have also been found in FRDA autopsy brain, cerebellum, and heart [49], the tissues that are affected in FRDA. In other studies, H3K27 was found to be highly methylated within the FXN coding region in patient cells [51, 52]. H3K27me3 recruits the polycomb repressive complex (PRC), thus providing another layer of repression to pathogenic FXN alleles. Thus, the histone posttranslational modification state within the coding region of inactive FXN alleles is consistent with a chromatin-mediated mechanism as the cause of gene silencing in FRDA [43]. Comprehensive reviews of the available ChIP data on histone posttranslational modifications in FXN gene silencing have been presented [47, 48].
Studies have also examined DNA methylation on active versus inactive FXN alleles in FRDA cell lines [29, 46], in human FRDA lymphocytes and tissue samples [53, 54], and in a mouse model [49], with the finding that DNA methylation at particular residues adjacent to the GAA·TTC repeats, adjacent to the CpG island (CGI) at the 5′ end of the FXN gene (in a region known as the CGI shore) is enriched in FRDA samples over controls. This CGI shore methylation, typically 70–95% in FRDA compared to < 10% in non-FRDA controls, is also observed in various patient-derived cells, including iPSC-derived neurons (S. Bidichandani and E. Soragni, personal communication, April 219 by email and verbal). Methylation of CGI shores is a recognized regulator of gene expression [55, 56], and the level of DNA methylation in the FXN CGI shore strongly correlates with gene silencing in FRDA. Pook and coworkers reported that the majority of methylation marks in FRDA are actually 5-hydoxymethylcytosine rather than 5-methylcytosine [39], but no follow-up studies have elucidated the significance of this finding for FXN gene regulation. In general, DNA methylation is a mark for recruitment of repressor complexes, containing histone deacetylases (HDACs), mediated by methyl-CpG binding proteins [57]. Once targeted for silencing, HDACs are recruited to FXN gene chromatin, perhaps through DNA structure, DNA methylation, or RNA-mediated mechanisms (Fig. 1); these enzymes deacetylate the amino-terminal tails of the histones on FXN gene chromatin, histone methyltransferases (HMT) then methylate H3K9 and/or H3K27 [52], and repressive complexes containing HP1 (H3K9me2/3) and/or polycomb proteins (H3K27me3 [58]) are recruited to expanded FXN gene chromatin. All of these events lead to chromatin condensation and FXN gene silencing. It is also likely that the repeats induce repositioning of FXN gene chromatin to the nuclear periphery in association with the nuclear lamins [59]. It is intriguing that related DNA sequences such as GAGA repeats induce chromatin repositioning to lamina associated domains in cells, resulting in heterochromatin-mediated gene silencing [60].
Studies have also addressed how histone posttranslational modifications affect transcription of the FXN gene by RNA polymerase II (pol II). Several approaches have been used to assess whether the initiation or elongation phases (or both) are affected by the repeats and concomitant histone modifications. These include ChIP experiments for histone modifications that are indicative of steps in the transcription cycle, ChIP for RNA pol II along the gene, nuclease sensitivity of the promoter region, and measurements of transcription rates/levels along the gene. Evidence has been presented both for and against a defect in transcription initiation, and some studies point to defects exclusively in transcription elongation. In one of the first such studies, Usdin and colleagues examined chromatin histone modifications that are associated with active pol II initiation (H3K4me3 at the promoter) and productive RNA elongation by pol II (H3K36me3 within the gene body), with the result that both histone modifications were depleted in pathogenic FXN alleles compared to unaffected alleles, suggesting that both initiation and elongation are affected by the repeats [61]. In contrast, Punga and Buhler reported that only H3K36me3 in the gene body was affected in FRDA cells, whereas H3K4me3 levels were largely unchanged between FRDA and unaffected cells at the promoter [28]. There is a general consensus in the field that histone marks associated with productive elongation by pol II (H3K36me3 and H3K79me2) are depleted in the FXN coding region in FRDA cells, indicating a defect in elongation [28, 51, 61].
Studies directly monitoring pol II occupancy at the FXN promoter and gene body generally show a decrease in FRDA compared to unaffected cells, and this has also been reported for the initiating form of pol II (phosphorylated at serine 5 within the carboxyl-terminal domain of the large pol II subunit, pS5 [61]) and total pol II [35, 51], but another study found no difference in pol II pS5 occupancy at the FXN promoter between affected and nonaffected cells [28]. However, several other lines of evidence point to promoter silencing in FRDA. Nascent RNA levels are reduced both upstream and downstream of the repeats [35], suggesting defects at the promoter. Bidichandani and colleagues showed that heterochromatin marks extend to the promoter in FRDA cells, and using a nuclease-accessibility assay, they showed that a nucleosome likely occupies the FXN transcription start site in FRDA cells, with an open chromatin region at this location in unaffected cells [62]. Consistent with a defect in transcription initiation, metabolic labeling studies also pointed to decreased initiation events in FRDA cells [62, 63] and in various tissues in the YG8R mouse model [64]. In contrast, deep RNA sequencing revealed a defect in elongation in patient fibroblasts compared to unaffected cells [65]. Single-molecule studies from Silva and colleagues showed that both transcription initiation and elongation were affected in reporter genes harboring long GAA·TTC repeats and at the endogenous FXN locus in FRDA lymphoblasts [59]. Thus, although there is a consensus in the field that downregulation of RNA synthesis in FRDA cells is associated with repeat-induced chromatin structural changes, whether the defect is due to blocks in transcript elongation or initiation or both remains an unsettled question. Nevertheless, numerous therapeutic strategies are aimed at overcoming the epigenetic changes due to repeat expansion at the FXN locus.
Therapeutic Approaches for FRDA
Figure 2 summarizes the cellular consequences of the GAA·TTC repeat expansion in FRDA and reduced levels of frataxin protein and outlines various therapeutic strategies. At the core of the disease lies the repeat expansion itself, and with genome engineering methods, removal of the repeats from the gene is an attractive approach (see Fig. 2, box, approach 1). Since we know that the repeats induce epigenetic silencing, therapies aimed at reversing gene silencing are being pursued by various approaches (approach 2). Alternatively, restoring normal levels of frataxin protein, by protein replacement therapy, gene therapy, stabilization of existing frataxin, or increased translation are all being considered (approach 3). Therapies aimed at countering the downstream consequences of frataxin deficiency, such as oxidative stress, are being pursued by a number of approaches (approach 4), and lastly, other therapies are aimed at countering FRDA symptomatically (approach 5). The Friedreich’s Ataxia Research Alliance (FARA) has summarized the current therapeutic pipeline, which is shown in Fig. 3.

Cellular dysfunction in FRDA and possible therapeutic opportunities. A cartoon representation of a FRDA patient cell is shown, along with the consequences of the GAA·TTC repeat expansion at the levels of the DNA (1), nuclear chromatin (2), low levels of frataxin in mitochondria (3), and cellular (4) and organismic (5) consequences of reduced frataxin. Possible therapeutic approaches targeting each of these defects are shown in the box at the right

Therapeutic pipeline for FRDA, including the clinical development stage for each approach as well as the organizations performing research, as of April 2019. Figure provided by J. Farmer of the Friedreich’s Ataxia Research Alliance
Approach 1: Genetic Correction
Although early studies suggested that it might be possible to modulate repeat expansions in affected tissues as a therapeutic approach (reviewed in [66] and see [67]), current excitement in the field now focuses on the possibility of physically removing repeats from pathogenic FXN alleles in affected tissues. With the advent of genome engineering technologies, such as zinc finger nucleases, TALENS, and CRISPR-Cas9 methods, one option for treating FRDA is genetic excision or correction of the repeats. Excision of the repeats refers to introducing DNA breaks flanking the repeats, using these genome engineering methods, leading to elimination of the repeat region [68, 69], whereas correction refers to using homologous recombination with a DNA template containing FXN intron 1 sequences with unaffected repeat numbers [70]. Previous studies have shown that correction or excision of the repeats restores normal transcription to the FXN gene, thus providing unequivocal evidence that it is the repeat sequence that is entirely responsible for epigenetic gene silencing in FRDA [68,69,70]. Tremblay and coworkers have used CRISPR-Cas9 to excise the GAA·TTC expansion from FXN intron 1, with concomitant restoration of FXN transcriptional activity and protein levels in the YG8R and YG8sR FRDA mouse models and in cell lines derived from these mice [68]. Although in vivo genetic correction is an attractive therapeutic option, successful application in humans will require effective delivery of gene editing machinery to cells and tissues by a gene therapy approach and precise and effective gene editing without off-target effects. One further consideration for genetic excision is whether deletion of DNA sequences within FXN intron 1 flanking the repeats will negatively affect transcriptional regulation or RNA processing. These issues will need to be addressed prior to clinical application of CRISPR-Cas9 methods to treat FRDA. Nonetheless, CRISPR Therapeutics (Zug, Switzerland and Cambridge, MA) has shown interest in applying this technology to FRDA.
Approach 2: Upregulation of FXN Gene Transcription
Based on the fact that the repeats are located in an intron do not change the coding potential of the frataxin gene, upregulation of the FXN gene in patient cells has long been considered as a therapeutic option [71]. Since the repeats cause epigenetic FXN gene silencing, most efforts have been aimed at countering these epigenetic changes; however, other therapeutics have been considered, such as artificial transcription factors and screening chemical libraries, including FDA-approved drug libraries, for compounds that increase FXN mRNA levels, independent of known mechanisms of action.
Epigenetic Drugs to Relieve FXN Transcriptional Repression
Several approaches have been taken to reverse epigenetic silencing of the frataxin gene. These include the sirtuin (class III HDAC) inhibitor nicotinamide, benzamide-type HDAC inhibitors, the sirtuin activator resveratrol, and more recently, antisense oligonucleotides to counter RNA-mediated silencing. Another reasonable approach would be to reverse histone methylation at H3K9 and H3K27 with histone methyltransferase (HMT) inhibitors. However, the common HMT inhibitor BIX-01294 was without effect on FXN mRNA in FRDA patient cells, although this compound did decrease FXN H3K9me2 and H3K9me3 levels [28]. Although DNA methylation appears to be linked to gene silencing, no studies on DNA methyltransferase (DMT) inhibitors in FRDA cells have been presented as of this writing. Combination therapies including DMT inhibitors and HDAC inhibitors might also be possible (S. Bidichandani, personal communication April 2019 by email).
Nicotinamide
Festenstein and colleagues reported that the class III HDAC or sirtuin inhibitor vitamin B3 (nicotinamide) activates the silenced FXN gene in FRDA lymphoblast cells lines, in primary lymphoid cells, and in various tissues in the YG8R mouse model [52]. These authors reported that concomitant with increases in FXN mRNA, nicotinamide caused a reduction in H3K9me3 and H3K27me3, an increased accessibility to DNase I, and an increase in H3/H4 acetylation at the FXN gene in patient PBMCs. Transcription analysis revealed that nicotinamide corrected the expression of 67% of genes found to be dysregulated in FRDA PBMCs. Although these results are impressive, no data are currently available that provides evidence for a direct role of sirtuins in FXN gene expression, and other data in the literature question the role of Sirt 1 (and possibly Sirts 2 and 3) in FRDA as a more specific inhibitor of these enzymes (EX527, 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide, [72]) fails to upregulate FXN gene expression in various FRDA patient cells (lymphoblasts, PBMCs, and fibroblasts [73]). Given the low avidity of nicotinamide for the sirtuins, requiring millimolar concentrations for inhibition, the mechanism of action of nicotinamide in FXN gene expression requires further investigation.
Nevertheless, positive preclinical results with nicotinamide have been followed up with an open-label, dose-escalation study in adult FRDA patients [74]. Although there were some adverse effects of high dose treatment (mainly nausea), the authors reported that nicotinamide was generally well-tolerated, and adverse effects were resolved after treatment or dose reduction. In terms of frataxin mRNA and protein levels, the authors reported that daily dosing at 3.5–6 g resulted in a sustained and significant upregulation of frataxin expression, which was accompanied by a reduction in heterochromatin modifications at the FXN locus in PBMCs. However, in this short duration study, clinical measures showed no significant changes. Further assessment of nicotinamide is being supported by E-Rare and the German Research Foundation (Fig. 3).
Histone Deacetylase Inhibitors
Three groups have identified HDAC inhibitors as possible therapeutics for FRDA. Utilizing a full-length FXN reporter cell line, Sarsero and colleagues found that the HDACi sodium butyrate had a small effect on frataxin expression [71]. Separately, Lufino and colleagues screened a collection of potential therapeutic compounds and found an amino alcohol called C5 (1-(3,4-dimethylphenoxy)-3-(4-morpholinyl)-2-propanol hydrochloride) that upregulates FXN gene expression in reporter screens and in patient cells, and C5 changed the histone acetylation status of pathogenic alleles, suggesting a mode of action through HDAC inhibition [75]; however, no follow-up studies of these findings have been presented. This author’s group asked whether commercially available HDAC inhibitors could affect FXN mRNA levels and acetylation at the FXN locus in FRDA lymphoblast cell lines [45]. Although many of the common HDAC inhibitors showed global increases in histone H3 and H4 acetylation, only a particular benzamide-type HDAC inhibitor BML-210 increased FXN gene acetylation and FXN mRNA levels in patient lymphoblasts and in PBMCs. In subsequent studies, we examined both the mechanism of action of the active benzamide HDAC inhibitors and their targets within the class I HDACs. Only HDAC inhibitors that function by a slow-on/slow-off inhibitory mechanism show activity in increasing FXN mRNA levels [58, 76], and inhibition of both HDACs 1 and 3 are necessary for upregulation [58, 73]. Improvements to the benzamide HDAC inhibitor scaffold have been reported [77] as well as efficacy in both the KIKI (repeat knock-in in both mouse Fxn alleles) [50, 78] and YG8R mouse models [79]. These results have also been extended to human neuronal cells derived from FRDA patient induced pluripotent stem cells [29, 70, 80], in which a benzamide HDACi was shown to increase FXN mRNA levels [29], correct many of the transcription defects associated with low levels of frataxin [70], and restored the levels of several Fe–S cluster proteins and protected FRDA neurons from oxidative stress-mediated cell death [80]. Transcription studies showed that many of the gene expression changes due to loss of frataxin can be restored by HDACi treatment both in the KIKO mouse model [50] and in patient iPSC-derived neurons [70]. Bidichandani and colleagues also showed that a benzamide HDACi opened the chromatin structure at the FXN promoter and increased transcription initiation events in patient lymphoid cells [81]. One member of this compound class (HDACi 109/RG2833) was taken into a phase Ib human clinical study [29]. No drug-related adverse effects were observed with RG2833 (up to 240 mg/day through an oral formulation). Efficacy of the drug was assessed in blood samples obtained from control and treated patients, with the result that doses of > 180 mg/day yield ~ 1.5-fold increases in FXN mRNA levels in patient PBMCs, along with increases in frataxin protein in a subset of patients and increased histone H3K9 acetylation at the FXN locus [29]. Although these data provided a proof-of-concept that HDACi treatment is a viable therapeutic approach for the disease, the original pimelic 2-aminobenzamide class of compounds suffered from poor brain penetration in animal models and poor metabolic stability in the clinical study [29]. Thus, RG2833 was not taken forward to later stage trials. Our laboratory identified modifications to the pimelic 2-aminobenzamide scaffold with improved metabolic stability and brain penetration [77]. A new generation of compounds are currently being assessed by BioMarin Pharmaceutical (Novato, CA), with a clinical candidate named BMN290 currently being pursued in preclinical studies.
Resveratrol
Using a high-throughput screen of a library of natural products and FDA-approved therapeutics, Li and coworkers identified a number of inducers of FXN gene expression [82]. Their primary screen utilized a HeLa cell line harboring the entire unaffected FXN gene fused to EGFP and hits from this screen were subsequently evaluated for effects on FXN mRNA and frataxin protein in FRDA patient lymphoid cells and fibroblasts. One compound that was prioritized for further study was resveratrol, which has been reported to be an activator of the sirtuin class of HDACs as well as an antioxidant. Other hits were anti-cancer agents and iron chelators, which could likely stimulate FXN gene expression either through a cellular stress response or the iron regulatory activity of frataxin. Li and coworkers reported an effect of resveratrol on FXN mRNA levels (~ 1.5-fold) in the brain of YG8R mice, and based on this finding, resveratrol was taken forward to an open-label clinical study in FRDA patients [83]. In this 12-week study, patient PBMCs were tested for frataxin levels and clinical measures of disease severity were monitored. Although no effects on frataxin were found, patients in a high dose group (5 g daily) showed improvement in neurological function, but gastrointestinal side effects were noted in this group. It is curious how a sirtuin activator (resveratrol) and a sirtuin inhibitor (nicotinamide) could both have positive effects on FXN gene expression, suggesting that these drugs may be working through distinct mechanisms that have yet to be elucidated. Nevertheless, based on these promising neurological findings [83], further clinical assessment of resveratrol seems warranted and Jupiter Orphan Therapeutics (Jupiter, FL) is engaged in such studies.
Oligonucleotide Approaches
In a series of recent papers, Corey and colleagues reported that oligonucleotides complementary to the GAA·TTC repeats can relieve FXN gene repression in patient fibroblasts [41, 84, 85]. The range of activating oligonucleotides includes standard double stranded siRNAs, locked nucleic acid motifs, and various chemically modified double- and single-stranded oligonucleotides [41, 84, 85], some with potency in the low nanomolar range [84, 85]. Corey and colleagues presented evidence that a likely mode of action of these compounds is interference with an RNA-silencing mechanism that induces heterochromatin at the GAA·TTC repeats, possibly by interfering with R-loops at the repeats [41]. ChIP experiments indicate that the activating siRNAs induce small changes in chromatin histone posttranslational modifications on the FXN gene, but have no effect on the recruitment of RNA polymerase II, suggesting that the oligonucleotides relieve a block to transcription elongation through the repeats rather than by increasing pol II occupancy at the promoter [41]. A recent study has also shown efficacy of duplex RNAs and antisense oligonucleotides in neural progenitor cells derived from FRDA induced pluripotent stem cells [86]; however, electroporation of these cells was necessary to see FXN mRNA induction. Future studies will undoubtedly test this approach in animal models for the disease, but a major hurdle will be delivery of nucleic acid therapeutics to the desired target tissues.
Upregulation of FXN Gene Expression with Synthetic Transcription Factors
A form of gene therapy for FRDA that is being considered is the application of TALE transcription activator fusion proteins to upregulate FXN gene expression in affected tissues ([87] and references cited therein). TALEs (transcription activator-like effectors) are bacterial proteins that can be engineered to bind preselected DNA sequences, and when fused to potent eukaryotic transcriptional activator sequences (such as VP64 or p300), these TALE activators have been shown to upregulate cognate genes [88]. Tremblay and colleagues have applied this technology to FRDA and shown that TALE activator fusion proteins targeted to the FXN promoter or intron 1 can increase FXN gene expression in FRDA primary fibroblasts [87]. To target the endogenous FXN gene in vivo, these authors incorporated the DNA sequence encoding their most active TALE activator into an adenovirus associated virus (AAV) vector and showed that expression of the TALE activator upregulated the endogenous FXN gene and frataxin protein in the YG8R mouse heart by 10-fold and in skeletal muscle by up to 35-fold. As a secondary consequence of frataxin expression, these authors showed that aconitase enzymatic activity was also upregulated on FXN expression in this system [87]. Although these results are impressive and provide an in vivo proof-of-concept, it is difficult to argue that TALE activators offer a superior approach to gene therapy compared to conventional FXN gene therapy, as outlined below.
Another approach taken to activate FXN transcription is through DNA-binding small molecules. Our laboratory previously described a synthetic pyrrole-imidazole (Py-Im) polyamide that specifically bound to GAA·TTC repeat DNA sequences and partially reversed FXN gene repression in FRDA cultured B-lymphoblasts and patient PBMCs [32]. Py-Im polyamides can be designed to bind predetermined DNA sequences (see references in [32]), and in the case of homopurine-homopyrimidine sequences such as the GAA·TTC repeats in FRDA, linear β-alanine linked polyamides have been shown to be highly effective in targeting such sequences, both in vitro with purified DNA [89] and in cell culture and in vivo studies in Drosophila ([90] and references therein). The likely mechanism of action of this molecule in reversing FXN epigenetic repression is displacement of heterochromatin proteins, such as HP1 [90], and concomitant chromatin opening. However, it was found that the GAA·TTC-selective polyamide did not cross the blood brain barrier in mice [91], and failed to show positive results in the KIKO FRDA mouse model (M. Rai and M. Pandolfo, personal communication April 2019 by email), and hence, Py-Im polyamides were abandoned by our group as a therapeutic for FRDA. Nevertheless, recent studies from Ansari and colleagues improved on the original GAA·TTC-selective Py-Im polyamide by coupling a bromodomain binding small molecule (JQ1) to the polyamide scaffold [92]. Bromodomain proteins, such as BRD4, are involved in transcription elongation, and by coupling JQ1 to the GAA·TTC DNA-binding polyamide, the transcription elongation machinery is brought to the epigenetically silenced FXN gene, and blocked RNA polymerase II is allowed to proceed through the repeats. This hybrid molecule, called Syn-TEF1, was active in primary lymphocytes from a series of FRDA patients, as well as in FRDA iPSC-derived cardiomyocytes and neuronal cells, and in an artificial mouse xenograft model [92]. Although the results of this study are impressive, no information on blood–brain penetration of the compound was presented, and such information will be essential to take Syn-TEF1 into further preclinical studies.
Other Drugs That Increase FXN mRNA
Interferon Gamma
A study from Testi and colleagues [93] reported that the cytokine interferon gamma (INFγ) can upregulate frataxin protein levels in a variety of nonaffected cultured cancer cell lines (HeLa, U937, U119) and in peripheral blood mononuclear cells from unaffected individuals as well as fibroblasts and lymphoid cells from numerous FRDA patients. This effect was due to increases in frataxin mRNA and was abolished by treatment with actinomycin D, suggesting a direct effect on transcription of the FXN gene. These authors also showed that INFγ was effective in the YG8R FRDA mouse model, showing increases in frataxin expression in dorsal root ganglia (DRG) neurons, as well as preventing pathological degeneration of these neurons and INFγ treatment improved locomotor performance in FRDA mice [93]. These positive results provided the impetus for an open-label clinical study of INFγ in patients [94]. Small but significant changes in frataxin levels were observed in red blood cells, PBMCs, and platelets after 12 weeks of treatment. However, a longer 6-month clinical study failed to find statistically significant neurological improvement or increases in frataxin protein in buccal cells or whole blood [95] and Horizon Pharmaceuticals (Dublin, Ireland) has abandoned INFγ as a therapeutic for FRDA.
Nrf2 Activators
Studies in several groups have shown that frataxin deficiency in FRDA decreases expression of the antioxidant transcription factor NF-E2-related factor (Nrf2) in FRDA cells, cultured motor neurons and in disease relevant tissues in mouse models for FRDA [96, 97]. Lack of Nrf2 has been proposed to lead to sensitivity to oxidative stress in FRDA, especially in affected tissues such as the DRG and cerebellum. Hence, efforts have focused on identification of compounds that increase Nrf2 levels or activity/availability of Nrf2 as potential therapies for FRDA [98,99,100]. From these efforts, a series of compounds have been identified that show activity in FRDA-relevant cell or animal models, including dyclonine [98], sulforaphane [99], dimethyl fumarate [101, 102], and omaveloxolone [100]. Some of these compounds have been shown to directly increase FXN mRNA and protein, likely by gene activation through an Nrf2 binding site in the FXN gene promoter [98], although direct evidence for the role of Nrf2 in FXN transcriptional regulation has not as yet been presented. Dimethyl fumarate has recently been shown to increase FXN expression by ~ 85% in peripheral blood mononuclear cells in a human clinical study in multiple sclerosis patients [103]. Nrf2 activators/inducers are under clinical study for FRDA, with positive results recently reported for omaveloxolone (RTA-408) in a phase 2 human clinical study [104]. Omaveloxolone was well tolerated and improved neurological function in a 12-week study in a cohort of FRDA patients [104]. These promising results encourage further clinical assessment of Nrf2 activators, with omaveloxolone being the most advanced. These efforts are being pursued by Reata Pharmaceuticals (Irving, TX).
Approach 3: Increasing Frataxin Protein
Several approaches have been taken to find therapies that increase frataxin protein levels, including screening compound libraries or FDA-approved therapeutics, protein replacement therapy, gene therapy, or finding compounds that either stabilize frataxin protein from degradation or increase the translation of the low levels of FXN mRNA in patient cells. Alternatively, recent studies also show that both mesenchymal stem cells and hematopoietic and progenitor stem cells (HPSCs) can transfer frataxin protein from stem cells to affected cells, at least in FRDA mouse models.
Screening-Approved Therapeutics
Several groups have taken the approach of screening FDA-approved therapeutics for molecules that increase frataxin protein in vitro. This section will provide a few examples of this approach.
Erythropoietin
An early study from Strum and coworkers reported that recombinant human erythropoietin (rhu-EPO) can induce frataxin protein levels in a variety of human cell types, including primary lymphocytes from FRDA patients [105]. This finding has been confirmed in multiple studies, and notably Acquaviva and colleagues reported that this effect was at the level of frataxin protein, with no effects on FXN mRNA observed in FRDA patient fibroblasts on treatment with rhu-EPO [106]. These findings and the fact that EPO can cross the blood–brain barrier [107] formed the impetus for clinical studies in FRDA patients, with early positive results in an 8-week study in which increases in frataxin protein were found along with reduction in oxidative stress markers [108]. However, no effects on frataxin or downstream markers of frataxin deficiency or neurological function were found in longer, randomized placebo-controlled, and double-blind trials [109, 110]. Although these clinical results cast doubt on the in vivo efficacy of rhu-EPO in FRDA (or modified forms of EPO such as carbamylated EPO [111]), the biopharmaceutical company STATegics (Sunnyvale, CA) is taking another complementary approach of screening small molecule agonists of the tissue-protective EPO receptor as therapeutics for FRDA. In a recent study, it was shown that these molecules increase FXN mRNA and protein in primary human cortical cells, in unaffected and FRDA patient PBMCs, and in the brain and heart of FRDA KIKO mice [112]. These positive results, and a better preclinical safety profile for EPO receptor agonists over rhu-EPO [112], will no doubt encourage further development of these compounds.
Etravirine
Using a cell-based screen for compounds that increase frataxin protein levels, Alfredi and colleagues identified etravirine, an antiviral drug that is in use in anti-HIV therapy, among a library of 853 FDA-approved drugs [113]. Etravirine was shown to function by increasing translation of existing FXN mRNA, rather than by increasing FXN gene expression, but just how etravirine functions to increase frataxin levels was not addressed in this study. Etravirine elevated aconitase activity in FRDA cells and conferred resistance to oxidative stress in these cells, consistent with an increase in frataxin; however, no in vivo efficacy studies were presented and data in the literature suggest that etravirine might not cross the blood–brain barrier [114], making it less attractive for addressing the neurological aspects of the disease. Fratagene Therapeutics (Rome, Italy) is developing this approach.
Protein Replacement Therapy
Protein or enzyme replacement therapy has been highly effective in a number of monogenic diseases and is widely used in routine clinical practice. For FRDA, direct delivery of frataxin protein to affected tissues is also being considered as a therapeutic option (Chondrial Therapeutics; Bala Cynwyd, PA). Previous studies from Payne and colleagues have shown that fusion of human frataxin with the HIV TAT (transactivator of transcription) cell-penetrating peptide sequence afforded cellular and mitochondrial uptake in human cells and in a FRDA mouse model [115]. This study showed that TAT-frataxin bound iron in vitro, localized into mitochondria in FRDA patient fibroblasts, and reduced caspase-3 activation in response to an exogenous iron-oxidant stress. Using a FXN conditional knockout mouse model, these authors showed that TAT-frataxin increased lifespan and cardiac output, as well as restoring aconitase activity and normal mitochondrial proliferation and ultrastructure in the heart. These positive results encourage further development of a protein replacement therapy for FRDA.
Gene Therapy
Gene therapy is another approach to increasing frataxin protein levels in affected tissues. Two major areas of investigation for frataxin delivery are under investigation and focus on either adeno-associated virus (AAV) gene delivery or direct delivery of FXN mRNA. Encouraging results from both approaches have been presented in the literature. Since the coding sequence for frataxin is quite small, AAV vectors can easily accommodate the full FXN mRNA sequence (devoid of intronic sequences). Several FXN conditional knockout mouse models have been used to test efficacy of AAV-FXN, with positive results in both cardiac knockouts [116, 117] and neurological knockouts [118, 119]. Puccio and colleagues reported a conditional mouse model with complete frataxin deletion in cardiac and skeletal muscle and prevention of cardiac symptoms in this model by intravenous injection of AAV-FXN. Most impressively, AAV-FXN administration after the onset of disease fully reversed cardiomyopathy within a few days [116]. A recent study has shown full rescue of cardiomyopathy with only one half of cardiomyocytes receiving AAV-FXN vector [117]. Puccio and colleagues developed a second conditional mouse model with complete frataxin deletion in parvalbumin-expressing cells that recapitulates FRDA sensory ataxia and neuropathy [118]. Similar to the cardiac model, postsymptomatic delivery of AAV-FXN provided complete rescue of sensory ataxia in this model. Other efforts toward FXN gene therapy include HSV vectors [120], but these studies are not as far advanced as the AAV approaches. Taken together, results with AAV vectors provide strong preclinical proof-of-concept for gene therapy in both the cardiac and neurological aspects of FRDA, and these results have inspired a number of biotechnology and pharmaceutical companies to develop programs in FRDA gene therapy (Fig. 3). For gene therapy to be realized as a treatment for a chronic disease such as FRDA, viruses for gene delivery need to be optimized to evade neutralizing antibodies and these vectors need to be targeted to the affected tissues in the disease, including both the central and peripheral nervous system and the heart. Numerous academic and industrial efforts are aimed at such improvements in the field of AAV gene therapy (reviewed in [121]).
A second approach to gene therapy is the direct delivery of FXN mRNA to affected tissues rather than the gene encoding this mRNA [122]. Efforts at Pfizer (Cambridge, MA) have focused on the use of lipid nanoparticles (LNP) as a means of mRNA delivery, using a codon-optimized FXN mRNA. Nabhan and colleagues showed that LNP-FXN mRNA is processed to mature frataxin protein when administered intravenously to adult mice and was detected up to 7 days after administration in the liver [122]. When this mRNA was administered into the spinal cord of adult mice via intrathecal injection, the recombinant human protein was detectable in the DRG. Whether LNP-FXN mRNA will reverse neurological or cardiac symptoms in FRDA mouse models has yet to be reported.
Stabilizing Frataxin and Increasing Frataxin Protein Expression
Several approaches have been taken to find molecules that will either stabilize existing frataxin protein from turn-over, thereby increasing its intracellular concentration, or molecules that will increase the cell’s ability to synthesize frataxin protein from the diminished amount of FXN mRNA present in patient cells.
Frataxin Stabilization
If one could identify the mechanism(s) whereby frataxin protein is degraded in cells, it should be possible to design or identify compounds that will prevent frataxin turn-over and hence increase the steady-state levels of the protein in FRDA cells and hopefully in human patients. To this end, Testi and colleagues reported studies that identify the sites of ubiquitinylation on frataxin, the ubiquitin ligases involved in degradation, and compounds that stabilize and increase frataxin levels in cells [123,124,125]. Rufini et al. showed that frataxin is degraded by the ubiquitin-proteasome system in cells and that frataxin K147 is the primary site for ubiquitinylation [123]. Mutation of K147 to arginine resulted in accumulation of significantly higher levels of frataxin in cells. Computational studies lead to the identification of compounds from a chemical library that bind the frataxin K147 cleft and thereby block ubiquitinylation and turn-over, leading to higher protein levels in FRDA cells, with a concomitant recovery of aconitase and ATP levels [123]. In a second publication, more active molecules with similar activity in FRDA patient cells were reported [124]. Having identified the major site for ubiquitinylation and regulation of frataxin levels, Testi and colleagues went on to identify RNF126 as the ubiquitin ligase involved in this process [125]. Thus, small molecule inhibitors that either target frataxin itself at the ubiquitinylation site or RNF126 are being considered as therapeutics by Fratagene Therapeutics (Rome, Italy). Moreover, when designing strategies for either protein replacement therapy or viral gene therapy, perhaps frataxin K147R should be considered.
Src Inhibitors
Inhibition of Src protein kinases is also being considered as a therapy for FRDA. Since Src protein kinases are themselves regulated by redox and cellular ATP levels, Cherubini and colleagues asked whether Src kinases regulate frataxin levels in cells and whether Src protein kinase inhibitors modulate frataxin protein levels in FRDA cells [126]. These authors reported that frataxin can be phosphorylated at position Y118, and such phosphorylation promotes the ubiquitinylation and degradation of frataxin protein. Therefore, inhibition of this phosphorylation event was considered as a means to increase intracellular levels of frataxin protein. As predicted, Src inhibitors, including the FDA-approved therapeutic dasatinib, induced accumulation of frataxin protein but were ineffective on a Y118F mutant frataxin that cannot be phosphorylated. Importantly, some of these Src inhibitors rescued the aconitase defect in FRDA patient cells. Along with etravirine and frataxin stabilizers, Src inhibitors are being pursued as FRDA therapeutics by Fratagene Therapeutics (Rome, Italy).
Stem Cell-Based Therapies
A number of studies have considered human stem cells as therapeutics for FRDA, including mesenchymal stem cells ([127] and references therein), mesenchymal stem cell-derived factors [128], and hematopoietic stem and progenitor cells (HSPCs [129]). Intrathecal delivery of bone marrow-derived mesenchymal stem cells, both autologous and allogeneic, to the YG8R FRDA mouse model has been reported to increase frataxin expression in the DRG, to increase the levels of antioxidant enzymes, and to improve motor function [127]. Similar findings have been reported by Kemp and colleagues [130], and these investigators have also shown that stem cell-derived factors [128], as well as the cytokines granulocyte-colony stimulating factor (G-CSF) and stem cell factor (SCF) [131] have similar effects of ameliorating the disease phenotype in the YG8R FRDA mouse model, and each of these treatments induced frataxin expression in affected tissues. As to how stem cells increase frataxin levels, Rocca and colleagues recently reported the transfer of wild-type frataxin and Cox8 mitochondrial proteins from HSPC-derived microglia/macrophages to neurons and muscle myocytes in the YG8R mouse model [129]. In this study, the transplanted HSPCs engrafted and then differentiated into microglia in the brain and spinal cord and into macrophages in the DRGs, heart, and muscle of YG8R FRDA mice, ameliorating muscle weakness and locomotor deficits and degeneration of large sensory neurons in the DRG. The results of these stem cell transplantation studies will certainly encourage further development of this approach for FRDA.
Approach 4: Correction of Downstream Consequences of Frataxin Deficiency
Peroxidation-Resistant Polyunsaturated Fatty Acids (dPUFAs)
Since lipid peroxidation is one of the downstream consequences of frataxin deficiency [132], treatment with peroxidation-resistant polyunsaturated fatty acids is currently being assessed as an FRDA therapeutic by Retrotope (Los Altos, CA, USA). Cottacelli and colleagues assessed the role of oxidative stress in lipid peroxidation by treating FRDA patient cells with polyunsaturated fatty acids (PUFAs) deuterated at bis-allylic sites [133]. Such molecules are known to be far less susceptible to peroxidation than their nondeuterated counterparts (reviewed in [133]). The authors showed that deuterated linoleic and α-linolenic acids reduced lipid peroxidation in FRDA cells and rescued oxidative stress in these cells. Zesiewicz and colleagues evaluated the safety, pharmacokinetics, and efficacy (cardiopulmonary exercise testing and timed 25-ft walk) of a deuterated ethyl linoleate (RT001) in FRDA patients in a phase I/II double-blind placebo-controlled study [134]. Encouraging results were obtained in this study, such as an improvement in peak workload in the drug-treated group compared to placebo as well as an improvement in peak oxygen consumption. These results and the safety and tolerability of the compound should provide impetus for further clinical assessment of RT001.
Phosphodiesterase Inhibitors
Recently Molla and colleagues performed a proteomic study of DRGs comparing the YG8R FRDA mouse model to control C57BL/6J mice and found decreased expression of electron transport chain proteins, components of the oxidative phosphorylation system, antioxidant enzymes, and decreased expression of proteins involved in Ca2+ signaling and G protein-coupled receptors [135]. Remarkably, phosphodiesterase (PDE) inhibitors including sildenafil were able to restore cytosolic Ca2+ levels and revert the axonal dystrophy found in FRDA YG8R DRG neurons [135]. Although PDE inhibitors are widely used clinically, the authors did not address the consequences of their use in treatment of a chronic disease such as FRDA and this will need to be carefully assessed prior to consideration of open-label clinical trials.
Antioxidants
There is a long history of pursuing anti-oxidants as therapeutics for FRDA, and many such compounds have been evaluated in FRDA clinical trials (reviewed in [136]). Perhaps the most extensively evaluated compound is the co-enzyme Q10 derivative idebenone. Although early results in mouse models [137] and in a 12-month clinical study in FRDA pediatric patients [138] were encouraging, full clinical assessment failed to show neurological or cardiac improvement (reviewed in [139]) and the sponsoring pharmaceutical company Santhera Pharmaceuticals (Prattein, Switzerland) has abandoned idebenone for FRDA. Other antioxidants, such as EPI-743 (BioElectron Technology Corporation, Mountain View, CA), are also in development for FRDA. EPI-743 targets oxidoreductase enzymes, such as 15-lipoxygenase, that control the redox state in cells, controlling oxidative stress and cell death. Zesiewicz and colleagues recently reported the results of a multicenter clinical trial of EPI-743 during a 6-month placebo-controlled phase, followed by an 18-month open-label phase. EPI-743 was demonstrated to be safe and well tolerated, and most importantly, EPI-743 treatment was associated with a statistically significant improvement in neurological function and disease progression at 24 months, relative to a natural history patient cohort, suggesting that further evaluation of this compound is warranted.
Other antioxidant approaches have been taken by numerous investigators over the years (reviewed in [136]), but notable recent results from Hecht and colleagues ([140] and references therein) have shown that lipophilic methylene violet (MV) and methylene blue (MB) analogues can reverse ROS accumulation, maintain mitochondrial membrane potential, and increase ATP production in FRDA patient cells. MB analogues were also shown to promote increased frataxin levels and mitochondrial biogenesis [140] and Hecht and colleagues have reported the synthesis of numerous derivatives with improved potency and reduced cytotoxicity, making these compounds suitable for animal studies and perhaps clinical evaluation.
Estrogen and Related Compounds
Richardson and colleagues [141] reported that phenolic estrogens, which function as anti-oxidants independent of estrogen receptor targeting activity, are able to reverse many of the oxidative stress-related consequences of frataxin depletion in FRDA cells. Estrogen derivatives were able to prevent lipid peroxidation and mitochondrial membrane potential defects, maintain ATP at near control levels, increase oxidative phosphorylation, and restore aconitase activity. Another study from the same authors [142] showed that the estrogen receptor agonists R- and S-equol have similar cytoprotective effects in FRDA fibroblasts challenged with the oxidant L-buthioine (S,R)-sulfoximine (BSO), reducing ROS and oxidative cell damage. Although these results were promising, no further studies have been published on either estrogen or estrogen-related compounds in FRDA.
PPAR-Gamma Agonists
Through a search of publicly available gene expression data, Marmolino and colleagues found that the peroxisome proliferator-activated receptor gamma (PPARγ) agonist rosiglitazone is an inducer of FXN mRNA [143]. PPARγ is a key regulator of the antioxidant response and mitochondrial biogenesis. Based on this observation, Marmolino et al. tested another PPARγ agonist Azelaoyl PAF and found similar increases in frataxin mRNA and protein in both unaffected cells and FRDA patient fibroblasts. Another study [144] confirmed the dysregulation of the PPARγ pathway in FRDA, both by gene expression analysis in the heart and skeletal muscle from the KIKO FRDA mouse model and by siRNA-mediated downregulation of frataxin. These authors found downregulation of the PPARγ coactivator PGC-1α and transcription factor Srebp1 in cellular and animal models of frataxin deficiency and in cells from FRDA patients. Similarly, siRNA downregulation of PGC-1α also results in reduced levels of frataxin, suggesting a feedback loop between frataxin expression and the PPARγ pathway. PGC-1α activation in cells or in the KIKO mouse model with the PPARγ agonist pioglitazone or with a cAMP-dependent protein kinase agonist restored a marker of frataxin deficiency, SOD2, suggesting modulation of the PPARγ pathway as a therapeutic approach for FRDA. A phase III trial of pioglitazone was completed but the results remain unpublished (reviewed in [145]). Nevertheless, another orally available PPARγ agonist MIN-102 is under investigation by Minoryx Therapeutics (Barcelona, Spain).
Approach 5: Other and Symptomatic Approaches
A detailed description of many of the completed and ongoing clinical trials for FRDA has been presented [145]. Additionally, other approaches are outlined in Fig. 3 and include (+)epicatechin (Cardero Therapeutics, Sunnyvale, CA) to increase mitochondrial biogenesis and reduce oxidative stress; methylprednisone as an anti-inflammatory to reduce signs of inflammation observed in FRDA [146, 147]; and the D-amino acid oxidase inhibitor TAK-831 (Takeda Pharmaceuticals), both as symptomatic treatments. TAK-831 has been shown to increase motor performance in other neurological diseases and hence may offer benefit in FRDA. Lynch and colleagues recently assessed the effect of methylprednisolone on safety, tolerability, and clinical measures in both adult and pediatric FRDA patients [148]. Comparing baseline scores with scores at 26 weeks of treatment, pediatric patients showed statistically significant improvement in gait distance in a one-minute walk test, but not in other outcome measures. The authors suggest that methylprednisolone should be considered in future studies of ambulatory FRDA patients. As of this writing, no other primary publications for these therapeutic approaches have appeared in the literature.
Conclusions and Future Prospects
It has now been over two decades since the discovery of the genetic basis for Friedreich ataxia [10] and over a decade since it was established that the repeats silence transcription through a chromatin-based mechanism [43, 45]. Given that FRDA is a rare disease, likely affecting fewer than 20,000 individuals worldwide, it is heartening that the scientific community has devoted such attention to this disease, with over 3300 publications in PubMed as of this writing. Early efforts toward therapies for the disease largely focused on reversing the consequences of diminished frataxin, including iron overload and oxidative cell damage. Although these efforts continue, a new era in therapeutic development has begun with therapies aimed at upregulating frataxin protein in affected tissues, either by reversing gene silencing or delivery of frataxin protein itself (either through protein replacement or gene therapy). There is tremendous excitement in both the patient and research communities looking forward to clinical trials of these therapies (see Fig. 3) and recent clinical studies with Nrf2 activators provide hope that treatments are on the horizon [104]. It is still not clear why particular cell types and tissues are most vulnerable to decreased frataxin, so future studies will undoubtedly be focused on why these cells/tissues are most affected and means to target therapies to the affected tissues.
References
La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11(4):247–58.
Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annual Review of Neuroscience. 2007;30:575–621.
Paulson H. Repeat expansion diseases. Handbook of Clinical Neurology. 2018;147:105–23.
Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 2016;17(6):383–95.
Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, et al. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One. 2012;7(11):e49083.
Zhang N, Ashizawa T. RNA toxicity and foci formation in microsatellite expansion diseases. Curr Opin Genet Dev. 2017;44:17–29.
Du J, Aleff RA, Soragni E, Kalari K, Nie J, Tang X, et al. RNA toxicity and missplicing in the common eye disease Fuchs endothelial corneal dystrophy. J Biol Chem. 2015;290:5979–90.
Cleary JD, Pattamatta A, Ranum LPW. Repeat-associated non-ATG (RAN) translation. J Biol Chem. 2018;293:16127–41.
Nageshwaran S, Festenstein R. Epigenetics and triplet-repeat neurological diseases. Front Neurol. 2015;6:262.
Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996;271(5254):1423–7.
Bidichandani SI, Ashizawa T, Patel PI. Atypical Friedreich ataxia caused by compound heterozygosity for a novel missense mutation and the GAA triplet-repeat expansion. Am J Hum Genet. 1997;60(5):1251–6.
Gellera C, Castellotti B, Mariotti C, Mineri R, Seveso V, Didonato S, et al. Frataxin gene point mutations in Italian Friedreich ataxia patients. Neurogenetics 2007;8(4):289–99.
Lamba LD, Ciotti P, Giribaldi G, Di Maria E, Varese A, Di Stadio M, et al. Friedreich’s ataxia: a new mutation in two compound heterozygous siblings with unusual clinical onset. European neurology. 2009;61(4):240–3.
Anheim M, Mariani LL, Calvas P, Cheuret E, Zagnoli F, Odent S, et al. Exonic deletions of FXN and early-onset Friedreich ataxia. Arch Neurol. 2012;69(7):912–6.
Burk K. Friedreich ataxia: current status and future prospects. Cerebellum & ataxias. 2017;4:4.
Pianese L, Turano M, Lo Casale MS, De Biase I, Giacchetti M, Monticelli A, et al. Real time PCR quantification of frataxin mRNA in the peripheral blood leucocytes of Friedreich ataxia patients and carriers. J Neurol Neurosurg Psychiatry. 2004;75(7):1061–3.
Bidichandani SI, Ashizawa T, Patel PI. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am J Hum Genet. 1998;62(1):111–21.
Pandey A, Gordon DM, Pain J, Stemmler TL, Dancis A, Pain D. Frataxin directly stimulates mitochondrial cysteine desulfurase by exposing substrate-binding sites, and a mutant Fe-S cluster scaffold protein with frataxin-bypassing ability acts similarly. J Biol Chem. 2013;288(52):36773–86.
Bulteau AL, O’Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, Szweda LI. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 2004;305(5681):242–5.
Seznec H, Simon D, Bouton C, Reutenauer L, Hertzog A, Golik P, et al. Friedreich ataxia: the oxidative stress paradox. Hum Mol Genet. 2005;14(4):463–74.
Calabrese V, Lodi R, Tonon C, D’Agata V, Sapienza M, Scapagnini G, et al. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J Neurol Sci. 2005;233(1–2):145–62.
Lynch DR, Farmer JM, Wilson RB. Mortality in Friedreich’s ataxia. Tex Heart Inst J. 2007;34(4):502–3; author reply 3-4.
Ohshima K, Montermini L, Wells RD, Pandolfo M. Inhibitory effects of expanded GAA.TTC triplet repeats from intron I of the Friedreich ataxia gene on transcription and replication in vivo. J Biol Chem. 1998;273(23):14588–95.
De Michele G, Filla A, Criscuolo C, Scarano V, Cavalcanti F, Pianese L, et al. Determinants of onset age in Friedreich’s ataxia. J Neurol. 1998;245(3):166–8.
Baralle M, Pastor T, Bussani E, Pagani F. Influence of Friedreich ataxia GAA noncoding repeat expansions on pre-mRNA processing. Am J Hum Genet. 2008;83(1):77–88.
Xia H, Cao Y, Dai X, Marelja Z, Zhou D, Mo R, et al. Novel frataxin isoforms may contribute to the pathological mechanism of Friedreich ataxia. PLoS One. 2012;7(10):e47847.
Sznajder LJ, Thomas JD, Carrell EM, Reid T, McFarland KN, Cleary JD, et al. Intron retention induced by microsatellite expansions as a disease biomarker. Proc Natl Acad Sci U S A. 2018;115(16):4234–9.
Punga T, Buhler M. Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation. EMBO Mol Med. 2010;2(4):120–9.
Soragni E, Miao W, Iudicello M, Jacoby D, Demercanti S, Clerico M, et al. Epigenetic therapy for Friedreich’s ataxia. Ann Neurol. 2014;76:489–508.
Sakamoto N, Chastain PD, Parniewski P, Ohshima K, Pandolfo M, Griffith JD, et al. Sticky DNA: self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol Cell. 1999;3(4):465–75.
Sakamoto N, Ohshima K, Montermini L, Pandolfo M, Wells RD. Sticky DNA, a self-associated complex formed at long GAA*TTC repeats in intron 1 of the frataxin gene, inhibits transcription. J Biol Chem. 2001;276(29):27171–7.
Burnett R, Melander C, Puckett JW, Son LS, Wells RD, Dervan PB, et al. DNA sequence-specific polyamides alleviate transcription inhibition associated with long GAA.TTC repeats in Friedreich's ataxia. Proc Natl Acad Sci U S A. 2006;103(31):11497–502.
Grabczyk E, Usdin K. Alleviating transcript insufficiency caused by Friedreich’s ataxia triplet repeats. Nucleic Acids Res. 2000;28(24):4930–7.
Grabczyk E, Usdin K. The GAA*TTC triplet repeat expanded in Friedreich’s ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 2000;28(14):2815–22.
Groh M, Lufino MM, Wade-Martins R, Gromak N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014;10(5):e1004318.
Dion A, Wilson JH. Instability and chromatin structure of expanded trinucleotide repeats. Trends Genet. 2009;25:288–97.
De Biase I, Chutake YK, Rindler PM, Bidichandani SI. Epigenetic silencing in friedreich ataxia is associated with depletion of CTCF (CCCTC-binding factor) and antisense transcription. PLoS One 2009;4(11):e7914.
He Y, Vogelstein B, Velculescu VE, Papadopoulos N, Kinzler KW. The antisense transcriptomes of human cells. Science 2008;322:1855–7.
Al-Mahdawi S, Sandi C, Mouro Pinto R, Pook MA. Friedreich ataxia patient tissues exhibit increased 5-hydroxymethylcytosine modification and decreased CTCF binding at the FXN locus. PLoS One 2013;8(9):e74956.
Mikaeili H, Sandi M, Bayot A, Al-Mahdawi S, Pook MA. FAST-1 antisense RNA epigenetically alters FXN expression. Sci Rep. 2018;8(1):17217.
Li L, Matsui M, Corey DR. Activating frataxin expression by repeat-targeted nucleic acids. Nat Commun. 2016;7:10606.
Eimer H, Sureshkumar S, Singh Yadav A, Kraupner-Taylor C, Bandaranayake C, Seleznev A, et al. RNA-dependent epigenetic silencing directs transcriptional downregulation caused by intronic repeat expansions. Cell 2018;174:1095–105.
Saveliev A, Everett C, Sharpe T, Webster Z, Festenstein R. DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature 2003;422(6934):909–13.
Elgin SC, Grewal SI. Heterochromatin: silence is golden. Curr Biol. 2003;13(23):R895–8.
Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol. 2006;2(10):551–8.
Greene E, Mahishi L, Entezam A, Kumari D, Usdin K. Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucl Acids Res. 2007;35:3383–90.
Sandi C, Sandi M, Virmouni SA, Al-Mahdawi S, Pook MA. Epigenetic-based therapies for Friedreich ataxia. Front Genet. 2014;5:1–12.
Yandim C, Natisvili T, Festenstein R. Gene regulation and epigenetics in Friedreich’s ataxia. J Neurochem. 2013;126 Suppl 1:21–42.
Al-Mahdawi S, Pinto RM, Ismail O, Varshney D, Lymperi S, Sandi C, et al. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum Mol Genet. 2008;17(5):735–46.
Rai M, Soragni E, Jenssen K, Burnett R, Herman D, Gottesfeld JM, et al. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS ONE. 2008;3:e1958 doi:https://doi.org/10.1371/journal.pone.0001958.
Kim E, Napierala M, Dent SY. Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich’s ataxia. Nucleic Acids Res. 2011;39(19):8366–77.
Chan PK, Torres R, Yandim C, Law PP, Khadayate S, Mauri M, et al. Heterochromatinization induced by GAA-repeat hyperexpansion in Friedreich’s ataxia can be reduced upon HDAC inhibition by vitamin B3. Hum Mol Genet. 2013;22(13):2662–75.
Evans-Galea MV, Carrodus N, Rowley SM, Corben LA, Tai G, Saffery R, et al. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann Neurol. 2012;71(4):487–97.
Castaldo I, Pinelli M, Monticelli A, Acquaviva F, Giacchetti M, Filla A, et al. DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J Med Genet. 2008;45(12):808–12.
Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41(12):1350–3.
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178–86.
El-Osta A, Wolffe AP. DNA methylation and histone deacetylation in the control of gene expression: basic biochemistry to human development and disease. Gene Expr. 2000;9:63–75.
Soragni E, Chou CJ, Rusche JR, Gottesfeld JM. Mechanism of action of 2-aminobenzamide HDAC inhibitors in reversing gene silencing in Friedreich’s ataxia. Front Neurol. 2015;6:44.
Silva AM, Brown JM, Buckle VJ, Wade-Martins R, Lufino MM. Expanded GAA repeats impair FXN gene expression and reposition the FXN locus to the nuclear lamina in single cells. Hum Mol Genet. 2015;24(12):3457–71.
Zullo JM, Demarco IA, Pique-Regi R, Gaffney DJ, Epstein CB, Spooner CJ, et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 2012;149(7):1474–87.
Kumari D, Biacsi RE, Usdin K. Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells. J Biol Chem. 2011;286(6):4209–15.
Chutake YK, Costello WN, Lam C, Bidichandani SI. Altered nucleosome positioning at the transcription start site and deficient transcriptional initiation in Friedreich ataxia. J Biol Chem. 2014;289(22):15194–202.
Chutake YK, Lam C, Costello WN, Anderson M, Bidichandani SI. Epigenetic promoter silencing in Friedreich ataxia is dependent on repeat length. Ann Neurol. 2014;76(4):522–8.
Chutake YK, Costello WN, Lam CC, Parikh AC, Hughes TT, Michalopulos MG, et al. FXN promoter silencing in the humanized mouse model of Friedreich ataxia. PLoS One. 2015;10(9):e0138437.
Li Y, Lu Y, Polak U, Lin K, Shen J, Farmer J, et al. Expanded GAA repeats impede transcription elongation through the FXN gene and induce transcriptional silencing that is restricted to the FXN locus. Hum Mol Genet. 2015;24(24):6932–43.
Lopez Castel A, Cleary JD, Pearson CE. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol. 2010;11(3):165–70.
Du J, Campau E, Soragni E, Ku S, Puckett JW, Dervan PB, et al. Role of mismatch repair enzymes in GAA.TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells. J Biol Chem. 2012;287(35):29861–72.
Ouellet DL, Cherif K, Rousseau J, Tremblay JP. Deletion of the GAA repeats from the human frataxin gene using the CRISPR-Cas9 system in YG8R-derived cells and mouse models of Friedreich ataxia. Gene Ther. 2017;24(5):265–74.
Li Y, Polak U, Bhalla AD, Rozwadowska N, Butler JS, Lynch DR, et al. Excision of expanded GAA repeats alleviates the molecular phenotype of Friedreich’s ataxia. Mol Ther. 2015;23(6):1055–65.
Lai J-I, Nachun D, Petrosyan L, Throesch B, Campau E, Gao F, et al. Transcription profiling of isogenic Friedreich ataxia neurons and effect of an HDAC inhibitor on disease signatures. J Biol Chem. 2019;294:1846–59.
Sarsero JP, Li L, Wardan H, Sitte K, Williamson R, Ioannou PA. Upregulation of expression from the FRDA genomic locus for the therapy of Friedreich ataxia. J Gene Med. 2003;5(1):72–81.
Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol. 2006;26(1):28–38.
Xu C, Soragni E, Chou CJ, Herman D, Plasterer HL, Rusche JR, et al. Chemical probes identify a role for histone deacetylase 3 in Friedreich’s ataxia gene silencing. Chem Biol. 2009;16:980–9.
Libri V, Yandim C, Athanasopoulos S, Loyse N, Natisvili T, Law PP, et al. Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich’s ataxia: an exploratory, open-label, dose-escalation study. Lancet 2014; 384:504–13.
Lufino MM, Silva AM, Nemeth AH, Alegre-Abarrategui J, Russell AJ, Wade-Martins R. A GAA repeat expansion reporter model of Friedreich’s ataxia recapitulates the genomic context and allows rapid screening of therapeutic compounds. Hum Mol Genet. 2013;22(25):5173–87.
Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem. 2008;283:35402–9.
Xu C, Soragni E, Jacques V, Rusche JR, Gottesfeld JM. Improved histone deacetylase inhibitors as therapeutics for the neurodegenerative disease Friedreich’s ataxia: a new synthetic route. Pharmaceuticals 2011;4(12):1578–90.
Rai M, Soragni E, Chou CJ, Barnes G, Jones S, Rusche JR, et al. Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich's ataxia patients and in a mouse model. PLoS One. 2010;5(1):e8825.
Sandi C, Pinto RM, Al-Mahdawi S, Ezzatizadeh V, Barnes G, Jones S, et al. Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol Dis. 2011;42(3):496–505.
Codazzi F, Hu A, Rai M, Salerno Scarzella F, Mangiameli E, Pelizzoni I, et al. Friedreich ataxia induced pluripotent stem cell-derived neurons show a cellular phenotype that is corrected by a benzamide HDAC inhibitor. Hum Mol Genet. 2016;25:4847–55.
Chutake YK, Lam CC, Costello WN, Anderson MP, Bidichandani SI. Reversal of epigenetic promoter silencing in Friedreich ataxia by a class I histone deacetylase inhibitor. Nucleic Acids Res. 2016.
Li L, Voullaire L, Sandi C, Pook MA, Ioannou PA, Delatycki MB, et al. Pharmacological screening using an FXN-EGFP cellular genomic reporter assay for the therapy of Friedreich ataxia. PLoS One. 2013;8(2):e55940.
Yiu EM, Tai G, Peverill RE, Lee KJ, Croft KD, Mori TA, et al. An open-label trial in Friedreich ataxia suggests clinical benefit with high-dose resveratrol, without effect on frataxin levels. J Neurol. 2015;262(5):1344–53.
Li L, Shen X, Liu Z, Norrbom M, Prakash TP, O’Reilly D, et al. Activation of frataxin protein expression by antisense oligonucleotides targeting the mutant expanded repeat. Nucleic Acid Ther. 2018;28(1):23–33.
Shen X, Kilikevicius A, O’Reilly D, Prakash TP, Damha MJ, Rigo F, et al. Activating frataxin expression by single-stranded siRNAs targeting the GAA repeat expansion. Bioorg Med Chem Lett. 2018;28(17):2850–5.
Shen X, Beasley S, Putnam JN, Li Y, Prakash TP, Rigo F, et al. Efficient electroporation of neuronal cells using synthetic oligonucleotides: identifying duplex RNA and antisense oligonucleotide activators of human frataxin expression. Rna. 2019.
Cherif K, Gerard C, Rousseau J, Ouellet DL, Chapdelaine P, Tremblay JP. Increased frataxin expression induced in Friedreich ataxia cells by platinum TALE-VP64s or platinum TALE-SunTag. Molecular Therapy Nucleic Acids. 2018;12:19–32.
Waryah CB, Moses C, Arooj M, Blancafort P. Zinc fingers, TALEs, and CRISPR Systems: a comparison of tools for epigenome editing. Methods Mol Biol. 2018;1767:19–63.
Urbach AR, Love JJ, Ross SA, Dervan PB. Structure of a beta-alanine-linked polyamide bound to a full helical turn of purine tract DNA in the 1:1 motif. J Mol Biol. 2002;320(1):55–71.
Blattes R, Monod C, Susbielle G, Cuvier O, Wu JH, Hsieh TS, et al. Displacement of D1, HP1 and topoisomerase II from satellite heterochromatin by a specific polyamide. Embo J. 2006;25:2397–408.
Harki DA, Satyamurthy N, Stout DB, Phelps ME, Dervan PB. In vivo imaging of pyrrole-imidazole polyamides with positron emission tomography. Proc Natl Acad Sci USA. 2008;105:13039–44.
Erwin GS, Grieshop MP, Ali A, Qi J, Lawlor M, Kumar D, et al. Synthetic transcription elongation factors license transcription across repressive chromatin. Science 2017;358(6370):1617–22.
Tomassini B, Arcuri G, Fortuni S, Sandi C, Ezzatizadeh V, Casali C, et al. Interferon gamma upregulates frataxin and corrects the functional deficits in a Friedreich ataxia model. Hum Mol Genet. 2012;21(13):2855–61.
Seyer L, Greeley N, Foerster D, Strawser C, Gelbard S, Dong Y, et al. Open-label pilot study of interferon gamma-1b in Friedreich ataxia. Acta Neurologica Scandinavica 2015;132(1):7–15.
Lynch DR, Hauser L, McCormick A, Wells M, Dong YN, McCormack S, et al. Randomized, double-blind, placebo-controlled study of interferon-gamma 1b in Friedreich ataxia. Annals of Clinical and Translational Neurology 2019;6(3):546–53.
Shan Y, Schoenfeld RA, Hayashi G, Napoli E, Akiyama T, Iodi Carstens M, et al. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid Redox Signal. 2013.
D’Oria V, Petrini S, Travaglini L, Priori C, Piermarini E, Petrillo S, et al. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. International Journal of Molecular Sciences. 2013;14(4):7853–65.
Sahdeo S, Scott BD, McMackin MZ, Jasoliya M, Brown B, Wulff H, et al. Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich’s ataxia. Hum Mol Genet. 2014;23(25):6848–62.
Petrillo S, Piermarini E, Pastore A, Vasco G, Schirinzi T, Carrozzo R, et al. Nrf2-inducers counteract neurodegeneration in frataxin-silenced motor neurons: disclosing new therapeutic targets for Friedreich’s ataxia. International Journal of Molecular Sciences. 2017;18(10).
Abeti R, Baccaro A, Esteras N, Giunti P. Novel Nrf2-inducer prevents mitochondrial defects and oxidative stress in Friedreich’s ataxia models. Frontiers in Cellular Neuroscience. 2018;12:188.
Hayashi G, Jasoliya M, Sahdeo S, Sacca F, Pane C, Filla A, et al. Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum Mol Genet. 2017;26(15):2864–73.
Hayashi G, Cortopassi G. Lymphoblast oxidative stress genes as potential biomarkers of disease severity and drug effect in Friedreich’s ataxia. PLoS One. 2016;11(4):e0153574.
Jasoliya M, Sacca F, Sahdeo S, Chedin F, Pane C, Brescia Morra V, et al. Dimethyl fumarate dosing in humans increases frataxin expression: a potential therapy for Friedreich’s ataxia. PLoS One. 2019;14(6):e0217776.
Lynch DR, Farmer J, Hauser L, Blair IA, Wang QQ, Mesaros C, et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Annals of Clinical and Translational Neurology. 2019;6(1):15–26.
Sturm B, Stupphann D, Kaun C, Boesch S, Schranzhofer M, Wojta J, et al. Recombinant human erythropoietin: effects on frataxin expression in vitro. Eur J Clin Invest. 2005;35(11):711–7.
Acquaviva F, Castaldo I, Filla A, Giacchetti M, Marmolino D, Monticelli A, et al. Recombinant human erythropoietin increases frataxin protein expression without increasing mRNA expression. Cerebellum 2008;7(3):360–5.
Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci U S A. 2000;97:10526–31.
Boesch S, Sturm B, Hering S, Goldenberg H, Poewe W, Scheiber-Mojdehkar B. Friedreich’s ataxia: clinical pilot trial with recombinant human erythropoietin. Ann Neurol. 2007;62(5):521–4.
Mariotti C, Fancellu R, Caldarazzo S, Nanetti L, Di Bella D, Plumari M, et al. Erythropoietin in Friedreich ataxia: no effect on frataxin in a randomized controlled trial. Mov Disord. 2012;27(3):446–9.
Sacca F, Puorro G, Marsili A, Antenora A, Pane C, Casali C, et al. Long-term effect of epoetin alfa on clinical and biochemical markers in Friedreich ataxia. Mov Disord. 2016;31(5):734–41.
Boesch S, Nachbauer W, Mariotti C, Sacca F, Filla A, Klockgether T, et al. Safety and tolerability of carbamylated erythropoietin in Friedreich’s ataxia. Mov Disord. 2014;29(7):935–9.
Miller JL, Rai M, Frigon NL, Pandolfo M, Punnonen J, Spencer JR. Erythropoietin and small molecule agonists of the tissue-protective erythropoietin receptor increase FXN expression in neuronal cells in vitro and in FXN-deficient KIKO mice in vivo. Neuropharmacology 2017;123:34–45.
Alfedi G, Luffarelli R, Condo I, Pedini G, Mannucci L, Massaro DS, et al. Drug repositioning screening identifies etravirine as a potential therapeutic for Friedreich’s ataxia. Mov Disord. 2019;34(3):323–34.
Bertrand L, Dygert L, Toborek M. Antiretroviral treatment with efavirenz disrupts the blood-brain barrier integrity and increases stroke severity. Sci Rep. 2016;6:39738.
Vyas PM, Tomamichel WJ, Pride PM, Babbey CM, Wang Q, Mercier J, et al. A TAT-frataxin fusion protein increases lifespan and cardiac function in a conditional Friedreich’s ataxia mouse model. Hum Mol Genet. 2012;21:1230–47.
Perdomini M, Belbellaa B, Monassier L, Reutenauer L, Messaddeq N, Cartier N, et al. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat Med. 2014;20(5):542–7.
Belbellaa B, Reutenauer L, Monassier L, Puccio H. Correction of half the cardiomyocytes fully rescue Friedreich ataxia mitochondrial cardiomyopathy through cell-autonomous mechanisms. Hum Mol Genet. 2018.
Piguet F, de Montigny C, Vaucamps N, Reutenauer L, Eisenmann A, Puccio H. Rapid and complete reversal of sensory ataxia by gene therapy in a novel model of Friedreich ataxia. Mol Ther. 2018;26(8):1940–52.
Gerard C, Xiao X, Filali M, Coulombe Z, Arsenault M, Couet J, et al. An AAV9 coding for frataxin clearly improved the symptoms and prolonged the life of Friedreich ataxia mouse models. Molecular Therapy Methods & Clinical Development 2014;1:14044.
Ventosa M, Wu Z, Lim F. Sustained FXN expression in dorsal root ganglia from a nonreplicative genomic HSV-1 vector. J Gene Med. 2017;19(11):376–86.
Barnes C, Scheideler O, Schaffer D. Engineering the AAV capsid to evade immune responses. Current opinion in biotechnology. 2019;60:99–103.
Nabhan JF, Wood KM, Rao VP, Morin J, Bhamidipaty S, LaBranche TP, et al. Intrathecal delivery of frataxin mRNA encapsulated in lipid nanoparticles to dorsal root ganglia as a potential therapeutic for Friedreich’s ataxia. Sci Rep. 2016;6:20019.
Rufini A, Fortuni S, Arcuri G, Condo I, Serio D, Incani O, et al. Preventing the ubiquitin-proteasome-dependent degradation of frataxin, the protein defective in Friedreich’s ataxia. Hum Mol Genet. 2011;20(7):1253–61.
Rufini A, Cavallo F, Condo I, Fortuni S, De Martino G, Incani O, et al. Highly specific ubiquitin-competing molecules effectively promote frataxin accumulation and partially rescue the aconitase defect in Friedreich ataxia cells. Neurobiol Dis. 2015;75:91–9.
Benini M, Fortuni S, Condo I, Alfedi G, Malisan F, Toschi N, et al. E3 ligase RNF126 directly ubiquitinates frataxin, promoting its degradation: identification of a potential therapeutic target for Friedreich ataxia. Cell Rep 2017;18(8):2007–17.
Cherubini F, Serio D, Guccini I, Fortuni S, Arcuri G, Condo I, et al. Src inhibitors modulate frataxin protein levels. Hum Mol Genet. 2015;24(15):4296–305.
Jones J, Estirado A, Redondo C, Pacheco-Torres J, Sirerol-Piquer MS, Garcia-Verdugo JM, et al. Mesenchymal stem cells improve motor functions and decrease neurodegeneration in ataxic mice. Mol Ther. 2015;23(1):130–8.
Kemp K, Dey R, Cook A, Scolding N, Wilkins A. Mesenchymal stem cell-derived factors restore function to human frataxin-deficient cells. Cerebellum. 2017;16(4):840–51.
Rocca CJ, Goodman SM, Dulin JN, Haquang JH, Gertsman I, Blondelle J, et al. Transplantation of wild-type mouse hematopoietic stem and progenitor cells ameliorates deficits in a mouse model of Friedreich’s ataxia. Sci Transl Med. 2017;9(413).
Kemp KC, Hares K, Redondo J, Cook AJ, Haynes HR, Burton BR, et al. Bone marrow transplantation stimulates neural repair in Friedreich’s ataxia mice. Ann Neurol. 2018;83(4):779–93.
Kemp KC, Cerminara N, Hares K, Redondo J, Cook AJ, Haynes HR, et al. Cytokine therapy-mediated neuroprotection in a Friedreich’s ataxia mouse model. Ann Neurol. 2017;81(2):212–26.
Abeti R, Parkinson MH, Hargreaves IP, Angelova PR, Sandi C, Pook MA, et al. Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia. Cell death & disease. 2016;7:e2237.
Cotticelli MG, Crabbe AM, Wilson RB, Shchepinov MS. Insights into the role of oxidative stress in the pathology of Friedreich ataxia using peroxidation resistant polyunsaturated fatty acids. Redox Biology 2013;1:398–404.
Zesiewicz T, Heerinckx F, De Jager R, Omidvar O, Kilpatrick M, Shaw J, et al. Randomized, clinical trial of RT001: early signals of efficacy in Friedreich’s ataxia. Mov Disord. 2018;33(6):1000–5.
Molla B, Munoz-Lasso DC, Calap P, Fernandez-Vilata A, de la Iglesia-Vaya M, Pallardo FV, et al. Phosphodiesterase inhibitors revert axonal dystrophy in Friedreich’s ataxia mouse model. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2019.
Strawser CJ, Schadt KA, Lynch DR. Therapeutic approaches for the treatment of Friedreich’s ataxia. Expert Rev Neurother. 2014;14(8):949–57.
Seznec H, Simon D, Monassier L, Criqui-Filipe P, Gansmuller A, Rustin P, et al. Idebenone delays the onset of cardiac functional alteration without correction of Fe-S enzymes deficit in a mouse model for Friedreich ataxia. Hum Mol Genet. 2004;13(10):1017–24.
Meier T, Perlman SL, Rummey C, Coppard NJ, Lynch DR. Assessment of neurological efficacy of idebenone in pediatric patients with Friedreich’s ataxia: data from a 6-month controlled study followed by a 12-month open-label extension study. J Neurol. 2012;259:284–91.
Cook A, Boesch S, Heck S, Brunt E, Klockgether T, Schols L, et al. Patient reported outcomes in Friedreich’s ataxia after withdrawal from idebenone. Acta Neurologica Scandinavica. 2019.
Khdour OM, Bandyopadhyay I, Visavadiya NP, Roy Chowdhury S, Hecht SM. Phenothiazine antioxidants increase mitochondrial biogenesis and frataxin levels in Friedreich’s ataxia cells. MedChemComm. 2018;9(9):1491–501.
Richardson TE, Yu AE, Wen Y, Yang SH, Simpkins JW. Estrogen prevents oxidative damage to the mitochondria in Friedreich’s ataxia skin fibroblasts. PLoS One. 2012;7(4):e34600.
Richardson TE, Simpkins JW. R- and S-equol have equivalent cytoprotective effects in Friedreich’s ataxia. BMC pharmacology & toxicology. 2012;13:12.
Marmolino D, Acquaviva F, Pinelli M, Monticelli A, Castaldo I, Filla A, et al. PPAR-gamma agonist Azelaoyl PAF increases frataxin protein and mRNA expression: new implications for the Friedreich’s ataxia therapy. Cerebellum 2009;8(2):98–103.
Coppola G, Marmolino D, Lu D, Wang Q, Cnop M, Rai M, et al. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich’s ataxia. Hum Mol Genet. 2009;18(13):2452–61.
Aranca TV, Jones TM, Shaw JD, Staffetti JS, Ashizawa T, Kuo SH, et al. Emerging therapies in Friedreich's ataxia. Neurodegenerative disease management. 2016;6(1):49–65.
Nachun D, Gao F, Isaacs C, Strawser C, Yang Z, Dokuru D, et al. Peripheral blood gene expression reveals an inflammatory transcriptomic signature in Friedreich’s ataxia patients. Hum Mol Genet. 2018;27(17):2965–77.
Shen Y, McMackin MZ, Shan Y, Raetz A, David S, Cortopassi G. Frataxin deficiency promotes excess microglial DNA damage and inflammation that is rescued by PJ34. PLoS One 2016;11(3):e0151026.
Patel M, Schadt K, McCormick A, Isaacs C, Dong YN, Lynch DR. Open label pilot study of oral methylprednisolone for the treatment of patients with friedreich ataxia. Muscle & Nerve. 2019.
Acknowledgments
Research in the Gottesfeld lab is supported by grants from the National Institute for Neurological Disorders and Stroke (NINDS/NIH, 5R01NS062856), the National Eye Institute (NEI/NIH, 1R01EY026490 and 1R01EY029166), the Friedreich’s Ataxia Research Alliance, and BioMarin Pharmaceutical, Inc. The author thanks Jennifer Farmer of the Friedreich’s Ataxia Research Alliance for the permission to use the FARA therapeutic pipeline graphic (Fig. 3), Dr. Elisabetta Soragni for her input on the manuscript, and Dr. Sanjay Bidichandani for the communication of unpublished results on DNA methylation. The opinions presented in this review are solely those of the author, and apologies must be given to the many scientists whose work could not be covered.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author serves as a consultant to BioMarin Pharmaceutical and is an inventor on patents licensed by The Scripps Research Institute to BioMarin Pharmaceutical.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 1111 kb)
Rights and permissions
About this article

Cite this article
Gottesfeld, J.M. Molecular Mechanisms and Therapeutics for the GAA·TTC Expansion Disease Friedreich Ataxia. Neurotherapeutics 16, 1032–1049 (2019). https://doi.org/10.1007/s13311-019-00764-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00764-x