
Abstract
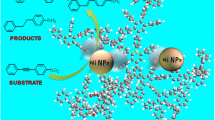
New bead-shaped heterogeneous nanoparticle catalysts viz., amino-terminated poly(amidoamine) (PAMAM) grafted on poly(styrene)-co-Poly(vinylbenzylchloride) (PS-Poly(VBC)) matrices immobilized/stabilized with palladium nanoparticle were prepared by simplified procedure. The first step is the preparation of PS-Poly(VBC) beads by suspension polymerization method. Second, the PAMAM G(0) G(1) and G(2) dendrimers were grafted individually onto the PS-Poly(VBC) matrices via divergent method by repeating two reactions, i.e., Michael addition of methyl acrylate to surface amino groups of aminomethylated PS-Poly(VBC) matrixes followed by amidation of the resulting esters with ethylene diamine. The resulting three types of PAMAM G(0), G(1) and G(2) grafted on PS-Poly(VBC) matrices were complexed individually with PdCl2 and thus yielded the corresponding new bead-shaped heterogeneous nanoparticle catalyst immobilized with PdNPs. The appearance of surface plasmon resonance band noticed at 547 nm in UV confirms the formation of PdNPs. The SEM result shows that the intensity of white patches due to immobilization of PdNPs increases with generation number and XRD reveals that the crystalline nature was decreased against generation number of the PAMAM. The catalytic efficiency of PS-Poly(VBC)-NH2-PdNPs-G(0), G(1) and G(2) catalysts were examined by Suzuki coupling reaction performed in mixture of water/ethanol. The observed reaction yield reveals that the activity was proportional to the generation number of PAMAM grafted onto the PS-Poly(VBC) matrices. The percentage of reaction yield (biphenyl) is sustained to ≈70 % even up to five cycles and this in turn confirms the stability of the catalysts. These catalysts can be used to conduct the Suzuki-coupling reaction in continuous mode operation in industrial scale.
Similar content being viewed by others

Introduction
The Suzuki coupling reaction between arylboronic acids and aryl halides catalyzed by Pd(0)/(II), and producing C–C bond-based product is one of the most widely employed organic transformation performed by industrial communities for the synthesis of many fine chemicals and life-saving drugs (Vilaro et al. 2008). The synthesis and utilization of biaryl compounds are implicated in key steps for building of numerous conducting polymers, molecular wires, liquid crystals, agrochemicals, pharmaceuticals and chiral skeleton for many asymmetric catalysts (Hassan et al. 2002; Bolm et al. 2001). Various C–C coupling reactions such as Suzuki, Heck and Still reactions generally proceed in the presence of homogeneous Pd species, with a wide range of ligands, which makes the catalyst recovery a tedious operation and might result in unacceptable Pd contamination of the product (Jiang et al. 2007). Therefore, removal of residual Pd is a challenging task for chemists and in the pharmaceutical industries that involves Pd-catalyzed process (Garrett and Prasad 2004). For these reasons, recently there has been increasing interest in the development of heterogeneous Pd catalysts which can be recovered easily by simple filtration and reused. Besides, inorganic supports like zeolites, metal oxides, supported alkali metal catalyst and silica supports (Kockritz et al. 2006), polymeric supports have gained attention because they are inert, non-toxic, non-volatile, insoluble and often recyclable. Chloromethylated poly(styrene) cross-linked with divinylbenzene (DVB) support is most widely used due to their flexibility, chemical versatility and the ease with which their physical properties can be varied.
Metal nanoparticles (NPs) are attractive catalysts due to their large-surface to volume ratio and consequently large fractions of the metal atoms are accessible to reactant molecules and available for catalysis (Fendler 1998). However, due to the very active surface atoms, the unprotected metallic nanoparticles in solution are kinetically unstable with respect to aggregation. The stabilization of nanoparticles in solution can be achieved by adding stabilizers like surfactants, soluble polymers, quaternary ammonium salts, polyoxoanions or by immobilization onto various supports such as alumina, metal oxides, carbon or polymeric materials including poly(styrene) microspheres (Astruc et al. 2005), polyelectrolyte brushes (Dtzauer et al. 1994), cyclodextrin (Strimbu et al. 2003), poly(N-vinyl-2-pyrrolidone) (Narayanan and El-Sayed 2003) and dendrimers (Pittelkow et al. 2003). Dendrimers have a number of advantages over conventional organic or polymeric ligands (Helms and Frechet 2006). The controlled architectures of dendrimers make them useful in several areas of research such as host–guest chemistry (Aulenta et al. 2003), drug delivery (Twyman et al. 1999) and self-assembly (Zhou et al. 2004); especially transition metal nanoparticle catalyst derived from dendrimers has promising applications (Ooosterom et al. 2001). Although several advantages have been gained already in the dendrimer-based homogenous catalyst, they often restrict the process of packing the catalyst material in column reactor or continuous flow reactors to conduct the multi-step synthesis in continuous mode operation and it needs special separation techniques like nano filtration (Ribaudo et al. 2006).
Further, in the recent years, studies pertinent to dendrimer grafting onto the solid matrix have attracted considerable attention. Especially, dendrimers grown on solid materials such as polymer beads (Pan et al. 2005; Tsubokawa et al. 2001), magnetic nanoparticles (Antebi et al. 2002), carbon black (Reynhardt et al. 2004) and silica (Abu-Reziq et al. 2006; Bourque et al. 1999) are the recently reported techniques for the design of innovative insoluble dendritic materials with variety of advanced applications. Similarly, numerous solid-supported palladium catalysts have been reported under mild/environmentally benign reaction conditions. These supported catalysts were prepared by immobilizing the palladium (II) on supported ligands (Phosphine, amines, N-heterocyclic carbines) (Cho et al. 2006) or ligand-free palladium(0) nanoparticles on various solid supports like polystyrene (Dantas Ramos et al. 2004), carbon (Okumur et al. 2005), zeolites (Choudary et al. 2002), silica (Sachin et al. 2007), cellulose (Reddy et al. 2006), cornstarch (Gronnow et al. 2005), polymethyl methacrylate (Song and Yi 2008), chitosan (Eunyoung Sin et al. 2010) and Perovskites (Andrews et al. 2005). But, unfortunately in many cases, including polymer-supported PdNPs (Evagelisti et al. 2009), the catalysts quite often prove to be ineffective method due to leaching of Pd0 in small amount into the solution (Ichinose et al. 2003). Exceptionally, only few studies are reported with leach-free heterogeneous Pd systems; they include Pd salts on Zeolites (Corma et al. 2004), PdNPs deposited on particular layered double hydroxide (Choudary et al. 2002), or on suitable functionalized zeolites containing primary amino group (Mandal et al. 2004) and thio groups (Shimizu et al. 2004). Polymer-supported catalysts are extensively used in both industry and research institutes owing to its advantages as compared with inorganic-based heterogeneous catalysts. While accepting the merits, in certain polymeric matrices, the polymer-supported catalysts have some drawbacks also. Particularly, leaching of catalytic metal may frequently occur from the catalyst centers which in turn contaminate the valuable products. In addition, there may be a reduction or complete loss of selectivity of the catalysts when they get attached to an insoluble support. Several attempts have already been made to solve these problems and to develop a stable polymer-supported catalyst with high activity and selectivity. More specifically, to conduct the reactions in aqueous media, Yang et al. (2006) reported a PEG-coated resin-supported palladium catalyst for Suzuki-coupling reaction. Uozumi et al. (2005) reported that their amphiphilic resin-supported palladium nanoparticles exhibit an excellent catalytic activity for cross-coupling reactions. It was also shown that Pd/C catalyst was found to be active in Suzuki reactions for bromoarenes in iPrOH/H2O, EtOH/H2O mixtures or neat iPrOH (Kitamura et al. 2007; Maegawa et al. 2007) and catalyzes the cross-coupling of chloroarenes with various arylboronic acids in N,N-dimethylacetamide/H2O or N-methyl-2-pyrrolidone/H2O mixtures. However, reusability of the catalysts was not tested for the reaction (Leblond et al. 2001; Heidenreich et al. 2002).
Although different dendrimer-grafted inorganic and organic matrix has been used for complexation/stabilization of metal catalyst, there are only few studies reported for grafting of dendrimer onto the bead-shaped polymer-supported metal nanoparticle catalysts. But still, these types of insoluble heterogeneous catalysts have their own inconveniences and practical difficulties. To circumvent this problem, a convenient form of economically affordable, stable, flexible, insoluble, active heterogeneous catalyst needs to be developed to conduct the Suzuki coupling reactions with continuous mode and enhanced yield. Dendrimer-functionalized polymer supports combine the advantages of the dendrimers (high density of active sites) with the ease of separation of solid-phase catalysts from the reaction mixture by simple filtration. In view of these background, in this work, we have made a novel attempt for the synthesis, characterization and catalytic properties of bead-shaped insoluble PS-Poly(VBC) matrix grafted with poly(amidoamine) PAMAM dendrimer–palladium nanoparticle catalyst for Suzuki coupling reaction. That is, three different types of catalysts viz., PS-Poly(VBC)-NH2-PdNPs-G(0), PS-Poly(VBC)-NH2-PdNPs-G(1) and PS-Poly(VBC)-NH2-PdNPs-G(2) were synthesized using aminomethylated PS-Poly(VBC) beads as common matrix through sequential growing of dendrimer by conducting the Michael addition and amidation reactions. These three types of PdNPs complexed catalysts were characterized beyond doubt using FTIR, XRD, SEM and EDAX and were employed individually to examine the catalytic activity through Suzuki coupling as a representative reaction.
Chemicals required
Monomers like styrene (Alfa-Aaeser), 4-vinylbenzyl chloride (Sigma-Aldrich) and divinyl benzene (Alfa-Aaeser) were used with prior purification. Other chemicals like gelatin, Poly(vinylalcohol), boric acid, azobisisobutyronitrile, sodium nitrite, sodium hydroxide, potassium phthalimide, hydrazine hydrate, methyl acrylate, ethylene diamine, Palladium Chloride (Sigma-Aldrich), phenyl boronic acid, chlorobenzene, cetyltrimethyl ammonium bromide (CTAB) and D.D water were used as received. Solvents like ethanol, methanol, tetrahydrofuran, dichloromethane, diethylether and acetone were distilled before use.
Experimental methods
Preparation of cross-linked poly(styrene)-co poly (vinylbenzylchloride) beads
The insoluble form of crosslinked poly(styrene)-co-poly(vinylbenzylchloride) (PS-PVBC) matrix was prepared as per methods prescribed in literature (Balakrishnan and Ford 1982). To describe briefly, the method involves the suspension co-polymerization of styrene (ST), vinylbenzylchloride (VBC) (functional-monomer) cross-linked with divinylbenzene (DVB) (Scheme 1). That is, by fixing 2 % cross-linking (amount of DVB) and 25 % active site (amount of VBC), the organic phase was maintained at 82.5 g and aqueous phase at 225 g. Exactly 1.35 g of gelatin, 2.55 g boric acid and 2.25 g poly(vinylalcohol) were thoroughly dissolved in 30, 60 and 130 ml of double-distilled hot water (50 °C). Then these solutions were mixed together and then the pH of the solution was maintained as ten through the addition of 25 % aqueous sodium hydroxide solution and 0.1 g sodium nitrite was added so as to maintain the conformation of gelatin. It was then transferred to 250-ml three-necked RB flask equipped with overhead mechanical stirrer and reflux condenser. Nitrogen was passed continuously and the temperature was maintained at 50 °C. After half an hour, the organic phase containing 3.3 g of DVB, 58.52 g of styrene and 0.4125 g of AIBN was thoroughly mixed and added to the reaction flask. Subsequently, the thermostat temperature was increased to 70°C and the stirring speed was maintained at 400 rpm using tachometer. Then after 3 h, i.e., after the partial polymerization time (PPT) of ST and DVB, functional monomer viz., vinylbenzylchloride was added and the ter-polymerization reaction was allowed to proceed to completion, for 48 h. The resulting ter-polymer beads were filtered and washed with hot water and cold methanol repeatedly until the wash solution did not turn cloudy upon the addition of water. Then, it was dried at 60 °C in a vacuum oven for 2 days and cross-linked poly(styrene)-co-poly(vinylbenzylchloride) beads (PS-PVBC) were thus obtained. The dried beads were sieved into different mesh size and the beads with representative mesh size of −100 + 120 were analyzed through FT-IR and SEM.

Preparation of PS-Poly(VBC) matrix
Functionalization of amino methyl onto the bead shaped PS-Poly(VBC) matrix
Functionalization of amino methyl onto the PS-Poly(VBC) beads involves two steps viz., (1) preparation of pthalimidomethyl beads followed by (2) conversion of pthalimide group into amino methyl poly(styrene) bead.
Preparation of phthalimidomethyl functionalized PS-Poly(VBC) beads
Beads containing chloride group viz., PS-Poly(VBC) (1 g) were taken in 150 ml R.B Flask and treated with 3 g of potassium phthalimide in 100 ml of dry DMF and the reaction mixture was stirred with magnetic stirrer for 18 h at 50 °C. Then after the completion of the reaction, the resultant beads were washed thrice with DMF, methanol, water and methanol individually. Then the filtered beads were dried in vacuum overnight at 60 °C for 24 h and thus phthalimidomethyl functionalized PS-Poly (VBC) beads were obtained. The resulting dried beads were analyzed with FT-IR and the obtained spectrum shown in Fig. 1b.

FT–IR spectra of PAMAM typed dendrimer grafted onto PS-Poly(VBC) matrices
Functionalization of amino methyl onto PS-Poly(VBC) beads
The phthalimidomethyl functionalized PS-Poly(VBC) beads (0.5 g) were treated with 5 ml of hydrazine hydrate and allowed for refluxing at 60 °C overnight with ethanol. Then the beads were filtered from the hot ethanol and washed again three times with ethanol followed by treatment with Aq. KOH, water and ethanol. The resulting beads were dried at 60 °C in vacuum oven for 1 day and amino methyl functionalized PS-Poly(VBC) matrix viz., AMPS-Poly(VBC) obtained. The dried beads were analyzed with FT-IR and the obtained spectrum is shown in Fig. 1c. The surface morphology of the dried beads was inspected with SEM analysis and the recorded images are shown in Fig. 4b.
Grafting of PAMAM dendrimer onto the surface of AMPS-Poly(VBC) matrix
Grafting of PAMAM dendrimer onto the surface of AMPS-Poly(VBC) beads was performed by sequential growing technique by adopting the Michael addition and amidation reactions (Scheme 2). Initially, 0.5 g of functionalized matrix viz., AMPS-Poly(VBC) was taken into 100 ml R.B flask; to that 5 ml of methyl acrylate and 15 ml of methanol was added and then the mixture was stirred for 4 days in the presence of nitrogen atmosphere at room temperature. After completion of the reaction, the functionalized beads were washed with methanol, dichloromethane (DCM) and acetone [5 ml × 3 times each]. Then the resulting beads were dried under vacuum oven for 1 day at 60 °C which yielded the PS-Poly(VBC) NH2-G(0.5). The dried beads were analyzed with FT-IR and the obtained spectrum is shown in Fig. 1d. In the second step, the amidation reaction was performed using the PS-Poly(VBC)NH2-G(0.5) matrix derived from the Michael addition reaction. That is, the transamination reaction was carried out by taking 0.4 g of PS-Poly(VBC)NH2-G(0.5) in 100 ml R.B flask to which ethylene diamine (EDA) (5 ml) and methanol (15 ml) were added separately at 0 °C in an ice bath and the mixture were stirred for 1 h. Then the temperature of the mixture was slowly increased to room temperature (30 °C) and was stirred for 4 days using magnetic stirrer in presence of nitrogen atmosphere. Then the resulting beads were washed with methanol, acetone and diethyl ether [5 ml × 3 times each] and dried under vacuum at 60 °C for 24 h and PS-Poly(VBC) NH2-G(1) was thus obtained. The amidation of PS-Poly(VBC) NH2-G(1) was analyzed with FT-IR and the obtained spectrum is shown in Fig. 1e. The surface morphology of the dried beads was analyzed with SEM and the recorded images shown in Fig. 4c. With a view to generate the second-generation dendrimer, the PS-Poly(VBC)NH2-G(1) matrix was employed again for Michael addition and amidation reactions individually by repeating the same experimental procedure viz., Michael addition and amidation reactions, respectively, which yielded the PS-Poly(VBC)NH2-G(2) matrixes. Then again generations/growth of (G2) on PS-Poly(VBC)NH2-G(2) matrix was studied with FT-IR and the recorded spectra shown in Fig. 1f, g, respectively. The surface morphology of the dried beads was analyzed with SEM and the recorded image shown in Fig. 4d.

Preparation of AMPS-Poly(VBC) supported dendrimer synthesis
Preparation of bead-shaped heterogeneous nanoparticle catalysts through stabilization/immobilization of PdNPs onto the PS-Poly(VBC) functionalized with PAMAM dendrimer matrix
The three different types of bead-shaped polymer grafted/functionalized dendrimer matrixes viz., PS-Poly(VBC)-NH2-G(0), PS-Poly(VBC)-NH2-G(1) and PS-Poly(VBC)-NH2-G(2) were made into catalysts by simple complexation reaction with palladium precursor viz., PdCl2. That is, 0.1 g of three types of PS -Poly(VBC)-NH2 matrices grafted/functionalized with PAMAM having generation number, namely G(0), G(1) and G(2) were taken individually in 100 ml R.B flask and swelled separately in 10 ml of acetone. 10 ml of an aqueous solution of (2 × 10−4 M) PdCl2 was added to each matrix and then stirred at room temperature using magnetic stirrer for 12 h and during the reaction, white-colored functionalized beads were turned into dark yellowish brown irrespective of types and generation number. Then the resulting dark yellowish brown bead matrices were filtered in G3 sintered crucible under vacuum and then washed with water (20 ml × 5 times), acetone (20 ml × 3 times) and methanol (20 ml × 3 times) and dried under vaccum at ambient temperature for 24 h and thus the respective complexed catalyst viz., PS-Poly(VBC)-NH2-G(0)-Pd, PS-Poly(VBC)-NH2-G(1)-Pd and PS-Poly(VBC)-NH2-G(2)-Pd were produced. Then the obtained respective PS-Poly(VBC)-NH2 beads stabilized with Pd2+ were taken in 50 ml R.B flask and allowed for reduction reaction by adding 1 mmol of cold NaBH4 (10 ml) and then the corresponding solution was stirred vigorously for about 2 h at room temperature. After the reduction of Pd2+, the yellow color of the beads had been changed completely into intense brown, which indicates the reduction of Pd2+ to Pd° and the production of PS-Poly(VBC)-NH2-PdNPs-G(0), PS-Poly(VBC)-NH2-PdNPs-G(1) and PS-Poly(VBC)-NH2-PdNPs-G(2) catalysts. The complexation of Pd with each matrix was examined through FT-IR and the obtained spectra shown in Fig. 2a for PS-Poly(VBC)-NH2-Pd-G(0), in Fig. 2b for PS-Poly(VBC)-NH2-Pd-G(1) and in Fig. 2c for PS-Poly(VBC)-NH2-Pd-G(2) catalyst. The surface morphology of the each catalyst was analyzed with SEM and the recorded images shown in Fig. 4e, f and g for PS-Poly(VBC)-NH2-PdNPs-G(0), G(1) and G(2), respectively.

FT-IR spectra for the complexes of a, b and c for PS- Poly(VBC)-NH2-Pd(II)-G(0), G(1) and G(2)
The influence of solvent in the formation of complex in catalyst preparation
Complexation behavior of Pd (II) ions with PAMAM functionalized PS-Poly(VBC) matrixes was studied. Earlier reports indicated that metal ion uptake during the complexation was poor if water had been used as a reaction medium. This is because of the poor swellability of matrix in water medium, but, in the case of mixture of polar organic solvent miscible with water, the metal ion uptake was proved to be better, because the dendronised (PAMAM) polymer was more solvated in organic/aqueous solvents and this in turn facilitates the complexation reaction (Krishnan and Sreekumar 2008). Therefore, in this study, we have used water/acetone with 1:1 mol ratio.
Suzuki coupling reactions
Phenyl boronic acid (1.5 mmol), aryl bromide (1 mmol), Na2CO3 (3 mmol) in water and ethanol (10 ml) were added to a 50-ml round-bottom flask. Then 0.5 mg of respective catalyst viz., PS-Poly(VBC)-NH2-PdNPs-G(0), Poly(VBC)-NH2-PdNPs-G(1) and Poly(VBC)-NH2-PdNPs-G(2) was added individually to the reaction mixture and the resulting solution stirred magnetically and heated to 100 °C for 12 h under nitrogen (Scheme 3). The occurrence of reaction, irrespective of the catalyst, was monitored in TLC at every 1 h using dichloromethane (DCM) and diethyl ether as eluting solvents in 1:1 ratio. After 12 h (complete reaction), the reaction mixtures irrespective of the catalyst were cooled to room temperature and were extracted using the mixture DCM (25 ml) and water (25 ml). The product formed from the reaction, i.e., bi-phenyl was analyzed using FT-IR. Further, the structural confirmation was done through NMR. All these three catalysts were filtered, dried and used again for the same reaction to study their stability and recycling efficiency.

Suzuki coupling reaction catalyzed by PS-Poly(VBC)-NH2-PdNPs-G(0), G(1) and G(2) catalysts
Results and discussions
Ftir
The bead-shaped PS-Poly(VBC) matrix was prepared using suspension polymerization technique and the resulting matrix was functionalized with –NH2 group using potassium pthalimide and hydrazine hydrate. Initially, the functionalization of phtalimidomethyl has been established by analyzing the PS-Poly(VBC) (control) and pthalimido functionalized PS-Poly(VBC) matrices through FT-IR and the corresponding spectra were shown in Fig. 1a, b, respectively. In Fig. 1b phthalimidomethyl group shows two characteristic peaks at 1,771 and 1,717 cm−1 which corresponds to C=O stretching of amide group and in contrast, these peaks were not noticed in PS-Poly(VBC) matrix (Fig. 1a). Similarly, another characteristics peak viz., C–Cl stretching was observed at 698 cm−1 in PS-Poly(VBC) matrix, whereas this peak was sharply reduced in the pthalimido functionalized PS-Poly(VBC) (Fig. 1b). Hence, the appearance of C=O amide stretching and the reduction of C–Cl stretching indicates the functionalization of pthalimide onto the PS-Poly(VBC) matrix. Further, the amino methyl funtionalization onto PS-Poly(VBC) matrix was performed by treating the phthalimide functionalized PS-Poly(VBC) with hydrazine hydrate and yielded the amino methyl functionalized PS-Poly(VBC) matrixes (PS-Poly(VBC)-NH2). That is, in the FT-IR spectrum (Fig. 1c), the phthalimide group was converted into aminomethyl group with the help of hydrazine hydrate and this is proved from the disappearance of characteristic peaks at 1,771 and 1,717 cm−1 (C=O stretching due to amide-I band) when compared with phthalimide functionalized PS-Poly(VBC) matrix followed by the appearance of bands around 3,000–3,350 cm−1 for primary amino groups.
The grafting of PAMAM dendrimer with generation number G(0), G(1) and G(2) onto PS-PVBC-NH2 matrix was carried out through Michael addition and amidation reactions by sequential growing technique. With a view to generate the Generation (1) the resulting aminomethyl functionalized beads viz., PS-Poly(VBC)-NH2 were taken and employed for Michael addition with the addition of methyl acrylate and thus yielded the PS-Poly(VBC)NH2-G(0.5). The solid-phase synthesis of PS-Poly(VBC) and PAMAM grafting was monitored by FTIR. The FTIR spectrum (Fig. 1d) shows two characteristic peaks at 1,735 cm−1 and 1,077 cm−1 for C=O and C–O stretching frequency which belongs to ester group generated after the addition of methyl acrylate, andalsothe disappearance of peak at 3,131 cm−1 due to amino groups. In the second step, the transamination reaction was carried out by taking PS-Poly (VBC)-NH2-G(0.5) with EDA and this in turn yielded the PS-Poly(VBC)-NH2-G(1). On comparing the 0.5 generation matrix (control) with their corresponding amidation functionalized matrixes, namely PS-Poly(VBC)-NH2-G(1), it is observed that the peaks noticed at 1,735 cm−1 (C=O stretching) and 1,077 cm−1 (C–O stretching) were found to have disappeared and in contrast a new peak at 1,640 cm−1 has appeared, and this is pertinent to the conversion of ester into amides. Similarly, another characteristic peak viz., NH2 group was noticed at 3,350 cm−1 which confirms the occurrence of amidation reaction. The resulting matrix was analyzed with FTIR and the observed spectrum was shown in Fig. 1e. Therefore, all these observations have undoubtedly lent support for the occurrence of Michael addition and amidation reaction and thus for generation of G(1) onto the PS-Poly(VBC)-NH2 matrix. Repetition of the above two steps viz., Michael addition and amidation reaction has produced the second-generation G(2) PAMAM dendrimers grafted onto the PS-Poly(VBC)-NH2 bead matrices. The observed FTIR spectrum for the resulting PS-Poly(VBC)-NH2-G(1.5) and G(2) matrices was shown in Fig. 1f and g, respectively.
The complexation of Pd2+ and its subsequent reduction was established through FTIR. From the FT-IR spectrum it is proved that irrespective of the spectrum the complexing functional group, namely –NH2 group noticed around 3,000–3,200 cm−1 was found to have disappeared after complexation in all the three respective catalysts, which indicates that complexation of Pd (II) has taken place with respective amino groups. This in turn confirms the formation of PdNPs onto PS-Poly(VBC)-NH2-G(0), G(1) and G(2) matrices. The corresponding FT-IR spectrum of each nanoparticle catalyst viz., PS-Poly(VBC)-NH2-PdNPs-G(0), PS-Poly(VBC)-NH2-PdNPs-G(1) and PS-Poly(VBC)-NH2-PdNPs-G(2) was shown in Fig. 2a–c, respectively.
UV–visible studies
Initially, the PS-Poly(VBC)-NH2-G(0), G(1) and G(2) matrices were swelled individually in acetone/water mixture; then the metal precursor solution viz., PdCl2 was added and stirred vigorously. As a result of this stirring, the solution containing beads were changed into yellow color and this in turn changed the beads (color) from white to yellow. The yellow color PS-Poly(VBC)-NH2-Pd2+-G(0), G(1) and G(2) matrices were washed repeatedly with water and methanol until the supernatant liquid gives pH to 7 and the obtained beads were dried in vaccum. Then these beads PS-Poly(VBC)-NH2-Pd2+-G(0), G(1) and G(2) were converted individually into Pd0 through addition of NaBH4 solution. The reduction of Pd2+ to Pd0 has been visualized from the change of yellow color to dark brown of the beads irrespective of the generation number and thus formed the corresponding PdNPs catalyst. Further, to confirm the formation/immobilization of PdNPs onto PS-Poly(VBC)-NH2 matrices with different generations G(0), G(1) and G(2), all the three PdNPs derived catalysts were treated with cetyltrimethyl ammonium bromide (CTAB) solutions in the presence of THF and sonicated for 1 h to extract PdNPs immobilized/stabilized onto the Poly(VBC)-NH2-Pd2+ G(0), G(1) and G(2) matrices. In fact, the extraction of PdNPs was completed within 45 min, after completion of 1 h sonication, the color of the solution was changed into dark brown, thus confirming the extraction of PdNPs from the matrices. Further, the resulting respective metal nanoparticle solution was analyzed with UV. From the UV–vis spectrum, it is observed that surface plasmon resonance (SPR) band appeared at 240 nm, irrespective of the catalyst solution and thus confirms the formation of PdNPs. However, for understanding the UV–vis spectrum of G(2) derived solution was shown in Fig. 3.

UV-Visible Spectrum of PdNPs extracted from PS-Poly (VBC)-NH2-PdNPs-G(2)- catalyst
SEM analysis
The functionalisation of PAMAM and complexation of Pd(II) onto the surface of G(0), G(1) and G(2) matrixes of PS-Poly(VBC)-NH2 were studied through SEM. From Fig. 4a, it is inferred that the surface morphology of the first-level matrix viz. PS-Poly(VBC) was homogenous and smooth with spherical shape, whereas, the surface of PS-Poly(VBC)-NH2 matrices with different generation number includes G(0), G(1) and G(2) before (Fig. 4b–d) and after immobilization of PdNPs (Fig. 4e–g) irrespective of that the types show heterogeneous and rough surface. Particularly, on increasing the generation from G(0) to G(2), the intensity of heterogeneity was found to progressively increase and thus reflect more grafting/functionlisation of PAMAM onto the respective matrix. Further, appearance of white patches on the surface of the spherical beads and their increased intensity with increase in generation number of PAMAM-typed dendritic moiety [from G(0) to G(2)] are due to contribution of PdNPs. The complexation of Pd onto the matrix increases with increase in the generation number (due to more availability of –NH2 group on the periphery of G(2) than G(0)). Moreover, it was also observed that the physical state of the matrix was maintained as it was observed in first-level matrix viz., PS-Poly(VBC) irrespective of the generation number even after the multi-step synthesis and complexation steps. In a nutshell, it is worth highlighting here that the appearance of heterogeneity and roughness is a clear evidence for the functionalization of PAMAM with G(0) to G(2), and the intensity of white patches noticed in all catalysts indicates the formation of palladium nanoparticles irrespective of the types of the catalysts.

SEM Images of a PS-Poly (VBC), b, c and d for PS-Poly(VBC)-NH2-G(0), G(1) and G(2) and e, f, and g for PS-Poly(VBC)-NH2-PdNPs-G(0), G(1) and G(2)
EDAX analysis
The grafting of PAMAM onto the PS-Poly(VBC) matrix and formation of PdNPs on all the three different types of matrix-based catalysts were studied extensively through EDAX. The characteristic elements such as C, N, O and Pd and their quantity (%) available before and after formation of PdNPs onto the matrixes are given in Table 1. From the percentage composition of elements, it is proved that the carbon and nitrogen have been found to increase on par with increasing the generation number from G(1) to G(2), irrespective of the types of the matrixes obtained before formation of nanoparticles. In the case of catalyst, reasonable amount of Pd % was detected in all the three types of catalysts. Therefore, the decreased/increased trend of C/N and the presence of Pd in different percentages undoubtedly support the functionalization of PAMAM G(0) to G(2) onto the respective matrix and complexation of Pd, whereas, the Pd has not been noticed in PS-Poly(VBC) (control).
X-ray diffraction study
X-ray diffraction study was performed to determine the structure of the polymer-supported matrix before and after the surface modification reactions, i.e., crystallinity of the first level common matrix viz., PS-Poly(VBC) and polymer grafted with PAMAM dendrimer catalysts viz., PS-Poly(VBC)-NH2-PdNPs G(0), PS-Poly(VBC)-NH2-Pd NPs G(1) and PS-Poly(VBC)-NH2-Pd NPs G(2). The XRD of PS-Poly(VBC) matrix showed two well-defined peaks at 2θ value of 7–10 and 19–23. These 2θ values evidenced that the matrix exists in crystallinity nature.
In contrast, the XRD patterns of PdNPs immobilized catalysts viz., PS-Poly(VBC)-NH2-PdNPs-G(0), PS-Poly(VBC)-NH2-PdNPs-G(1) and PS-Poly(VBC)-NH2-PdNPs-G(2) (Fig. 5a–c) shows the peak at the same 2θ values 7–10 and 19–23. However, the intensity of the peaks was found to be decreased as the generation number of the dendrimer grafted onto PS-Poly(VBC) matrix increases. That is, the sharp reduction of 2θ peak intensity in G(0), G(1) and G(2) complexed catalysts suggested the reduction of crystalline behavior on comparison with the first-level common matrix (i.e.) PS-Poly(VBC). The most significant fact to establish here is the presence of PdNPs on all the three types of catalysts. In addition to two well-defined peaks at 2θ values of 8.5 and 21.8, there is also appearance of new peaks at 38.5, 40.8 and 63.8 which corresponds to reflection of planes indexed to (111), (200) and (220), which in turn confirms the formation of PdNPs. This indicates that the crystallinity of the PS-Poly(VBC) matrix/bead remained intact upon immobilization/stabilization of PdNPs. Further, the basic structure of the parent PS-Poly(VBC) was not damaged in the whole process of catalyst preparation. However, the relative intensities of some of diffraction lines may undergo slight changes because of complexation and formation of PdNPs.

XRD Pattern of PS-Poly(VBC)-NH2-PdNPs-G(0), G(1) and G(2) catalyst
Comparative analysis of catalytic efficiency of all the 3-types of polymer-supported dendritic PdNPs catalysts using Suzuki coupling as a model reaction
The catalytic activity of the three different bead-shaped polymer-supported PS-Poly(VBC)-NH2PdNPs catalysts viz., PS-Poly(VBC)-NH2-PdNPs G(0), PS-Poly(VBC)-NH2-PdNPs G(1) and PS-Poly(VBC)-NH2-PdNPs G(2) was investigated individually for the Pd-catalyzed Suzuki coupling reaction under identical reaction condition. The reaction was usually performed in an organic solvent or in a mixture of water and polar organic solvent such as ethanol, which provides salvation of the organic substrates and the inorganic bases. Analogue solvent mixture study was already reported by (Ren and Meng 2008) for Suzuki coupling reaction using poly(N-ethyl-4-vinylpyridinium)bromide stabilized PdNPs catalyst. Although few studies that performed the reaction in water alone have been reported already, in our study we observed that due to the poor swellability of the insoluble cross-linked PS-Poly(VBC) beads in water alone the reaction has not proceeded effectively. Therefore, we have used water/EtOH mixture and performed the Suzuki coupling reaction effectively. Further, it was also predicted that the nature of the base that was used in the reaction has also contributed greatly and produced the enhanced yield of the biphenyls. It is already reported by (Lyubimov et al. 2009) that the efficiency of the carbonate bases depends on their basicity, i.e., most basic carbonates afford highest yield. Even though we used the least basic carbonate (Na2CO3) to the Suzuki coupling reaction, we could achieve more than 70 % of biphenyl yield irrespective of the catalyst. The formation of Bi-phenyl product was quantitatively inspected with TLC analysis under specific condition for each catalyst and it was observed that the amount of the product formed was in the order of G(2) > G(1) > G(0) (Table 2). The observed order reveals that the activity of the catalyst was proportional to the generation number of the dendrimer, i.e., PAMAM. The product obtained in the Suzuki coupling reaction was determined by FT-IR. The product (biphenyl) gives the characteristic peaks at 3,076, 3,040 cm−1 for aromatic C–H(Str), 1,598, 1,486 cm−1 for phenyl ring substitution overtones(finger print region) and 737 cm−1 for phenyl ring substitution bending vibration, which in turn confirms the C–C bond formation between the two reactants viz., Chlorobenzene and PhB(OH)3. The recycling efficiency of all the three catalysts PS-Poly(VBC)-NH2-PdNPs G(0), G(1) and G(2) was also examined. That is, as usual at the end of the reaction, the respective catalyst was recovered by filtration and washed thoroughly with water and ethanol, dried at room temperature and reused again for five times for the same reaction under same reaction conditions. The observed yield was not decreased compared with the first-cycle catalyst. Although the catalytic activity gradually decreases (% of biphenyl), still the percentage of yield was maintained ≈70 % even after the fifth cycle (Table 2) and thus confirmed that all the three catalysts were found to be more stable even after the fifth cycle.
Conclusions
Three different types of insoluble, stable and convenient form of bead-shaped heterogeneous polymer-supported dendritic nanoparticle catalysts were prepared by simplified experimental procedure inclusive of ter-polymerisation, sequential growing of dendrimers by Michael addition and amidation and then complexation of Pd(II) followed by reduction using NaBH4. The functionalistion of PAMAM dendrimers with generation number G(0), G(1) and G(2) onto PS-Ploy(VBC) matrix and their formation/immobilization of PdNPs have been confirmed through FT-IR, UV–visible, SEM, EDAX and XRD analyses. From the UV–vis spectrum, the surface plasmon resonance (SPR) band appeared at 240 nm which confirms the formation of PdNPs. From the SEM images and XRD results, it is reported that the bead-shaped polymer-supported dendritic catalysts viz., PS-Poly(VBC)-NH2-PdNPs G(0), PS-Poly(VBC)-NH2-PdNPs G(1) and PS-Poly(VBC)-NH2-PdNPs G(2) derived from aminomethylated PS-Ploy(VBC) matrices have proved to be physically stable with heterogeneous surfaces and the crystalline nature decreased against generation number from G(0) to G(2). The catalytic activity of these three types of PdNPs catalysts were examined with respect to PAMAM generation number, which proves that the catalytic activity of C–C coupling in Suzuki reaction also increased on increasing the generation number. The recycling efficiency of all the three types of heterogeneous nanoparticle catalysts were inspected for five times and the obtained yield was found to be relatively maintained compared with first cycle irrespective of the catalyst and thus confirmed the stability and activity. The most important merit of this bead-shaped catalyst is, it can be very well packed into a column reactor and thus the coupling reaction can be conducted in continued mode operation which in turn can easily takes care of the industrial—scale process.
References
Abu-Reziq R, Alper H, Wang SD, Post ML (2006) Metal supported on dendronized magnetic nanoparticles: highly selective hydroformylation catalysts. J Am Chem Soc 128:5279
Andrews SP, Stephan AF, Tanaka H, Ley SV, Smith MD (2005) Heterogeneous or homogeneous? a case study involving palladium-containing perovskites in the Suzuki reaction. Adv Synth Catal 347:647–654
Antebi S, Arya P, Manzer LE, Alper H (2002) Carbonylation reactions of iodoarenes with PAMAM dendrimer-palladium catalysts immobilized on silica. J Org Chem 67:6623
Astruc D, Lu F, Aranzaes JR (2005) Nanoparticles as recyclable catalysts: the frontier between homogeneous and heterogeneous catalysis. Angew Chem Int Ed 44:7852–7872
Aulenta F, Hayes W, Rannard S (2003) Dendrimers: a new class of nanoscopic containers and delivery devices. Eur Polym J 39:1741–1771
Balakrishnan T, Ford WT (1982) Particle size control in suspension co-polymerization of styrene, chloro methylstyrene and divinylbenzene. J Appl Poly Sci 27:133
Bolm C, Hildebrand JP, Muniz K, Hermanno N (2001) Catalyzed asymmetric arylation reactions. Angewandte Chemie (Int edn) 40:3285–3308
Bourque SC, Maltais F, Xiao WJ, Tardifo O, Alper H, Arya P (1999) Hydroformylation reactions with rhodium-complexed dendrimers on silica. J Am Chem Soc 12:3035–3038
Cho JK, Najman R, Dean TW, Ichihara O, Muller C, Bradley M (2006) Captured and cross-linked palladium nanoparticles. J Am Chem Soc 128:6276
Choudary BM, Madhi S, Chowdary NS, Kantam ML, Sreedhar B (2002) Layered double hydroxide supported nanopalladium catalyst for heck-, suzuki-, sonogashira-, and stille-type coupling reactions of chloroarenes. J Am Chem Soc 124:14127–14136
Corma A, Garcia H, Levya A, Primo A (2004) Alkali-exchanged sepiolites containing palladium as bifunctional (basic sites and noble metal) catalysts for the Heck and Suzuki reactions. Appl Catal A 257:77
Dantas Ramos AL, da Diva Alves P, Aranda DAG, Schmal M (2004) Characterization of carbon supported palladium catalysts: inference of electronic and particle size effects using reaction probes. Appl Catal A 277:71–81
Dtzauer DM, Dai J, Sun L, Bruening-vandenberg ML, Meiger EW (1994) Encapsulation of guest molecules into a dendritic box. Science 266:1226–1229
Eunyoung S, Song-Se Y, Yoon-Sik L (2010) Chitosan-g-mPEG-supported palladium(0) catalyst for Suzuki cross-coupling reaction in water. J Mol Catal A Chem 315:99–104
Evagelisti C, Panziera N, Pertici P, Vitulli G, Salvadori P, Battocchio C, Polzonetti G (2009) Palladium nanoparticles supported on polyvinylpyridine: catalytic activity in Heck-type reactions and XPS structural studies. J Catal 262:287–293
Fendler JH (1998) Nanoparticles and nanostructured films: preparation, characterization and applications. Wiley-VCH, Weinheim
Garrett CE, Prasad K (2004) The art of meeting palladium specifications in active pharmaceutical ingredients produced by Pd-catalyzed reactions. Adv Synth Catal 346:889–900
Gronnow MJ, Luque R, Macquarrie DJ, Clark JH (2005) A novel highly active biomaterial supported palladium catalyst. Green Chem 7:552
Hassan J, Srignon M, Gozzi C, Schultz E, Lemaire M (2002) Aryl–aryl bond formation one century after the discovery of the Ullman reaction. Chem Rev 102:1359–1469
He JH, Ichinose I, Kunitake T, Nakao A, Shiraishi Y, Toshima N (2003) Facile fabrication of Ag–Pd Bimetallic nanoparticles in ultrathin TiO2-Gel films: nanoparticle morphology and catalytic activity. J Am Chem Soc 125:11034
Heidenreich RG, Kohler K, Krauter JGE, Pietsch J (2002) Pd/C as a highly active catalyst for Heck, Suzuki and Sonogashira reactions. Syn Lett 7:1118–1122
Helms B, Frechet JMJ (2006) The dendrimer effect in homogeneous catalysis. Adv Synth Catal 348:1125–1148
Jagtap SR, Raje VP, Shriniwas D, Bhanage BM (2007) Silica supported Polyvinyl pyridine as a highly active heterogeneous base catalysts for the synthesis of cyclic carbonates from carbondioxide and epoxides. J Mol Catal A Chem 266:69–74
Jiang XL, Sclafani J, Prasad K, Repi O, Blacklock TJ (2007) Pd-Smopex-111: a new catalyst for Heck and Suzuki cross-coupling reactions. Org Process Res Dev 11:769–772
Kitamura Y, Sako S, Udzu T, Tsutsui A, Maegawa T, Monguchi Y, Sajiki H (2007) Ligand-free Pd/C-catalyzed Suzuki–Miyaura coupling reaction for the synthesis of heterobiaryl derivatives. Chem Commun 47:5069
Kockritz A, Sebek M, Dittmar A, Radnik J, Bruckner A, Bentrup U, Pohl M, Hugl H, Agerlein W (2006) Ru-catalyzed oxidation of primary alcohols. J Mol Catal A Chem 246:85–99
Krishnan RG, Sreekumar K (2008) First example of organocatalysis by polystyrene-supported PAMAM dendrimers: highly efficient and reusable catalyst for Knovenagel condensation. Eur J Org Chem 28:4763–4768
Leblond CR, Andrews AT, Sun Y, Sowa R (2001) Activation of aryl chlorides for Suzuki cross-coupling by ligandless, heterogeneous palladium. Org Lett 13:1555–1557
Lyubimov SE, Vasil AA, Korlyukov AA, Iylin MM, Pisarev SA, Matveev VV, Chalykh AA (2009) Palladium containing hypercrosslinked polystyrene as an easy to prepare catalyst for Suzuki reaction in water and organic solvents. React Func Polym 69:755–758
Maegawa T, Kitamura Y, Sako S, Udzu T, Sakurai A, Tanaka A, Kobayashi Y, Endo K, Bora U, Kurita T, Kozaki A, Monguchi Y, Sajiki H (2007) Heterogeneous Pd/C-catalyzed ligand-free, room-temperature Suzuki–Miyaura coupling reactions in aqueous media. Chem Eur J 13:5937–5943
Mandal S, Roy D, Chaudhari RV, Sastry M (2004) Pt and Pd nanoparticles immobilized on amine-functionalized zeolite: excellent catalysts for hydrogenation and heck reactions. Chem Mater 16:3714
Nakao R, Rhee H, Uozumi Y (2005) Hydrogenation and dehalogenation under aqueous conditions with an amphiphilic-polymer-supported nanopalladium catalyst. Org Lett 7:163–169
Narayanan R, El-Sayed MA (2003) Effect of catalysis on the stability of metallic nanoparticles: Suzuki reaction catalyzed by PVP-Pd nanoparticles. J Am Chem Soc 125:8340–8347
Okumur K, Nota K, Yoshida K, Niwa M (2005) Catalytic performance and elution of Pd in the Heck reaction over zeolite-supported Pd cluster. J Catal 231:245–253
Ooosterom GE, Reek JMH, Kamer PCJ, Van Leeuwen PWNM (2001) Transition metal catalysis using functionalized dendrimers. Angew Chem Int Ed Engl 40:1828–1849
Pan BF, Gao F, Cur CH (2005) Dendrimer modified magnetite nanoparticles for protein immobilization. J. Collid Interface Sci 284:1–6
Pittelkow M, Moth-Poulsen K, Boas U, Christensen JB (2003) Poly(amidoamine)-dendrimer-stabilized Pd(0) nanoparticles as a catalyst for the Suzuki reaction. Langmuir 19:7682–7684
Qu RJ, Niu Y, Ji C, Wang C, Cheng G (2006) Synthesis, characterization and adsorption properties of metal ions of silica-gel functionalized by ester and amino-terminated dendrimer-like polyamidoamine polymer. Micropor Mesopor Mat 97:58–65
Reddy KR, Kumar NS, Reddy PS, Kantam ML, Sreedhar B (2006) Cellulose supported palladium(0) catalyst for Heck and Sonogashira coupling reactions. J.Mol.Catal.A:Chem 252:12
Ren LZ, Meng LJ (2008) Suzuki coupling reactions catalyzed by Poly(N-ethyl-4-vinylpyridium) bromide stabilized palladium nanoparticles in aqueous solutions. Express Polym Lett 2:251–255
Reynhardt JPK, Yang Sayari A, Alper H (2004) Eriodic mesoporous silica-supported recyclable rhodium-complexed dendrimer catalysts. Chem. Matter 16:4095
Ribaudo F, van Leeuwen PWNM, Reek JNH (2006) In: Gade LH (ed) Dendrimer catalysis, Springer, Berlin, pp 39–59
Shimizu K, Koizumi S, Hatamachi T, Yoshida H, Komai S, Kodama T, Kitayama Y (2004) Structural investigations of functionalized mesoporous silica-supported palladium catalyst for Heck and Suzuki coupling reactions. J Catal 228:141–151
Song D, Yi WB (2008) Polymethyl methacrylate micro-spheres supported palladium: a new catalyst for Heck and Suzuki reactions. J Mol Catal A Chem 280:20
Strimbu L, Liu J, Kaifer AE (2003) Cyclodextrin-capped palladium nanoparticles as catalysts for the Suzuki reaction. Langmuir 19:483–485
Tsubokawa N, Ichioka H, Satoh T, Murota M, Sato S, Shimizu H (2001) Grafting of hyperbranched poly(amidoamine) onto carbon black surfaces using dendrimer synthesis methodology. Poly Adv Technol 12:596–602
Twyman LJ, Beezer AE, Esfand R, Hardy MJ, Mitchell JC (1999) The synthesis of water soluble dendrimers, and their application as possible drug delivery systems. Tetrahedron Lett 40:1743–1746
Vilaro M, Arsequell G, Valencia G, Ballesteros A, Barluenga J (2008) Arylation of Phe and Tyr side chains of unprotected peptides by a Suzuki–Miyaura reaction in water. Org Lett 10:3243
Yang Q, Ma S, Li J, Xiao F, Xiong H (2006) A water-compatible, highly active and reusable PEG-coated mesoporous silica-supported palladium complex and its application in Suzuki coupling reactions. Chem Commun 23:2495–2497
Zhou M, Bently D, Ghosh I (2004) Helical supramolecules and fibers utilizing leucine zipper-displaying dendrimers. J Am Chem Soc 126:734–735
Acknowledgments
The authors EM and JNJ gratefully acknowledge the Council of Scientific and Industrial Research, New Delhi, for financial assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Murugan, E., Jebaranjitham, J.N. & Usha, A. Synthesis of polymer-supported dendritic palladium nanoparticle catalysts for Suzuki coupling reaction. Appl Nanosci 2, 211–222 (2012). https://doi.org/10.1007/s13204-012-0085-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13204-012-0085-9