
Abstract
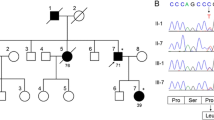
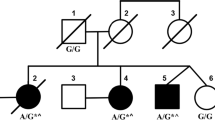
Spinocerebellar ataxias (SCAs) are inherited neurodegenerative diseases characterized by loss of balance, coordination, and slurred speech. Recently, a digenic mode of inheritance of TBP/STUB1 contributing to SCA was demonstrated. The clinical manifestations of SCATBP/STUB1 include not only ataxia but also obvious cognitive and behavioral impairment. Here, we describe a Chinese family with SCATBP/STUB1 and performed a literature search for similar cases. We identified a Chinese family with SCATBP/STUB1 and compare our clinical findings with other cases described in the literature so far. Four individuals in this family have been found to carry SCATBP/STUB1, of which three have clinical manifestations. A heterozygous deletion mutation in the STIP1-homologous and U-box containing protein 1 (STUB1) gene, NM_005861.4:c433_435del(p.K145del), was identified. The proband is a 34-year-old female with progressive dementia and dysarthria. The mother and uncle of the proband first presented with motor abnormalities and gradually developed cognitive impairment. The proband and her uncle showed cerebellar atrophy on MRI. The proband’s brother carried digenic variants but was asymptomatic. SCATBP/STUB1 is a novel SCA subtype. The main clinical manifestations are motor, cognitive, and behavioral abnormalities. Brain MRI shows significant cerebellar atrophy and cortical thinning. The independent segregation of TBP and STUB1 alleles should be considered when evaluating patients with cognitive impairment and ataxia.



Similar content being viewed by others

Data Availability
The original data presented in this study have been included in this article. Further inquiries can be directed to the corresponding authors.
Change history
24 February 2024
A Correction to this paper has been published: https://doi.org/10.1007/s12311-024-01672-3
Abbreviations
- CCAS:
-
Cerebellar cognitive affective syndrome
- CHIP:
-
HSP70-interacting protein
- HSP:
-
Heat shock protein
- MMSE:
-
Mini-Mental State Examination
- MoCA:
-
Montreal Cognitive Assessment
- MRI:
-
Magnetic resonance imaging
- poly-Q:
-
Polyglutamine
- SARA:
-
Scale for the Assessment and Rating of Ataxia
- SCA:
-
Spinocerebellar ataxia
- STUB1 :
-
STIP1-homologous and U-box containing protein 1
- TBP :
-
TATA-box binding protein
References
Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia[J]. Nat Rev Dis Primers. 2019;5(1):24.
Coarelli G, Brice A, Durr A. Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. F1000Res. 2018;7:F1000 Faculty Rev–1781.
Toyoshima Y, Takahashi H. Spinocerebellar ataxia type 17 (SCA17). Adv Exp Med Biol. 2018;1049:219–31.
Nanetti L, Magri S, Fichera M, Castaldo A, Nigri A, Pinardi C, Mongelli A, Sarro L, Pareyson D, Grisoli M, Gellera C, Di Bella D, Mariotti C, Taroni F. Complex ataxia-dementia phenotype in patients with digenic TBP/STUB1 spinocerebellar ataxia. Movement Disorders. 2023;38(4):665–75.
Magri S, Nanetti L, Gellera C, Sarto E, Rizzo E, Mongelli A, Ricci B, Fancellu R, Sambati L, Cortelli P, Brusco A, Bruzzone MG, Mariotti C, Di Bella D, Taroni F. Digenic inheritance of STUB1 variants and TBP polyglutamine expansions explains the incomplete penetrance of SCA17 and SCA48. Genetics Med. 2022;24(1):29–40.
Genis D, Ortega-Cubero S, San Nicolás H, Corral J, Gardenyes J, de Jorge L, López E, Campos B, Lorenzo E, Tonda R, Beltran S, Negre M, Obón M, Beltran B, Fàbregas L, Alemany B, Márquez F, Ramió-Torrentà L, Gich J, Volpini V, Pastor P. Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48)[J]. Neurology. 2018;91(21):e1988–98.
Shi CH, Schisler JC, Rubel CE, Tan S, Song B, McDonough H, Xu L, Portbury AL, Mao CY, True C, Wang RH, Wang QZ, Sun SL, Seminara SB, Patterson C, Xu YM. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet. 2014;23(4):1013–24.
Roux T, Barbier M, Papin M, Davoine CS, Sayah S, Coarelli G, Charles P, Marelli C, Parodi L, Tranchant C, Goizet C, Klebe S, Lohmann E, Van Maldergem L, van Broeckhoven C, Coutelier M, Tesson C, Stevanin G, Duyckaerts C, Brice A, Durr A. Clinical, neuropathological, and genetic characterization of STUB1 variants in cerebellar ataxias: a frequent cause of predominant cognitive impairment. Genetics Med. 2020;22(11):1851–62.
Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain. 1998;121(Pt 4):561–79.
Schmahmann JD. The cerebellum and cognition. Neurosci Lett. 2019;688:62–75.
Reetz K, Costa AS, Mirzazade S, Lehmann A, Juzek A, Rakowicz M, Boguslawska R, Schöls L, Linnemann C, Mariotti C, Grisoli M, Dürr A, van de Warrenburg BP, Timmann D, Pandolfo M, Bauer P, Jacobi H, Hauser TK, Klockgether T, Schulz JB. Genotype-specific patterns of atrophy progression are more sensitive than clinical decline in SCA1, SCA3 and SCA6. Brain. 2013;136(Pt 3):905–17.
Fujigasaki H, Martin JJ, De Deyn PP, Camuzat A, Deffond D, Stevanin G, Dermaut B, Van Broeckhoven C, Dürr A, Brice A. CAG repeat expansion in the TATA box-binding protein gene causes autosomal dominant cerebellar ataxia. Brain. 2001;124(Pt 10):1939–47.
Toyoshima Y, Onodera O, Yamada M, Tsuji S, Takahashi H. Spinocerebellar Ataxia Type 17. 2005 Mar 29 [updated 2022 Jul 28]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024.
Heimdal K, Sanchez-Guixé M, Aukrust I, Bollerslev J, Bruland O, Jablonski GE, Erichsen AK, Gude E, Koht JA, Erdal S, Fiskerstrand T, Haukanes BI, Boman H, Bjørkhaug L, Tallaksen CM, Knappskog PM, Johansson S. STUB1 mutations in autosomal recessive ataxias — evidence for mutation-specific clinical heterogeneity. Orphanet J Rare Dis. 2014;9:146.
Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19(6):4535–45.
Nikolay R, Wiederkehr T, Rist W, Kramer G, Mayer MP, Bukau B. Dimerization of the human E3 ligase CHIP via a coiled-coil domain is essential for its activity. J Biol Chem. 2004;279(4):2673–8.
Ronnebaum SM, Patterson C, Schisler JC. Emerging evidence of coding mutations in the ubiquitin-proteasome system associated with cerebellar ataxias. Human Genome Variation. 2014;1:14018.
Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Höhfeld J, Patterson C. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem. 2001;276(46):42938–44.
Kampinga HH, Kanon B, Salomons FA, Kabakov AE, Patterson C. Overexpression of the cochaperone CHIP enhances Hsp70-dependent folding activity in mammalian cells[J]. Mol Cell Biol. 2003;23(14):4948–58.
Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ, Davidson BL, Rebagliati MR, Paulson HL. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci. 2005;25(40):9152–61.
Al-Ramahi I, Lam YC, Chen HK, de Gouyon B, Zhang M, Pérez AM, Branco J, de Haro M, Patterson C, Zoghbi HY, Botas J. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem. 2006;281(36):26714–24.
Williams AJ, Knutson TM, Colomer Gould VF, Paulson HL. In vivo suppression of polyglutamine neurotoxicity by C-terminus of Hsp70-interacting protein (CHIP) supports an aggregation model of pathogenesis. Neurobiol Dis. 2009;33(3):342–53.
Barbier M, Davoine CS, Petit E, Porché M, Guillot-Noel L, Sayah S, Fauret AL, Neau JP, Guyant-Maréchal L, Deffond D, Tranchant C, Goizet C, Coarelli G, Castrioto A, Klebe S, Ewenczyk C, Heinzmann A, Charles P, Tchikviladzé M, Van Broeckhoven C, Brice A, Durr A. Intermediate repeat expansions of TBP and STUB1: genetic modifier or pure digenic inheritance in spinocerebellar ataxias? Genetics Med. 2023;25(2):100327.
Acknowledgements
The authors thank the Infinite Med Co., Ltd. for performing genetic testing and gene structure analysis in our patients.
Author information
Authors and Affiliations
Contributions
LL and JC wrote the manuscript, performed literature retrieval and utilization, contributed to figure and table preparation, and helped with the diagnostic process. JH is the corresponding author of this study. JC, JH, ZL, GZ and DC were involved in patient care. JC assisted with the diagnostic process, supported the interpretation, and critically revised the manuscript. All authors contributed to the manuscript and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics Statement
This study was reviewed and approved by the Medical Ethics Committee of Peking University, Shenzhen Hospital.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
Cite this article
Liu, L., Chen, J., Zhang, G. et al. A Chinese Family with Digenic TBP/STUB1 Spinocerebellar Ataxia. Cerebellum (2024). https://doi.org/10.1007/s12311-024-01664-3
Accepted:
Published:
DOI: https://doi.org/10.1007/s12311-024-01664-3