
Abstract
Aeromonas hydrophila is a gram-negative fish pathogenic bacterium, also responsible for causing opportunistic pathological conditions in humans. It causes a number of diseases in fish due to which the fish industry incurs huge economic losses annually. Due to problems of antibiotic resistance, and the rapidity with which the infection spreads among fishes, vaccination remains the most effective strategy to combat this infection in fish populations. Among various virulence factors associated with bacterial virulence, outer membrane proteins have been widely evaluated for their vaccine potential owing to their surface exposure and related role in pathogenicity. In the present study, we have investigated the immunogenic potential of a non-specific porin, outer membrane protein C (OmpC) whose expression is regulated by the two-component regulatory system and plays a major role in the survival of A. hydrophila under different osmolaric conditions. The full-length gene (~1 kb) encoding OmpC of A. hydrophila was cloned, characterized and expressed in E. coli. High yield (~112 mg/L at shake flask level) of the recombinant OmpC (rOmpC) (~40 kDa) of A. hydrophila was obtained upon purification from inclusion bodies using Ni2+-NTA affinity chromatography. Immunization with purified rOmpC in murine model generated high endpoint (>1:40,000) titers. IgG isotyping, ELISA and ELISPOT assay indicated mixed immune response with a TH2 bias. Also, the anti-rOmpC antibodies were able to agglutinate A. hydrophila in vitro and exhibited specific cross-reactivity with different Aeromonas strains, which will facilitate easy detection of different Aeromonas isolates in infected samples. Taken together, these data clearly indicate that rOmpC could serve as an effective vaccine against different strains of Aeromonas, a highly heterogenous group of bacteria.
Similar content being viewed by others

Introduction
Aeromonas, a motile gram-negative ubiquitous bacterium, belonging to the Aeromonadaceae family, is a major fish pathogen. Diseases caused by A. hydrophila in fish include hemorrhagic septicemia, dermal and ocular ulceration, tail or fin rot, red-sore disease, erythrodermatitis and scale protrusion [1 and references therein]. An opportunistic pathogen in humans, Aeromonas causes gastroenteritis, wound infections, blood-borne dyscrasias and systemic illness [1, 2].
Current strategies to control A. hydrophila infection include use of avirulent strains and heat-killed cells as vaccines, and antibiotics like chloramphenicol, oxytetracycline, chlortetracycline [3, 4]. However, these are not very effective in contained fish cultures. Also the inconsistency in preparation and culturing of pathogenic bacteria are few major concerns that impede their development as effective vaccines. In addition, use of whole bacterium results in increased antigenic load and non-specific immune response upon immunization. On the other hand, use of virulent factors of the pathogen for vaccination allows generation of focused immune response against that specific antigen only, enhancing the efficacy of the immune response. The major virulence factors associated with A. hydrophila infection are surface polysaccharides, toxins (exotoxins and enterotoxins), S-layer and A-layer, proteases, secretion systems and several outer membrane proteins (OMPs) involved in adhesion and multidrug-resistance [5]. Despite successful identification of various virulence factors, a defined vaccine using these virulent factors against the pathogen has not yet been developed. A number of studies have established the role of OMPs in pathogenicity in different bacterial pathogens, and for this reason, they have emerged as attractive candidates for vaccine development due to their exposed antigenic determinant region (epitopes) on the bacterial cell surface [6].
Porins that form channels that are specific for certain molecules such as sugars, or non-specific for the transportation of small solutes, constitute a significant percentage of the outer membrane proteins [7]. The porins that control the transport of solutes across the membrane thus serve a primary barrier for the unwanted agents. Sequence homology among porins of different bacterial study has revealed them to be highly conserved [8]. The OmpF and OmpC are two of the major porins that are involved in transport of molecules across bacterial cell membrane [1]. Expression of the ompC and ompF is differentially regulated by the two-component regulatory system OmpR and envZ in response to change in osmolarity of the environment [9]. Certain E. coli strains that lack some of the major porins such as LamB, OmpA, OmpC and OmpF were found to be more susceptible to β-lactams in comparison to wild type strains, suggesting that these porins also play a role in antibiotic resistance [10].
Role of the EnvZ-OmpR of two-component regulatory system and OmpR-dependent genes has been shown to be instrumental in establishing bacterial virulence in many pathogens [11, 12]. Deletion or mutation of the OmpR and OmpR-dependent genes resulted in avirulent Salmonella typhimurium, respectively, rendered the bacteria avirulent [13, 14]. Vaccination with the OMPs of various organisms has been shown to provide protection in host upon challenge with many pathogenic bacteria including A. hydrophila [15–17]. These studies indicate that vaccine preparations employing porins/OMPs elicit strong immunogenic reactions, directing the generation of antibodies and humoral immune response to prevent the further infection.
Due to their highly conserved nature, role in adhesion, virulence and abundance (~2 % of the total cellular protein), the OmpC and OmpF can be considered as potential contenders for vaccine development against pathogenic bacteria. We have earlier reported that immunization of mice with rOmpF of A. hydrophila produced high titer agglutinating antisera [18]. In the present study, we report recombinant expression, and immunogenic potential of OmpC of A. hydrophila. If found effective, both the rOmpF and rOmpC can be used in combination to elicit effective immune response against A. hydrophila.
Materials and methods
Materials
Escherichia coli DH5α and E. coli BL21 (λDE3) strains were obtained from GIBCO BRL, USA and Novagen, USA, respectively. Swiss albino mice (Female, 4–6 weeks, weighing 15–20 g) were procured from animal house facility of JNU. Mice were given sterile water and fed ad libitum. The usage of animals for the purpose was approved by the Institutional Animal Ethics Committee of the University (IAEC approval # 7/2009).
Aeromonas hydrophila (isolate EUS112) was a kind gift from Dr. I. Karunasagar, College of Fisheries, Mangalore, and other Aeromonas strains were purchased from Microbial Tissue Culture Collection, Chandigarh, India [18]. Expression vector pET28a (+) and pGEMT easy vector were procured from Novagen (USA) and Promega (Germany), respectively. Chemicals required for DNA modification and restriction enzymes were purchased from New England Biolabs, USA. All other chemicals were of analytical grade and were obtained from Sigma-Aldrich Chemical Co., USA, unless stated otherwise. Nitrocellulose membrane (0.45 µM) was procured from Millipore (USA). Primers and oligonucleotides used in the present study were obtained from Sigma-Aldrich Chemical Co., USA.
Generation of 6× His-tagged rOmpC expression construct
Primers for cloning the ompC (without the signal sequence) were designed on the basis of the putative ompC sequence of A. hydrophila strain ATCC 7966 (NCBI Acc. No. CP000462.1). PCR amplification was carried out using the genomic DNA of A. hydrophila (EUS112) as a template and gene-specific primers (Forward 5′ CCAGGATCCACCGTCTACAACCAGAACGACACCAAAC 3′ and reverse 5′ CCAAAGCTTTT AGAAGTTGTACTGCAGGGCCAC 3′) containing BamHI and HindIII restriction enzyme sites (underlined), respectively, and Taq DNA polymerase. The reaction was performed at the following conditions: initial denaturation at 95 °C for 5 min, followed by 30 cycles of thermal denaturation at 95 °C for 1 min; annealing at 55 °C for 1 min; and extension at 72 °C for 1.5 min. Final extension reaction was performed for 7 min at 72 °C. The amplified ompC PCR product was cloned into pGEMT easy vector (Promega, USA), and the putative recombinants were confirmed by restriction enzyme digestion and DNA sequencing (DNA sequencing facility, University of Delhi, South Campus, New Delhi). Subsequently, the ompC insert released by digestion with BamHI and HindIII was cloned into pET 28a (+) pre-digested with BamHI and HindIII. Positive recombinant, confirmed by restriction enzyme digestion of the plasmid DNA and automated DNA sequencing for in frame cloning, was designated as pETAhompC. The recombinant plasmid thus consists of mature ompC gene under the control of T7 promoter.
Analysis of expression and purification of the rOmpC
Expression analysis of the rOmpC was performed essentially as described earlier [18, 19]. Briefly, E. coli BL21 (λDE3) cells harboring pETAhompC were grown till A 600 = 0.8 and induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). The induced culture of E. coli BL 21 (λDE3) cells harboring pETAhompC was harvested at the indicated post-induction time at 8000 rpm for 10 min at 4 °C. The supernatant served as the extracellular fraction. The cell pellet suspended in chilled sucrose buffer (50 mM Tris–HCl, pH 7.5, 10 mM EDTA, and 10 % sucrose) was incubated on ice for 15 min followed by centrifugation at 5000 rpm for 10 min at 4 °C. Pellet obtained was gently resuspended in ice-cold sterile double-distilled water, incubated on ice for 15 min and centrifuged at 5000 rpm for 10 min at 4 °C. Supernatant thus obtained corresponded to the periplasmic fraction, and the pellet was used to prepare the cytoplasmic fraction. For this, the pellet was resuspended in sonication buffer (100 mM Tris–HCl, pH 8.0, 1 mM PMSF and 500 mM NaCl), sonicated at 250 W (30 s pulses) for 5 min using Sonicator (Model XL2020, Misonix Inc., USA) and centrifuged at 13,000 rpm for 30 min at 4 °C. Supernatant collected from this step served as the cytoplasmic fraction containing the soluble proteins. For preparation of the membrane fraction, the pellet obtained in the last step was dissolved in lysis buffer (100 mM Tris–HCl, pH 8.0, 0.1 % Triton X-100) and centrifuged at 5000 rpm at 4 °C for 10 min. The supernatant thus obtained was denoted as membranous fraction. The inclusion bodies present in the pellet were solubilized in 8 M urea buffer (8 M urea, 20 mM Tris–HCl, pH 8.0 and 500 mM NaCl) and collected by centrifugation at 13,000 rpm for 15 min at room temperature (RT). All the fractions were analyzed by SDS-PAGE (12 %).
For purification of rOmpC, inclusion bodies were prepared from the induced E. coli BL21 (λDE3) cells harboring pETAhompC as described earlier [18], solubilized in 6 M GdmCl in Tris-NaCl buffer (10 mM Tris–HCl, pH 8.0, 500 mM NaCl) and centrifuged at 13,000 rpm for 20 min at 4 °C. The supernatant containing solubilized inclusion bodies was subjected to binding with Ni2+-NTA Sepharose slurry at 4 °C for 1 h. After removing the non-specific proteins by washing with wash buffer-I (8 M urea, 20 mM Tris–HCl, pH 8.0, 500 mM NaCl), the bound rOmpC was eluted with elution buffer (wash buffer containing different concentrations of imidazole). The eluted fractions were analyzed by SDS-PAGE (12 %). Fractions showing the presence of rOmpC were pooled and subjected to dialysis against 1× phosphate-buffered saline (PBS) using urea gradient dialysis method [18] and stored in small aliquots at −80 °C until further use. Endotoxin (LPS) levels in the purified rOmpC were determined using Litmus amebocyte lysate using Pierce LAL chromogenic endotoxin quantitation kit (Thermo Scientific, USA) as per the manufacturer’s protocol.
The protein concentration was estimated using BCA protein estimation kit (G Biosciences, USA) as per the manufacturer’s directions.
Western blot analysis
The authenticity of purified rOmpC and the specificity of the anti-rOmpC antisera was determined by Western blot analysis as described earlier [18]. Protein samples resolved on 12 % SDS-PAGE were electrophoretically transferred onto a nitrocellulose membrane (Advanced microdevices, India) in electrode transfer buffer (25 mM Tris–HCl, pH 8.3, 192 mM glycine, and 20 % methanol). The membrane was blocked overnight (O/N) with 2 % BSA in 1× PBS containing 0.05 % Tween-20 (1× PBST). The blot was subjected to three 1× PBST washes of 10 min each at RT between each treatment. The membrane was incubated with primary antibody (anti-His antibody or anti-rOmpC antibody generated in mice) at a dilution of 1:10,000 for 1 h at RT, followed by PBST washes. The blot was further incubated in 1× PBS containing alkaline phosphatase-conjugated secondary antibody (1:5000) for 1 h. After PBST washes, the blot was developed using Western blue-stabilized substrate solution (nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolyl-phosphate p-toluidine salt; Promega, USA). Double-distilled water was used to stop further color development.
Immunization of mice to raise antisera against rOmpC
Female Swiss albino mice (4–6 weeks, n = 5 per group) were used for immunization studies. Pre-immune (control) serum was collected from the retro-orbital plexus vein of eye prior to immunization. Mice were immunized intraperitoneally (i.p.) with 10 and 20 µg of rOmpC (in 100 µl of 1× PBS) emulsified in Complete Freund’s adjuvant, while subsequent boosters with the same dose of antigen were given in Incomplete Freund’s adjuvant on day 14, day 28 and day 42 of primary immunization. Control mice were immunized with corresponding volume of 1× PBS. Mice were bled 1 week after each booster, i.e., on day 21, day 35 and day 49, and antisera collected by centrifugation at 5000 rpm for 10 min at 4 °C were stored in small aliquots at −20 °C until further use.
Enzyme-linked immunosorbant assay (ELISA)
ELISA was performed to determine antibody titer and antibody isotyping, essentially as described earlier [18]. Round-bottom 96-well plates (Nunc, USA) were coated with rOmpC (500 ng/100 µl) and incubated overnight at 4 °C, followed by blocking with 2 % BSA (in 1× PBST) for 2 h at 37 °C. Different dilutions of the anti-rOmpC antisera (prepared in 1× PBS) were added to the wells and incubated for 1 h at 37 °C followed by the addition of secondary antibody (alkaline phosphatase (AP) conjugated IgG). Plate was washed thrice with 1× PBST after each incubation. PNPP substrate (P-nitrophenylphosphate, 1 mg/ml) made in AP buffer (1 mM MgCl2 pH 9.8, 50 mM Na2CO3) was used for color development, and absorbance was read at 450 nm using ELISA reader (Tecan, USA).
The type of immune response generated was determined by antibody isotyping of the anti-rOmpC sera using anti-IgG1, anti-IgG2a, anti-IgG2b secondary antibodies, conjugated with horseradish peroxidase (1:5000). TMB substrate (3,3′, 5,5′-Tetramethylbenzidine, BD biosciences, USA) was used for color development, and the absorbance was measured at 405 nm. The IgM isotype levels in the anti-rOmpC antisera were measured by ELISA using anti-IgM isotype antibodies provided with the Mouse Immunoglobulin Isotyping ELISA Kit (Cat. No. 550487, BD Pharmingen™, USA) as per the manufacturer’s protocol.
Lymphocyte proliferation assay and cytokine ELISA
For lymphocyte proliferation assay, a separate set of animals (n = 3 per group) were subjected to immunization with rOmpC (20 µg/mouse, i.p.) with two booster doses on day 14 and day 28 of primary immunization. Lymphocyte proliferation assay was performed as described essentially by Sharma et al. [19]. Briefly, splenocytes isolated from the spleens of the mice immunized with rOmpC (20 µg/mouse), 7 days post second booster (i.e., on day 35), were treated with chilled ammonium chloride (0.9 %) to lyse the red blood cells. Cells were collected by centrifugation and washed with complete Dulbecco’s modified eagle medium (DMEM, Biological Industries, USA) and resuspended in DMEM medium. Live cell counting was performed using trypan blue dye exclusion assay. Splenocytes (1 × 105 cells/well in 96-well plate, in triplicate for each treatment) were stimulated with rOmpC (20 µg/ml) and incubated at 37 °C in a 5 % CO2 humidified incubator for different times (24–72 h) post-stimulation. Concanavalin A (ConA, 5 µg/ml)-stimulated splenocytes were included as a positive control, and PBS-stimulated splenocytes isolated from both unimmunized and rOmpC-immunized mice comprised the negative control. Lymphocyte proliferation at different time points was measured using MTT assay.
Culture supernatants collected at different time intervals (24, 48 and 72 h) post-treatment from the lymphocyte proliferation assay plates were subjected to cytokine ELISA to assess the levels of secreted IL-4 and IFN-γ using BD cytokine ELISA kit (Becton–Dickinson Pharmingen) as per the manufacturer’s instructions.
Enzyme-linked immunosorbent spot (ELISPOT) assay
One set of splenocytes from the proliferation assay [stimulated with PBS or rOmpC (20 µg/ml) or ConA (5 µg/ml)] was also subjected to ELISPOT assay to determine the numbers of cells secreting IL-4 and IFN-γ using BD pharmingen ELISPOT kit.
Agglutination assay
Qualitative analysis of the agglutination ability of rOmpC antisera with live A. hydrophila cells was performed essentially as described by Sharma et al. [19]. Cultures (O/N) of different bacterial strains were grown at 37 °C. Secondary cultures of the same were inoculated next morning and grown for 5–6 h (till mid-log phase) at 200 rpm at 37 °C. A. hydrophila cells (5 × 108 CFU) from the secondary culture were incubated with the test antisera (pre-immune and anti-rOmpC antisera; 1:200 dilution in 1× PBS) for 1 h at 37 °C. Bacterial cells were collected by centrifugation at 5000 rpm for 10 min and resuspended in 1× PBS. Cell suspension was uniformly smeared on a clean glass slide and air-dried and heat-fixed by passing through flame transiently. The cells were then stained with methylene blue and visualized under microscope (Model Eclipse TE 2000S, Nikon, USA) after washing off the excess stain.
Slot-blot assay for crossreactivity analysis
Cross-reactivity analysis of anti-rOmpC antisera with lysates of different bacterial isolates was performed essentially as described earlier [18]. Purified rOmpC and cell lysate of E. coli BL21 (λ DE3) harboring pETAhompC were included as positive controls. Cell lysates of uninduced E. coli BL21 (λ DE3) harboring pETAhompC, E. coli DH5α cells and Chinese hamster ovary cells (CHO-K1) and BSA were included as negative controls. Cell lysates (1 µg each) of various strains slot blotted onto a nitrocellulose (NC) membrane (Slot blotter, Cleaver Scientific Ltd, UK) were subjected to Western blot analysis using anti-rOmpC antisera. After blocking the non-specific sites with BSA (2 % in 1× PBS) followed by 3 washes of 10 min each with wash buffer (150 mM PBS, pH 7.3, and 0.2 % Tween 20), the NC membrane was incubated with anti-rOmpC antisera (1:5000 in 1× PBS). This was followed by incubation with alkaline phosphatase-conjugated secondary antibody (AP anti-mouse IgG) for 1 h and three washes of 10 min each with wash buffer. Western blue substrate was used for color development.
Statistical analysis
Data were analyzed for statistical significance using Student’s t test, with p value ≤0.05 considered to be statistically significant.
Results
Cloning and sequence analysis of ompC
PCR amplification of the mature ompC using gene-specific primers resulted in the amplification of ~1 kb fragment. Release of an insert of ~1 kb fragment upon digestion of putative ompC clones with BamHI and HindIII confirmed successful cloning of the amplified ompC fragment in pET28a(+) vector. DNA sequencing confirmed the cloned PCR fragment to be ompC gene of A. hydrophila, and the sequence of the cloned ompC has been submitted to GenBank (GenBank Acc. No. HF546053.1).
Amino acid sequence analysis of the encoded product of the cloned ompC gene (1029 base pairs) confirmed the cloned insert to belong to OM-channels super-family of gram-negative bacteria.
The OmpC sequence was aligned with amino acid sequence of OmpC of different bacteria using MUSCLE alignment tool (Supplementary Fig. 1) [20]. Phylogenetic tree constructed using TreeTop-Phylogenetic Tree Prediction made server [21] clearly show three different branches suggesting three discrete clusters (Supplementary Fig. 2). The branch containing Cronobacter sakazakii, Enterobacter cloacae, E. coli, Citrobacter youngae, Buttiauxella agrestis, Trabulsiella guamensis, Shimwellia blattae and Raoultella planticola group of bacteria diverged much earlier and showed very less homology with the cloned OmpC of Aeromonas (<40 %). The cluster containing Aeromonads subspecies showed around 80–90 % similarity with A. hydrophila OmpC. The highest similarity of Aeromonas outside the genus was found with Plesiomonas shigelloides group of bacteria (~55 %). Although OmpC of Vibrio sp. is found to be only 20.68 % similar to OmpC of A. hydrophila, phylogenetic tree placed them very near to each other.
Expression and purification of the rOmpC
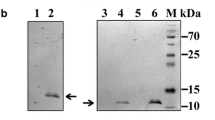
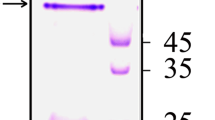
Induction of E. coli BL21 (λDE3) cells harboring pETAhompC with IPTG resulted in expression of the rOmpC in the induced cell lysates only (Fig. 1a). Molecular weight of the recombinant protein from SDS-PAGE was calculated to be ~40 kDa, which was close to predicted molecular weight of the histidine-tagged rOmpC (~41.4 kDa; 343 aa from the ompC gene and 34 aa from the vector). Detection of a single prominent band by immunoblot analysis using monoclonal anti-Histidine antibodies at the expected size in the induced cell lysates only confirmed the expressed protein to be the 6× histidine-tagged rOmpC (Fig. 1b). No band at this position was detected in the uninduced cell lysates. Optimization of inducer concentration and induction time was carried out to maximize the recombinant protein yield. IPTG concentration as low as 0.2 mM was able to induce the expression of rOmpC (Fig. 1c). Further increase in the inducer concentration did not significantly augment the recombinant protein’s expression. Maximum expression of the rOmpC was detected at 6 h post-induction which remained constant thereafter, thus alleviating the possibility of any toxicity associated with the recombinant protein expression (Fig. 1d).

a Analysis of rOmpC expression: Cell lysates (~50 µg each) of the uninduced (lane 1) and induced (lane 2) E. coli BL21(λDE3) cells harboring pETAhompC were analyzed by SDS-PAGE (12 %). M indicates protein molecular weight (kDa) marker. The arrow points to the rOmpC at ~40 kDa position, expressed only in the induced cell lysate (lane 2). b Immunoblot analysis of the rOmpC. The authenticity of the expressed rOmpC was established by Western blot analysis using anti-His antibody. Lanes 1 and 2 depict the induced and the uninduced cell lysates, respectively. A band of ~40 kDa (indicated by arrow) is visible in the induced cell lysate. c Optimization of IPTG concentration for rOmpC expression. Recombinant E. coli BL21 (λDE3) cells harboring pETAhompC were induced with different concentrations of IPTG (shown on top of the panel) for 4 h. Induced cell lysates (50 µg each) were analyzed by SDS-PAGE (12 %). IPTG concentration as low as 0.2 mM could also induce expression of rOmpC. d Time kinetics of the rOmpC expression. IPTG (1 mM) was used to induce the E. coli BL21 (λDE3) cells harboring pETAhOmpC for different time periods and the induced cell lysates (~50 µg each) were analyzed by SDS-PAGE (12 %). The rOmpC expression was found to be maximum at 6 h after which it plateaued. ‘UI’ in (c, d) indicates uninduced cell lysates. e Localization analysis of rOmpC expression. E. coli BL21 (λDE3) cells harboring the pETAhompC were induced with 1 mM IPTG and different cellular fractions were analyzed by SDS-PAGE(12 %). Lane 1 contains induced cell lysate. Lanes 2–6 indicate extracellular, periplasmic, cytoplasmic, membranous, and inclusion bodies fractions, respectively, prepared from the induced cell lysates. The rOmpC expressed predominantly in inclusion bodies (lane 6). f Purification of rOmpC using immobilized Ni2+-NTA affinity chromatography. Lane 1 shows purified rOmpC, eluted with 75 mM imidazole (indicated by the arrow)
Further different subcellular fractions were analyzed by SDS-PAGE to assess the localization of the expressed protein. The rOmpC exclusively expressed as inclusion bodies, and therefore it was purified from the insoluble fraction (inclusion bodies) using Ni2+-NTA affinity chromatography (Fig. 1e). SDS-PAGE analysis of the eluted fraction showed that the rOmpC eluted with 75 mM imidazole. The rOmpC was purified close to >90 % homogeneity (Fig. 1f). MALDI-TOF–MS analysis of the purified rOmpC further substantiated the recombinant protein to be OmpC of A. hydrophila (Supplementary Fig. 3). A substantially high yield of ~112 mg/L of purified rOmpC was obtained at shake flask level. The rOmpC was found suitable for immunization studies as the endotoxin level was determined to be 0.017 pg/µg of purified protein, which falls within the permissible limit of endotoxin in vaccine formulation for mouse immunization studies.
Analysis of immune response against rOmpC
Anti-rOmpC antibody titers were determined by ELISA in the antisera of mice immunized with different doses of rOmpC. The animals immunized with 10 and 20 µg of rOmpC showed an increase in immunoglobulin G (IgG) levels after the first booster followed by a decrease after the second booster (Fig. 2a, b, respectively). However, administration of the third booster dose again resulted in an increase in the IgG levels. The IgG levels were highest after the first booster at both the doses of rOmpC. The endpoint titer in anti-rOmpC antisera from mice immunized with both the doses were found to be >1:40,000.

Determination of antibody titer against rOmpC. Female Swiss albino mice were immunized with a 10 µg and b 20 µg rOmpC, and antisera drawn on different days post-immunization (D.P.I.) were analyzed for the presence of anti-rOmpC antibodies by ELISA. Endpoint titer was found to be >1:40,000 for anti-rOmpC antibodies in the antisera. Each data point represents mean ± standard deviation (SD) from pooled sera samples (n = 5 per experimental group) of two independent experiments, analyzed in triplicates. c Specificity of the anti-rOmpC antisera by immunoblot analysis. Proteins in the uninduced (UI) and induced culture (I) of E.coli BL21 (λDE3) cells harboring pETAhompC resolved by SDS-PAGE (12 %) were subjected to Western blotting using anti-rOmpC antisera (1:10,000). An intense band at the expected size detected only in the induced cell lysate only (lane I) reflects the specificity of anti-rOmpC antisera. M indicates migration of the protein molecular weight (kDa) markers
A prominent immunoreactive band of ~40 kDa was detected only in the induced cell lysates of E. coli BL21 (λDE3) transformed with pETAhompC in Western blot analysis using antisera raised against the rOmpC (Fig. 2c), confirming the specificity of anti-rOmpC antibodies in the antisera.
Antisera collected at different time points post-rOmpC immunization (with 10 and 20 µg) were analyzed for the levels of different isotypes of IgG produced in order to assess the type of immune response. As evident from the Fig. 3, levels of all the three immunoglobulins, namely IgG1, IgG2a and IgG2b, were significantly elevated (p ≤ 0.005) at both the doses. The ratio of IgG1:IgG2a/IgG2b levels was determined to be more than 1, indicating a bias toward TH2 type immune response. An increase in IgM levels was also observed in the rOmpC-immunized mice on different days post-immunization when compared to PBS-immunized mice and maintained positive titers throughout the experimental period. As shown in Fig. 4a, stimulation of sensitized lymphocytes from the spleen of rOmpC-immunized mice with rOmpC resulted in significant increase (p ≤ 0.005) in T-cell proliferation. The splenocytes isolated from control PBS-immunized mice did not show any increase in their proliferation upon stimulation with rOmpC. The proliferation index (PI) for rOmpC (PI = 2.1) was significantly greater than that of the control cells (PI = 1.3).

Antibody isotype profiling of anti-rOmpC antisera. Pooled anti-rOmpC antisera (n = 5 per group) collected at different time intervals post-immunization with a 10 µg and b 20 µg rOmpC were analyzed for the presence of different IgG isotypes and IgM using isotype-specific secondary antibodies. The data are presented as relative absorbance for different isotypes in experimental groups with respect to the control pre-immune sera, wherein the absorbance for pre-immune (control) sera at 405 nm has been normalized to 1. Each data point represents mean ± standard deviation (SD) from pooled sera of mice (n = 5) from each experimental group from two independent experiments, analyzed in triplicates

a In vitro T-cell proliferation by rOmpC. Splenocytes (1 × 105 cells/well, in triplicate) were isolated from Swiss albino mice immunized with rOmpC (20 µg/mouse), a week after the last booster dose and cultured either in the absence or presence of rOmpC (20 µg/ml, stimulated) for 72 h under 5 % CO2 humidified conditions at 37 °C. MTT assay was used to determine cell proliferation index. Cytokine ELISA to analyze T-cell response. IFN-γ (b) and IL-4 (c) levels in the culture supernatants of stimulated and unstimulated splenocytes (from a) were determined by cytokine ELISA at different time points. The data represent mean ± standard deviation (SD) of two independent experiments, performed in triplicates
Cytokine ELISA to analyze the levels of different cytokines (IL-4 and IFN-γ) secreted by the stimulated splenocytes of sensitized mice and control mice at different time points post-rOmpC stimulation showed both the IL-4 and IFN-γ levels to be significantly higher at all the study intervals in the culture supernatant of the sensitized splenocytes. IFN-γ levels in the culture supernatants increased gradually with time from ~7000, 8000 and ~15,000 pg/ml at 24, 48 and 72 h, respectively, while no significant increase was observed in the control cells stimulated with rOmpC (Fig. 4b). Similarly, IL-4 levels continued to rise gradually and were determined to be ~500 pg/ml, significantly higher than that of the control samples (~10 pg/ml) (Fig. 4c). These data suggest that though the antibody isotyping indicated a TH2-biased immune response, the rOmpC-immunized mice activated both the arms of immune response (humoral as well as cell-mediated) evident by a significant increase in the levels of both IFN-γ (TH1 marker) and IL-4 (TH2 marker) when compared to that of the control splenocytes.
These findings were further confirmed by analyzing the splenocyte cell population secreting IFN-γ and IL-4 using ELISPOT assay. ConA and unstimulated splenocytes were taken as positive and negative controls, respectively. A significant increase in the IFN-γ and IL-4 secreting cell populations in vitro was observed when the splenocytes isolated from sensitized mice (isolated from mice immunized with 20 µg rOmpC) were restimulated with the rOmpC (20 µg/ml) when compared to the control unstimulated cells (Table 1). Very high numbers of SFUs secreting IFN-γ (~31,203 ± 189/per 106 cells) were detected in the rOmpC-sensitized splenocytes after restimulation with the rOmpC. The cell population secreting IL-4 from rOmpC-sensitized splenocytes was determined to be 78.33 ± 6.8 SFUs/106 cells. No spots were observed in the unstimulated splenocytes isolated from rOmpC-immunized mice or rOmpC-stimulated splenocytes isolated from the control mice.
Agglutination capacity of anti-rOmpC antisera
Agglutination ability of anti-rOmpC antisera was evaluated by incubating live A. hydrophila cells with anti-rOmpC antisera. Figure 5a clearly depicts that the anti-rOmpC antisera were able to agglutinate the A. hydrophila cells efficiently, whereas no agglutination was observed with E. coli DH5α and Staphylococcus aureus. As expected, incubation of A. hydrophila cells with pre-immune sera did not show any agglutination. Further, loss of agglutinating activity was observed when anti-rOmpC antisera was incubated with rOmpC prior to addition to live A. hydrophila (EUS112) cells, validating specificity of the anti-rOmpC antisera for rOmpC.

a Agglutination assay using the anti-rOmpC antisera. Bacterial cells (5 × 108 CFU) from the live log phase cultures of A. hydrophila, E. coli DH5α and Staphyococcus aureus were incubated with pre-immune serum (PI), anti-rOmpC antisera (1:200) or anti-rOmpC antisera pre-incubated with rOmpC [1.5 µg incubated with 1 ml of antisera (1:200) for 30 min]. Agglutination is visible only in A. hydrophila cells that were incubated with anti-rOmpC antisera, whereas pre-incubation with rOmpC resulted in loss of agglutination activity of anti-rOmpC antisera. Images are taken at 40× magnification. b Cross-reactivity analysis of anti-rOmpC antisera with different Aeromonas isolates/strains: Different Aeromonas cell lysates (1 µg) were immunoblotted with anti-rOmpC antibody (1:5000). The positive control included rOmpC (b14) and induced culture of E.coli BL21 (λDE3) harboring pETAhompC (c7), whereas negative controls included uninduced cell lysate of E.coli BL21 (λDE3) harboring pETAhompC (c8), CHO-K1 cell lysate (c10), E. coli DH5α cell lysate (c11) and BSA (c12). All Aeromonas isolates and the positive control showed positive reaction while none of the negative controls exhibited any crossreactivity
Cross-reactivity analysis of anti-rOmpC antisera
In order to assess the ability of anti-rOmpC antisera to cross-react with different Aeromonas strains, slot-blot analysis was performed using whole cell lysates of different Aeromonas strains/isolates (supplementary Table 1). Figure 5b clearly demonstrates that the anti-rOmpC antiserum cross-reacted with all the A. hydrophila isolates tested. A very strong signal was seen in the positive control, rOmpC (slot b14), and the cell lysate of induced E. coli BL21 (λDE3) harboring pETAhompC (slot c7). No reaction was observed with negative controls including uninduced E. coli BL21 (λDE3) harboring pETAhompC (slot c8), CHO-K1 cell lysate (slot c10), E. coli DH5α cell lysate (slot c11) and BSA (slot c12). The intensity of immunoreactive band was less in some of the Aeromonas isolates, indicating the difference in surface epitopes or relatively less expression of the protein.
Discussion
Aeromonas hydrophila, a member of Aeromonad group, primarily causes systemic illness in poikilothermic animals such as fish. Potentially they could represent a serious problem causing food infections, as many strains are able to grow at temperatures suitable to that of a common refrigerator, at a pH of 4–10 and in presence of higher concentrations of salts [1 and references therein]. The bacterium is also responsible for a variety of infectious complications in immunocompetent and immunocompromised humans [22].
Pathogenicity of Aeromonas is attributed to its ability to produce an array of virulence factors and causing diseases ranging from gastroenteritis to systemic infections. A. hydrophila induces cytotoxicity and massive inflammation in the host during infection and necrosis of tissues [1, 23]. Extensive use of chemotheraputants and antibiotics to combat A. hydrophila infection has led to the development of antibiotic-resistant Aeromonas strains [24]. Though a number of vaccination strategies employing different bacterin preparations have been evaluated [25], no efficient vaccine is currently available to prevent or control the Aeromonas infection. Therefore, a detailed understanding of host responses to Aeromonas virulence factors is of prime importance for developing better treatment strategies.
A crucial step during developing a bacterial infection in primary host is the recognition of exposed antigens, followed by the ingestion of the pathogen by body immune cells. The outer membrane proteins situated on the bacterial cell surface are the primary host receptors recognized by the pathogen to generate an immune response and can therefore act as potential vaccine candidates. Immunogenicity of porins is attributed to various mechanisms such as inhibition of phagocytosis by activating the adenylate cyclase system [26], production of vast array of cytokines [27] and activating the complement system of host [28]. In agreement to this, OMPs from different bacterial strains such as outer membrane protein F (OprF) and outer membrane protein I (OprI) of P. aeruginosa [29], and OmpK of V. anguillarum [30] and Vibrio harveyi [31], have been reported to be highly immunogenic and capable of conferring protection against challenge with the corresponding bacteria.
We have also reported immunogenic and vaccine potential of the OmpF of A. hydrophila, regulated by two-component regulator system, of A. hydrophila. Here we report immunogenic potential of OmpC of A. hydrophila, yet another protein whose expression is under the control of two-component regulatory system in mouse model.
Since the signal sequence present at the N-terminus directs the nascent polypeptide chain to the periplasmic space through the translocon in the inner membrane [32], we cloned the ompC gene region without the region encoding the signal peptide (spanning residues 1–23 amino acids of the protein). The OMPs of bacteria are a highly conserved class of proteins among various bacterial species due to shared motifs of transmembrane β sheets embedded in the outer membrane [7, 33]. BlastP of the cloned mature OmpC showed that the amino acid sequence of OmpC contained several conserved domains common to the OM channel superfamily. Further extraction of the OM channel superfamily confirmed the encoded protein to be OmpC (Fig. 1b), thus authenticating the cloned gene to be ompC of A. hydrophila (EUS112). Outer membrane protein, such as OmpC, is exposed on surface, and multiple interactions with the environment to adapt to various ecological niches lead to genetic diversity among the OmpC of different bacteria. The clustal W alignment using MUSCLE program depicted conservation among OmpC of various Aeromonas subspecies. The OmpC of Plesiomonas group of bacteria showing highest homology with OmpC of A. hydrophila (outside the genus) shares similarity with each other in terms of their pathogenicity and disease symptoms. In spite of very less similarity (<23 %), Vibrio is situated in the same branch as of Aeromonads. Both the Aeromonas and Vibrio are similar in their habitat selection (autochthonous, can be isolated from various water resources) and season-dependent variation in their number (warmer temperature is optimum for their growth and reproduction) [34]. From the phylogenetic tree, it is apparent that the OmpC of A. hydrophila is more closely related to Vibrio family, thereby supporting its previous placement in the family Vibrionaceae [35].
To evaluate the vaccine potential of recombinant OmpC, the AhompC encoding mature OmpC was expressed in the heterologous host (E. coli). The presence of an intense band of the expected size of rOmpC (~40 kDa) exclusively in the induced fraction and its absence in the uninduced cell lysates suggest the absence of leaky expression, thereby indicating the tight regulation of expression associated with the T7 expression system. Removal of the signal peptide from rOmpC is expected to inhibit its translocation to the outer membrane, thus making purification of the recombinant protein much less cumbersome. A significantly high percentage of the rOmpC expression in the induced cell lysate points toward efficient transcription and translation of the cloned ompC gene by E. coli cells. As expected, the rOmpC expressed in the insoluble fraction, as over-expression of the OMPs without their signal sequence usually forms inclusion bodies due to improper folding in the cytoplasm [32]. Earlier studies have also reported the expression of OMPs in insoluble fraction due to their high expression or the absence of signal peptide [36, 37].
Outer membrane proteins purified from inclusion body fraction tend to aggregate easily due to exposed hydrophobic regions (and improper folding) [38]. Since urea gradient dialysis method could successfully refold rOmpF of A. hydrophila [18], same strategy was adopted for refolding of the purified rOmpC. This ensured proper folding of the protein with minimal possibility of reaggregation. Absence of any cysteine in the encoded OmpC minimized the probability of improper disulfide bond formation, and hence urea gradient dialysis without any redox couple (GSH-GSSG) was found to be suitable to restrain aggregation. The rOmpC protein was purified to almost 98 % near homogeneity with a yield of 37.3 mg/g wet cell weight (~112 mg/L) at shake flask level. The high yield of rOmpC observed was better than that of many T7 expression system-based purification [recombinant PorB (~1.1 mg/L) and PorA (30 mg/L or 30 mg/4.7 g wet cell wt) of Neisseria meningitidis [37, 39] ]. Long-term storage of the rOmpC both at 4 and −20 °C did not result in aggregation, which is a major bottleneck in the production of recombinant outer membrane protein due to the presence of hydrophobic region. This suggests that the protein has been refolded in a stable conformation. Thus, the strategy adopted to obtain maximum yield of rOmpC in soluble form in the present study can be employed for recombinant expression of other OMPs and thus can enhance the yield of recombinant protein, required in large amounts for vaccination purposes.
Evaluation of immunogenic and vaccine potential of the rOmpC in murine model clearly demonstrated it to be highly immunogenic, evident from a very high antigen-specific antibody response. These results are in line with earlier reports on highly immunogenic nature of OMPs [40–43]. A robust antibody response observed after primary booster as compared to secondary and tertiary booster can be attributed to the prevalent humoral immune response only after first booster, while subsequent boosters led to activation of cell-mediated immune response as well. Increase in antibody titer in response to immunization with outer membrane proteins Omp18 of Campylobacter jejuni and OmpW of V. alginolyticus has earlier been reported [15, 40]. Generation of protective antibodies reported upon passive immunization of OmpA of H. parasuis further potentiates the ability of outer membrane proteins in developing a state of protective immunity post-immunization [42]. In line with the above studies, high antibody titers obtained with rOmpC immunization clearly demonstrated it to be highly immunogenic and capable of invoking significant antigen-specific immune response.
Analysis of levels of different IgG isotypes [IgG1, indicative of humoral (TH2) immune response; IgG2a and IgG2b, indicative of cell-mediated (TH1) immune response] revealed generation of mixed immune response, with a bias toward humoral response, upon rOmpC immunization. Determination of IgM isotype also showed an increase in rOmpC-immunized mice. Since the isotype levels were measured only after first booster, the increase observed in IgM titers was not to the extent as observed with IgG1 and IgG2a. This is expected as IgM is produced in response to primary infection, and subsequent exposure to the pathogen (booster) results in memory response and high IgG titers resulting from switch from IgM to IgGs subtypes depending on the type of immune response elicited [44–46]. Similar IgG/IgM profiles have been reported by Chen et al. [47] in mice upon infection with Babesia microti, wherein IgM levels were at their maximum at initial stages of infection on day 10, which fell off thereafter but remained positive at low titer, whereas the IgG levels were found to be maximum at later time on day 24 post-infection.
This was further validated by evaluating the levels of representative cytokines for cellular immunity, namely IFN- γ (TH1) and IL-4 (TH2) by both cytokine ELISA and ELISPOT assay. An increased proliferation of the splenocytes isolated from the rOmpC-immunized mice upon in vitro restimulation with the rOmpC indicated that immunization with rOmpC was able to generate T-cell memory. Increased levels of both IFN-γ and IL-4 in the culture supernatants of the stimulated splenocytes of the rOmpC-immunized mice, and ELISPOT analysis of IFN- γ- and IL-4-secreting cell populations, further confirmed mixed immune response. Our findings are in line with the previous study carried out by various researchers. Schwenteit et al. [43] reported induction of TH2 response after infection with A. salmonicida subsp. achromogenes. The OmpX of E. coli exhibits immunogenic properties and induces a TH2-biased mixed immune response [41]. Similarly, the OmpA of many bacteria has been shown to activate various immune cells, such as dendritic cells and macrophages which act as antigen presenting cells and also activate a range of other cells to produce cytokines to bring out strong humoral response is well thought out to be an immunogenic protein [48] and references therein. Another study involving OmpC and OmpF of pathogenic E. coli showed that the immunization of mice with the formulations of these 2 porins elicited high titers of IgG2a which resulted in induction of a significant TH1/TH2 immune responses [10]. Studies have confirmed that extracellular pathogens are cleared efficiently by mixed or humoral immune response of the host [49]. Since Aeromonas is an extracellular pathogen, the generation of antibodies (part of humoral immunity/TH2 immune response) will undoubtedly help in the clearance of Aeromonas by opsonization, followed by phagocytosis of the said bacteria by macrophages and neutrophils. Other methods that can be adopted by host are co-association of antibodies with complement system leading to bacterial killing by antibody-dependent cell cytotoxicity (ADCC) or directly through opsonization and endocytosis by B-cells which can provide long-time immunity by activating effector and plasma cells. Earlier reports have shown that vaccines which activate both the arms of immune response in animal models provided better protection in comparison with those which elicited either TH1 or TH2 immune response [49]. Thus, the rOmpC which induce mixed immune response would make an efficient vaccine against A. hydrophila infection. On the other hand, another member of two-component regulatory system of A. hydrophila, recombinant outer membrane protein F, rOmpF, showed predominantly TH1 type of immune response in murine model [18]. The activation of different types of immune response with different outer membrane proteins clearly indicates the variation in antigen processing and presentation by host immune cells. Therefore, combination of both rOmpF and rOmpC can be assessed as an effective vaccine against A. hydrophila.
To assess the neutralizing ability of the anti-rOmpC antisera, in vitro agglutination assay using live A. hydrophila cells was performed. Agglutination assays have been used for the identification of bacterial strains [50, 51] during infectious/diseased conditions. In the host, agglutination of bacteria or any foreign organism is subjected to complement mediated lysis, thus pointing toward the role of agglutination in the host immune defense mechanism [52]. Positive and significant agglutination of the live A. hydrophila with the anti-rOmpC antisera clearly demonstrates neutralizing potential of the anti-rOmpC antibodies present in the antisera. Loss of agglutination ability of anti-rOmpC antisera upon pre-incubation with the rOmpC clearly indicated the specificity of interaction between the antibodies present in the antisera and the rOmpC on the bacterial cells. Specific agglutination of A. hydrophila cells by the anti-rOmpC antibodies also suggests its potential use in diagnosis. Since Aeromonas is a heterogeneous group of bacteria, it is desirable that the potential vaccine candidate generates an immune response which is able to protect against variety of strains of the bacterium. This is primarily demonstrated by the ability of the antisera to recognize antigens present on different strains of the bacterium. In the present study, the anti-rOmpC antisera were able to interact with the whole cell lysates of a number of A. hydrophila strains (as shown by the slot-blot analysis), indicating that the anti-rOmpC antisera were able to recognize different strains of Aeromonas, exhibiting broad cross-reactivity. Since A. hydrophila is a fish pathogen, and the immune system of mouse and fish differ with each other, preliminary analysis of the antigenic potential of the rOmpC in Labeo rohita (an Indian major carp) indicated that the rOmpC was able to generate significant immune response in L. rohita and the antisera were found to have endpoint titers of ~1:600 (data not shown). The difference in the antibody titers obtained in L. rohita and mice in the present study need to be analyzed in context of the differences in the immune system of the two organisms and investigated further.
Thus, the present study reports a detailed analysis of the immune response generated by rOmpC of A. hydrophila in murine model. High yield of purified rOmpC, its stability over long-term storage, mixed immune response generation, along with the high neutralizing potential of the antisera generated post-immunization in mice model indicate the potential of rOmpC of A. hydrophila to be used as a successful vaccine candidate. Since the rOmpC and rOmpF resulted in activation of TH2- and TH1-biased immune response, respectively, a vaccine preparation comprising both these antigens could be further evaluated. This would be expected to activate both arms of the immune system and thus is likely to be more efficacious.
References
Janda JM, Abbott SL. The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin Microbiol Rev. 2010;23:35–73.
Austin B, Altwegg M, Gosling PJ, Joseph S. The genus aeromonas. London: Wiley; 1996. p. 151–73.
Petersen A, Dalsgaard A. Antimicrobial resistance of intestinal Aeromonas spp. and Enterococcus spp. in fish cultured in integrated broiler-fish farms in Thailand. Aquaculture. 2003;19:71–82.
Rigos G, Troisi G. Antibacterial agents in Mediterranean finfish farming: a synopsis of drug pharmacokinetics in important Euryhaline fish species and possible environmental implications. Rev Fish Biol Fish. 2005;15:53–73.
Zhang YL, Ong CT, Leung KY. Molecular analysis of genetic differences between virulent and avirulent strains of Aeromonas hydrophila isolated from diseased fish. Microbiology. 2000;146:999–1009.
Achouak W, Heulin T, Pagès JM. Multiple facets of bacterial porins. FEMS Microbiol Lett. 2001;199:1–7.
Nikaido H. Porins and specific diffusion channels in bacterial outer membranes. J Biol Chem. 1994;296:3905–8.
Jeanteur D, Lakey JH, Pattus F. The bacterial porin superfamily: sequence alignment and structure prediction. Mol Microbiol. 1991;5:2153–64.
Stock JB, Ninfa AJ, Stock AM. Protein phosphorylation and regulation of adaptive responses in bacteria. Microbiol Rev. 1989;53:450–90.
Liu YF, Yan JJ, Lei HY, Teng CH, Wang MC, Tseng CC, Wu JJ. Loss of outer membrane protein C in Escherichia coli contributes to both antibiotic resistance and escaping antibody-dependent bactericidal activity. Infect Immun. 2012;80:1815–22.
Bernardini ML, Fontaine A, Sansonetti PJ. The two-component regulatory system ompR-envZ controls the virulence of Shigella flexneri. J Bacteriol. 1990;172:6274–81.
Pukklay P, Nakanishi Y, Nitta M, Yamamoto K, Ishihama A, Shiratsuchi A. Involvement of EnvZ-OmpR two-component system in virulence control of Escherichia coli in Drosophila melanogaster. Biochem Biophys Res Commun. 2013;438:306–11.
Dorman CJ, Chatfield S, Higgins CF, Hayward C, Dougan G. Characterization of porin and ompR mutants of a virulent strain of Salmonella typhimurium: ompR mutants are attenuated in vivo. Infect Immun. 1989;57:2136–40.
Turner AK, Terry TD, Sack DA, Londono-Arcila P, Darsley MJ. Construction and characterization of genetically defined aro omp mutants of enterotoxigenic Escherichia coli and preliminary studies of safety and immunogenicity in humans. Infect Immun. 2001;69:4969–79.
Qian R, Chu W, Mao Z, Zhang C, Wei Y, Yu L. Expression, characterization and immunogenicity of a major outer membrane protein from Vibrio alginolyticus. Acta Biochim Biophys Sin. 2007;39:194–200.
Guan R, Xiong J, Huang W, Guo S. Enhancement of protective immunity in European eel (Anguilla anguilla) against Aeromonas hydrophila and Aeromonas sobria by a recombinant Aeromonas outer membrane protein. Acta Biochim Biophys Sin. 2011;43:79–88.
Maiti B, Shetty M, Shekar M, Karunasagar I, Karunasagar I. Recombinant outer membrane protein A (OmpA) of Edwardsiella tarda, a potential vaccine candidate for fish, common carp. Microbiol Res. 2011;167:1–7.
Yadav SK, Sahoo PK, Dixit A. Characterization of immune response elicited by the recombinant outer membrane protein OmpF of Aeromonas hydrophila, a potential vaccine candidate in murine model. Mol Bio Rep. 2014;41:1837–48.
Sharma M, Dixit A. Identification and immunogenic potential of B cell epitopes of outer membrane protein OmpF of Aeromonas hydrophila in translational fusion with a carrier protein. Appl Microbiol Biotechnol. 2015;99:6277–91.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Brodsky LI, Ivanov VV, Kalaydzidis Ya L, Leontovich AM, Nikolaev VK, Feranchuk SI, Drachev VA. GeneBee-NET: internet-based server for analyzing biopolymers structure. Biochemistry. 1995;60:923–8.
Abuhammour W, Hasan RA, Rodgers D. Necrotizing fasciitis caused by Aeromonas in the immunocompromised individual. Pediatr Emerg Care. 2006;22:48–51.
Galindo CL, Gutierrez C Jr, Chopra AK. Potential involvement of galectin-3 and SNAP23 in Aeromonas hydrophila cytotoxic enterotoxin-induced host cell apoptosis. Microb Pathog. 2006;40:56–68.
Saavedra MJ, Guedes-Novais S, Alves A, Rema P, Tacao M, Correia A. Resistance to beta-lactam antibiotics in Aeromonas hydrophila isolated from rainbow trout (Oncorhynchus mykiss). Int Microbiol. 2004;7:207–11.
Poolman JT. In: Cohen S, Shafferman A, editors. Novel strategies in design and production of vaccines. New York: Plenum Press; 1996. p. 73–7.
Di Donato A, Draetta GF, Illiano G, Tufano MA, Sommese L, Galdiero F. Do porins inhibit the macrophage phagocyting activity by stimulating the adenylate cyclase? J Cyclic Nucleotide Protein Phosphor Res. 1986;11:87–97.
Galdiero F, L’ero GC, Benedetto N, Galdiero M, Tufano MA. Release of cytokines induced by Salmonella typhimurium porins. Infect Immun. 1993;61:155–61.
Galdiero F, Tufano MA, Sommese L, Folgore A, Tedesco F. Activation of complement system by porins extracted from Salmonella typhimurium. Infect Immun. 1984;46:559–63.
Von Specht BU, Domdey H, Schödel F, Blum B, Lücking C, Knapp B, Muth G, Hungerer KD, Bröker M. Outer membrane proteins of Pseudomonas aeruginosa as vaccine candidates. Behring Inst Mitt. 1994;95:85–96.
Hamod MA, Nithin MS, Shukur YN, Karunasagar I, Karunasagar I. Outer membrane protein K as a subunit vaccine against V. anguillarum. Aquaculture. 2012;354:107–10.
Li Ningqiu, Bai Junjie, Shuqin Wu, Xiaozhe Fu, Lao Haihua, Ye Xing, Shi Cunbin. An outer membrane protein, OmpK, is an effective vaccine candidate for Vibrio harveyi in Orange-spotted grouper (Epinephelus coioides). Fish Shellfish Immunol. 2008;25:829–33.
Bannwarth M, Schulz GE. The expression of outer membrane proteins for crystallization. Biochim Biophys Acta. 2003;1610:37–45.
Tommassen J. Assembly of outer-membrane proteins in bacteria and mitochondria. Microbiology. 2010;156:2587–96.
Evans AS, Brachman PS. Bacterial infections of humans: epidemiology and control. 3rd ed. New York: Springer; 1998.
Véron M La. position taxonomique des Vibrio et de certaines bactéries comparables. C R Acad Sci Paris. 1965;261:5243–6.
Jolley KA, Appleby L, Wright JC, Christodoulides M, Heckels JE. Immunization with recombinant Opc outer membrane protein from Neisseria meningitidis: influence of sequence variation and levels of expression on the bactericidal immune response against meningococci. Infect Immun. 2001;69:3809–16.
Wright JC, Williams JN, Christodoulides M, Heckels JE. Immunization with the recombinant PorB outer membrane protein induces a bactericidal immune response against Neisseria meningitidis. Infect Immun. 2002;70:4028–34.
Villaverde A, Carrio M. Protein aggregation in recombinant bacteria: biological role of inclusion bodies. Biotechnol Lett. 2003;25:1385–95.
Christodoulides M, Brooks JL, Rattue E, Heckels JE. Immunization with recombinant class 1 outer-membrane protein from Neisseria meningitidis: influence of liposomes and adjuvants on antibody avidity, recognition of native protein and the induction of a bactericidal immune response against meningococci. Microbiology. 1998;144:3027–37.
Burnens A, Stucki U, Nicolet J, Frey JJ. Identification and characterization of an immunogenic outer membrane protein of Campylobacter jejuni. Clin Microbiol. 1995;33:2826–32.
Maisnier-Patin K, Malissard M, Jeannin P, Jean-Francois H, Jean-Claude C, Hoeffel G, Jean-Francois G, Nguyen T, Saez JM, Delneste Y. The outer membrane protein X from Escherichia coli exhibits immune properties. Vaccine. 2003;2:3765–74.
Tian H, Fu F, Li X, Chen X, Wang W, Lang Y, Cong F, Liu C, Tong G, Li X. Identification of the immunogenic outer membrane protein A antigen of Haemophilus parasuis by a proteomics approach and passive immunization with monoclonal antibodies in mice. Clin Vaccine Immunol. 2011;18:1695–701.
Schwenteit JM, Breithaupt A, Teifke JP, Koppang EO, Bornscheuer UT, Fischer U, Gudmundsdottir BK. Innate and adaptive immune responses of Arctic charr (Salvelinus alpinus, L.) during infection with Aeromonas salmonicida subsp. achromogenes and the effect of the AsaP1 toxin. Fish Shellfish Immunol. 2013;35:866–73.
Brinkman DM, Jol-van der Zijde CM, ten Dam MM, Vossen JM, Osterhaus AD, Kroon FP, van Tol MJ. Vaccination with rabies to study the humoral and cellular immune response to a T-cell dependent neoantigen in man. J Clin Immunol. 2003;23:528–38.
Siegrist CA. Section 1: General aspects of vaccination. In: Plotkin SA, Orenstein WA, Offit PA, editors. Vaccine immunology. 5th ed. New York: Elsevier; 2008. p. 17–36.
Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–92 and reference therein].
Chen D, Copeman DB, Burnell J, Hutchinson GW. Helper T cell and antibody responses to infection of CBA mice with Babesia microti. Parasite Immunol. 2000;22:81–8.
Pore D, Mahata N, Pal A, Chakrabarti MK. Outer member protein A (OmpA) of Shigella flexneri 2a, induces protective immune response in a mouse model. PLoS ONE. 2011;6:e22663. doi:10.1371/journal.pone.0022663.
Spellberg B, Edwards JE. Type1/type2 immunity in infectious diseases. Clin Infect Dis. 2001;32:76–102.
Kronvall G. Rapid slide-agglutination method for typing Pneumococci by means of specific antibody adsorbed to protein A-containing staphylococci. J Med Microbiol. 1973;6:187–90.
Svenungsson B, Linberg AA. Identification of Salmonella bacteria by co-agglutination, using antibodies against synthetic disaccharide-protein antigens O2, O4 and O9, adsorbed to protein A-containing staphylococci. Acta Pathol Microbiol Scand B. 1978;86:283–90.
Goldsby RA, Kindt TJ, Kuby J, Osborne BA. Immunology, chapter 6. 5th ed. London: W. H. Freeman; 2002. p. 90–145.
Acknowledgments
The Council of Scientific and Industrial Research, New Delhi, and the University Grants Commission are gratefully acknowledged for research fellowship to SKY and MS. The work is supported by research grant from the Department of Biotechnology, New Delhi (BT/AAQ/Indo-Norway/183204/2007), and DBT-BUILDER grant to JNU (BT/PR5006/INF/22/153/2012). Authors acknowledge Dr. I. Karunasagar, College of Fisheries, Mangalore, India, and National Bureau of Fish Genetic Resources (NBFGR), Lucknow, for providing Aeromonas isolates.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yadav, S.K., Meena, J.K., Sharma, M. et al. Recombinant outer membrane protein C of Aeromonas hydrophila elicits mixed immune response and generates agglutinating antibodies. Immunol Res 64, 1087–1099 (2016). https://doi.org/10.1007/s12026-016-8807-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-016-8807-9